Abstract
Importance of the field: Chronic endobronchial infection in cystic fibrosis (CF) leads to progressive lung function loss and respiratory failure. Most adult CF patients are infected with Pseudomonas aeruginosa, an important predictor of mortality. Suppressing chronic P. aeruginosa infection with inhaled antibiotics is standard of care for CF patients.
Areas covered in this review: This review describes the development (2003 – 2010) of aztreonam lysine 75 mg powder and solvent for nebulizer solution (AZLI; Cayston®), an aerosolized formulation of the monobactam antibiotic aztreonam.
What the reader will gain: AZLI was studied in patients with CF and chronic P. aeruginosa airway infection. In placebo-controlled trials, AZLI improved respiratory symptoms, increased forced expiratory volume in 1 sec (FEV1), decreased sputum P. aeruginosa density, and was well tolerated. An open-label follow-on trial of nine ‘on/off’ courses showed that AZLI was safe and the effect durable with repeated administration. AZLI was recently approved for use in CF patients in Australia and the USA, and conditionally approved in Canada and the European Union. AZLI is given three times daily for 28 days (2 – 3 min/dose), followed by 28 days off-drug. AZLI is used only with the Altera Nebulizer System™, which provides appropriate particle size and small airway deposition, and has excellent portability.
Take home message: AZLI is a new therapy that is safe and effectively improves respiratory symptoms and FEV1 in patients with CF.
1. Introduction
Cystic fibrosis (CF) is a severe, autosomal-recessive, multi-system disease characterized by chronic, progressive sino-pulmonary disease, malabsorption and male infertility Citation[1]. Abnormal concentrations of sodium and chloride in the airway surface liquid impair mucociliary clearance, leading to inflammation, airway obstruction and chronic bacterial infections. Progressive bronchiectasis due to chronic infection is the primary cause of morbidity and early mortality Citation[2-6]. Although there is no therapy available to treat the underlying defect in CF, a better understanding of the disease and advances in therapies have increased the median age of death from < 5 years in the 1960s to the late 20s in 2007; however, age of death varies widely between countries Citation[2,6-8].
For individuals with CF, respiratory symptoms are frequent and include cough, production of thick, purulent sputum, chest tightness or wheezing, and limitation of exercise capacity. Intermittent pulmonary exacerbations are characterized by increased respiratory and systemic symptoms and a reduction in FEV1, only some of which is recovered with intensive treatment Citation[6,9,10]. Pulmonary exacerbations can occur many times a year, are more frequent as the disease progresses, particularly in individuals chronically infected with Pseudomonas aeruginosa and are the primary cause of hospitalization for CF patients Citation[6]. In 2007, 38% of CF patients in the USA experienced at least one acute pulmonary exacerbation requiring treatment with a course of intravenous (i.v.) antibiotics Citation[2]. Most CF patients in the USA are hospitalized in response to a worsening of symptoms. By contrast, some CF care centers (e.g., Copenhagen) hospitalize patients proactively even when symptoms are unchanged, to administer parenteral antibiotics to reduce bacterial load in the lung Citation[11]. Individuals with CF can be infected with a range of bacteria, viruses and fungi. There is increasing evidence that infection is polymicrobial in the majority of patients, but the infection most often associated with reduced survival is P. aeruginosa Citation[6,12-15].
Pseudomonas aeruginosa is a Gram-negative, biofilm-producing pathogen that occurs in more than 25% of US CF patients by the age of 6 years and in approximately 70% of patients over the age of 18 years Citation[2]. Chronic P. aeruginosa airway infections are rarely eradicated, although intensive treatment of new infections seems to delay chronic infection Citation[16-19]. Many other pathogens also chronically infect the airways of CF patients, including Haemophilus influenza, Staphylococcus aureus, Stenotrophomonas maltophilia and Burkholderia cepacia complex Citation[12-14].
2. Overview of antibiotic treatments for CF
Treatment of CF-related lung disease involves both chronic maintenance therapy and treatment of exacerbations. Current guidelines for chronic maintenance therapy include daily airway clearance, mucolytics such as dornase alpha and hypertonic saline, and anti-inflammatory medications such as daily azithromycin Citation[20,21]. In addition, recent US and European guidelines strongly recommend the use of chronic inhaled anti-pseudomonal antibiotics Citation[20,21]. Treatment guidelines for acute exacerbations differ from maintenance guidelines by emphasizing the use of parenteral antibiotics for exacerbations Citation[22,23].
Inhaled antibiotics are a keystone of maintenance treatment. The advantage of aerosolized antibiotics for the suppression of chronic airway infection is delivery to the site of infection in high concentrations and with minimal systemic absorption. Potential disadvantages of aerosolized antibiotics include an irritant effect on airways, uneven deposition in the lungs, and the time involved in preparing and administering them. In the 1990s, tobramycin inhalation solution (TIS, TSI, TNS, TOBI®; Novartis Pharmaceuticals Corp., East Hanover, NJ, USA; Bramitob®; Trinity-Chiesi Pharmaceuticals Ltd, Cheadle, UK) was introduced, accompanied by significant data to support its use as chronic, intermittent therapy when aerosolized via a jet nebulizer Citation[24-27]. Colistimethate sodium (CMS; Colomycin®; Forest Laboratories, UK; Promixin®; Profile Pharma Ltd, UK), an inactive prodrug that is hydrolyzed to colistin, was approved in 2003 as an inhaled formulation in the UK Citation[28-30]. Aerosolization of parenteral antibiotics such as gentamicin, ceftazidime or carbenicillin is used occasionally despite lack of multicenter studies to support this practice Citation[31]. In recent months, aztreonam lysine 75 mg powder and solvent for nebulizer solution (AZLI, Cayston®; Gilead Sciences, Inc., Foster City, CA, USA) was approved for use in patients with CF and chronic P. aeruginosa infection in Australia and the USA and conditionally approved in Canada and the European Union, pending the results of studies to verify clinical benefit () Citation[32-34].
Box 1. Drug summary.
3. Aztreonam lysine 75 mg powder and solvent for nebulizer solution (AZLI)
3.1 Chemistry and mechanism of action
The chemical formula of aztreonam is (Z)-2-[[[(2-amino-4-thiazolyl)[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]carbamoyl]methylene]amino]oxy]-2-methylpropionic acid Citation[35]. It is a white, crystalline, odorless powder with a molecular weight of 435.4 and is essentially insoluble in water Citation[36]. AZLI is lyophilized aztreonam lysine reconstituted with sterile 0.17% saline diluent. The parenteral formulation of aztreonam (Azactam®; Elan Pharmaceuticals, Inc., South San Francisco, CA, USA) contains arginine; this was substituted with lysine for AZLI because inhaled arginine results in severe cough and airway inflammation in patients with CF Citation[35,37].
Aztreonam binds to the penicillin-binding protein (PBP)-3 of aerobic Gram-negative bacteria, inhibits cell wall biosynthesis, and demonstrates time-dependent killing of sensitive Gram-negative bacteria Citation[35,38-40]. Thus the speed or degree of bacterial killing are not substantially improved by increasing the concentration of aztreonam above the minimum inhibitory concentration (MIC) for P. aeruginosa but are improved by increasing the total time that bacteria are exposed to the drug. Aztreonam does not induce chromosomally encoded beta lactamases and is stable to hydrolysis by most bacterially produced beta lactamases Citation[41,42]. There is a low incidence of allergic cross-reactivity with beta lactams such as penicillin, cephalosporins or carbapenems, and immunogenic reactions to aztreonam are rare Citation[43].
As an aerosol, AZLI has a relatively mono-dispersed particle spectrum well suited for lower airway drug deposition (mass median diameter: 3.8 μM). Drug delivery time in clinical trials averaged approximately 2 min for a 1-ml dose (75 mg AZLI). Aerosolized drugs should be used in devices in which they have been studied, as devices may differ in the amount of drug delivered and the deposition pattern within the airway Citation[44].
3.2 Pharmacokinetics and metabolism
AZLI is delivered directly to the airways, where high concentrations are achieved in sputum. Systemic absorption is low but occurs quickly. In pharmacokinetic studies, peak aztreonam plasma concentrations were observed 0.6 – 2.0 h after a 75-mg dose and plasma half-life was 2.1 h. Sputum aztreonam concentrations were approximately 1000-fold higher than the corresponding average plasma concentrations, reflecting direct deposition of aztreonam to the lung. Sputum concentrations exceeded the P. aeruginosa MIC50 (minimum concentration inhibiting 50% of P. aeruginosa isolates) for at least 4 h Citation[45]. The antimicrobial activity of aztreonam was not inhibited by sputum. In the Phase III placebo-controlled trials (AIR-CF1, AIR-CF2), the average sputum level 10 min after a single dose of 75 mg AZLI was 726 μg/g sputum; this was more than 10 times larger than the aztreonam MIC90 for all P. aeruginosa isolates obtained at baseline from the AZLI treatment group (64 μg/ml) Citation[46,47].
There was no evidence of aztreonam accumulation in sputum or plasma over 28 days of dosing; average peak plasma concentrations achieved after 75 mg AZLI administered three times daily were 0.55 μg/ml (day 0), 0.67 μg/ml (day 14) and 0.65 μg/ml (day 28; study AIR-CF1) Citation[46]. These levels were much lower than those achieved after i.v. infusion of the parenteral formulation of aztreonam arginine; peak serum concentrations of 54 μg/ml (500-mg dose), 90 μg/ml (1-g dose) and 204 μg/ml (2-g dose) were reported immediately after i.v. dosing Citation[35]. Parenterally administered aztreonam is primarily excreted in the urine (equal mix of active tubular secretion and glomerular filtration); in healthy subjects, excretion is complete within 12 h after dosing Citation[35].
3.3 Dosing and administration
The dose of AZLI is 75 mg, three times daily for 28 days, followed by 28 days off-drug Citation[32-34]. Doses of AZLI should be separated by at least 4 h. Use of a bronchodilator is recommended before AZLI administration Citation[32-34]. The drug is provided in powdered form, which is reconstituted immediately before use. AZLI is administered with the battery-powered, portable Altera Nebulizer System™ (PARI Innovative Manufacturers, Midlothian, VA, USA; PARI GmbH, Munich, Germany), which uses a vibrating perforated membrane to generate an aerosol. Total time for administration is 2 – 3 min per dose.
4. Clinical efficacy of AZLI
4.1 Overview of AZLI clinical trials
Administration of AZLI has been studied in five placebo-controlled and one open-label trial: two Phase I trials of safety and pharmacokinetics (CP-AI-001 and CP-AI-002) Citation[45,48], one Phase II trial of safety and efficacy (CP-AI-003) Citation[49], two Phase III trials of safety and efficacy (AIR-CF1 (CP-AI-007) and AIR-CF2 (CP-AI-005)) Citation[46,47]; and one open-label follow-on trial that enrolled patients from both Phase III placebo-controlled trials (AIR-CF3 (CP-AI-006)) () Citation[50-53].
Table 1. Summary of Phase II and Phase III AZLI clinical trials.
4.2 Efficacy end points employed in AZLI clinical trials
The Phase II and III AZLI clinical trials used the following outcome measures.
FEV1: In CF, forced expiratory volume in one second (FEV1) is a measure that reflects the severity of airway disease. Results are reported as percentage change from baseline values (L), which are measured before study drug initiation. Treatment effect is defined as the difference between AZLI and placebo treatment groups.
CFQ-R Respiratory Symptoms Score: The Cystic Fibrosis Questionnaire – Revised (CFQ-R) is a disease-specific questionnaire measuring health-related quality of life for children, adolescents and adults with CF Citation[54-56]. At present, it is the only validated patient-reported outcome (PRO) measure for both child and adult patients with CF Citation[57]. It has been validated across age groups, translated into more than 30 languages and meets the FDA 2009 guidance for PRO tools Citation[58]. The CFQ-R has 12 scales that measure different aspects of health-related quality of life, including respiratory symptoms, physical functioning and vitality. The minimal clinically important difference (MCID) is an indicator of the clinical relevance of change on measurement scales Citation[59-64]. For the CFQ-R, a difference of four points on the Respiratory Symptoms Scale was validated as the MCID; a more conservative MCID of five points was used to interpret the results of the placebo-controlled, Phase III AZLI trials Citation[46,47,65].
Time-to-need for inhaled or i.v. antipseudomonal antibiotics: This end point was used in the AIR-CF2 trial, which was an event-driven study Citation[47]. Investigators used the presence of one or more predefined symptoms predictive of pulmonary exacerbation to decide if patients needed additional antibiotics; symptoms included decreased exercise tolerance, increased cough, increased sputum production/chest congestion or decreased appetite Citation[9]. The start day of additional inhaled or i.v. antipseudomonal antibiotics defined the event.
Pseudomonas aeruginosa bacterial density in sputum: Pseudomonas aeruginosa bacterial density is calculated as the log10 value for the number of P. aeruginosa colony-forming units (CFUs) per gram of expectorated sputum.
Measures of safety and health: Safety data and general health markers, such as weight, were collected.
4.3 Phase III AZLI trials
4.3.1 AIR-CF1
AIR-CF1 (CP-AI-007) was a randomized, double-blind, placebo-controlled, international study of 28 days' treatment with 75 mg AZLI or placebo, dosed three times daily () Citation[46]. The primary end point was change in CFQ-R Respiratory Symptoms score. Mean age of the 164 randomized patients was 30 years (range: 7 – 74) and baseline FEV1 was 55% of predicted. Patients were generally receiving standard of care with two exceptions: TIS use in the previous 12 months averaged 1.8 courses rather than the expected six courses, and patients on azithromycin were excluded (see ). At study entry, patients had stable disease and had not received i.v., oral or inhaled antipseudomonal therapy for the previous 14 days. At day 28, the treatment difference (AZLI vs placebo) for mean change in CFQ-R Respiratory Symptoms scores was 9.7 points (p < 0.001; ). The treatment difference for mean percentage change in FEV1 (L) was 10.3%, and for change in sputum P. aeruginosa density was -1.45 log10 CFU/g (each p < 0.001; ). Increases in CFQ-R and FEV1 were seen across subgroups as defined by baseline disease severity, gender or age. AZLI treatment resulted in improvements on other CFQ-R scales ().
Figure 1. AIR-CF1 and AIR-CF2: change from baseline values for CFQ-R Respiratory Symptoms Scores, FEV1 (L), FEV1% predicted and Pseudomonas aeruginosa density in sputum.
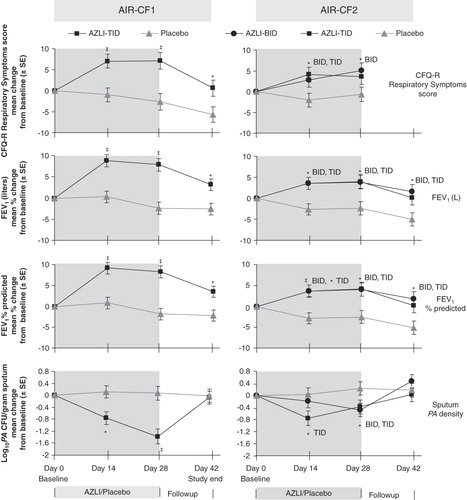
Table 2. Summary of CFQ-R results in Phase III AZLI trials: change from baseline at day 28.
4.3.2 AIR-CF2
AIR-CF2 (CP-AI-005) was a randomized, double-blind, placebo-controlled, event-driven study of 28 days' treatment with 75 mg AZLI or placebo given two or three times daily followed by no antibiotic treatment for up to 56 days Citation[47]. All patients received 28 days of TIS as a run-in to the study (). The primary end point was time-to-need for inhaled or i.v. antipseudomonal antibiotic therapy. Mean age of the 211 randomized patients who received AZLI or placebo was 26 years (range: 7 – 65) and baseline FEV1 was 55% of predicted. These patients were receiving more CF treatments at study entry than the subjects in AIR-CF1; all patients had used three or more courses of TIS in the 12 months before (mean 5.3) and ∼ 70% were receiving azithromycin. AZLI treatment increased median time-to-need for additional antipseudomonal antibiotics for symptoms of pulmonary exacerbation by 21 days, compared with placebo (p = 0.007). By the end of the 84-day study period, 32% of the AZLI-treated patients had an event leading to the use of additional inhaled or i.v. antipseudomonal antibiotics, compared with 50% of placebo-treated patients. At day 28, the treatment difference (pooled AZLI vs pooled placebo) for mean change in CFQ-R Respiratory Symptom scores was 5.01 points (p = 0.02), for percentage change in FEV1 (L) was 6.3% (p = 0.001), and for change in P. aeruginosa sputum density was -0.66 log10 CFU/g (p = 0.006; ). AZLI treatment also resulted in improvements on other CFQ-R scales (). The study was not powered to detect efficacy differences between two- and three-times-daily dosing.
4.3.3 AIR-CF3
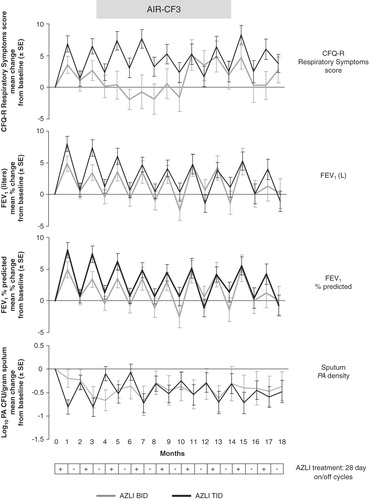
AIR-CF3 (CP-AI-006) was an open-label, follow-on study of patients who had participated in either AIR-CF1 or AIR-CF2; the study evaluated the safety and effect on disease-related end points of repeated exposure to 75 mg AZLI administered two or three times daily and included up to nine 28-day courses (on/off cycles) Citation[50-53]. Mean age of the 274 enrolled patients was 29 years (range: 8 – 74). Mean baseline FEV1 was 56% of predicted. Patients received the same dosing regimen that they had been assigned in their previous study (AIR-CF1 or AIR-CF2) Citation[46,47]. Low hospitalization rates were observed during this 18-month study and the adverse events were generally consistent with CF disease Citation[50,53,66]. During each of the nine 28-day courses of AZLI, FEV1 values and CFQ-R Respiratory Symptom scores improved and sputum P. aeruginosa density decreased, returning to near baseline values during the 28-day off-treatment periods (). Patients had increases in weight (gain ∼ 1.6 kg) over the 18 months, a favorable outcome in patients with CF. There was no evidence for the development of antibiotic resistance or the emergence of other pathogens. Adherence to treatment was approximately 90% and remained high across the 18-month study.
Figure 2. AIR-CF3: effect of multiple AZLI cycles on change from baseline values for CFQ-R Respiratory Symptoms scores, FEV1 (L), FEV1% predicted and Pseudomonas aeruginosa density in sputum.
5. Safety and tolerability of AZLI
The adverse event profile for AZLI was dominated by respiratory adverse events and was consistent with the signs and symptoms of CF. There were no statistically significant differences between treatment groups in any individual study for the proportion of patients reporting most categories of adverse events (i.e., drug-related adverse events, serious adverse events, severe adverse events, or adverse events resulting in withdrawal of study drug) Citation[46,47,50,53]. Bronchospasm, defined as a reduction of ≥ 15% in FEV1 immediately following administration of study medication at a study visit, was observed for 3% of AZLI-treated patients and 4% of placebo-treated patients in the double-blind studies, and for 8% of AZLI-treated patients in the 18-month open-label study. Adverse drug reactions (ADRs) were identified in the Phase III placebo-controlled trials; these events occurred for a higher percentage of AZLI-treated patients than placebo-treated patients, and the association of these events with use of AZLI was considered biologically plausible. shows the incidence of ADRs in the pooled, placebo-controlled Phase III studies (AIR-CF1, AIR-CF2) compared with the open-label, follow-on study (AIR-CF3). The incidence of ADRs is presented as the number of occurrences per patient-month in order to adjust for differences in study length. Pyrexia was the only ADR that showed a statistically significant difference between the AZLI and placebo arms in the placebo-controlled studies, and the incidence of pyrexia decreased with subsequent re-exposure in the open-label study.
Table 3. Adverse drug reactions adjusted for study duration: comparison of pooled AZLI placebo-controlled trials, AIR-CF1 and AIR-CF2, and the open-label AZLI trial, AIR-CF3.
6. Microbiological effects of AZLI
6.1 Sputum Pseudomonas aeruginosa density
The treatment difference (AZLI vs placebo) for mean sputum P. aeruginosa density was -1.45 log10 CFU/g (p < 0.001) in AIR-CF1 and -0.66 log10 CFU/g (p = 0.006) in AIR-CF2 () Citation[46,47]. Effects on sputum P. aeruginosa density were maintained across nine 28-day courses (on/off cycles) in AIR-CF3 () Citation[50,51,53]. The difference between the placebo-controlled trials was probably due to previous antibiotic use; in AIR-CF1, patients had been off antipseudomonal antibiotics for at least 14 days, and in AIR-CF2 patients had just completed a 28-day course of TIS.
6.2 Isolation of treatment-emergent pathogens
AZLI-treatment, compared with placebo, did not result in the acquisition of new pathogens or the emergence of a co-infecting pathogen Citation[67]. Prevalence of co-pathogens such as S. aureus, Achromobacter xylosoxidans or S. maltophilia in the study populations was consistent with published literature (Gilead, data on file).
6.3 Changes in Pseudomonas aeruginosa susceptibility
There were no clinically meaningful changes in the MIC50 or MIC90 of aztreonam for P. aeruginosa in the AIR-CF1, AIR-CF2 and AIR-CF3 studies Citation[46,47,52,53]. There were also no notable increases in the MIC50 or MIC90 values for P. aeruginosa of any other antibiotics tested, including beta lactams, aminoglycosides, and quinolones. Patients infected with P. aeruginosa isolates that seemed ‘resistant’ to aztreonam (with MIC values that exceeded the parenteral breakpoint) also had a meaningful clinical response to AZLI Citation[68].
7. Conclusions
AZLI therapy has demonstrated clinically important changes in respiratory symptoms (CFQ-R), FEV1 and additional antibiotic use in people with CF. There was also a measurable decrease in the bacterial density of P. aeruginosa in sputum. There were positive benefits on other CFQ-R scales and an increase in weight. Patients in subgroups defined by baseline disease severity, gender or age all demonstrated therapeutic benefit from therapy with 75 mg AZLI, administered three times daily. No change in the overall susceptibility of P. aeruginosa to aztreonam or other antibiotics was observed with 75 mg AZLI dosed three times daily, and no trends were observed in the treatment-emergent isolation of other organisms. AZLI is safe and generally well tolerated, and represents an important new therapy for patients with CF. It is administered with an aerosol device that provides appropriate particle size and small airway deposition, and has excellent portability and convenience for people with CF.
8. Expert opinion
The introduction of aztreonam lysine 75 mg powder and solvent for nebulizer solution (AZLI) is a welcome alternative to tobramycin inhalation solution (TIS) and colistin, which are at present the most commonly used inhaled antibiotics in the treatment of cystic fibrosis (CF) Citation[69]. Compared with TIS and colistin, AZLI represents a different class of antibiotic with a different mechanism of antimicrobial action that is complementary to the antibiotics now available. Aztreonam inhibits cell wall biosynthesis and demonstrates time-dependent bactericidal activity against susceptible organisms Citation[35,38-40]. Therefore, the total time during which sputum aztreonam concentrations remain above a particular susceptibility breakpoint is most likely to be relevant in predicting clinical response to AZLI. Aminoglycosides such as TIS, by contrast, inhibit protein synthesis and demonstrate concentration-dependent bactericidal activity. Peak sputum aminoglycoside concentration is most likely to be relevant in predicting clinical response to TIS and other aminoglycosides. Inhaled antibiotic therapy is used in 60 – 80% of CF patients chronically infected with Pseudomonas aeruginosa, and it is likely that AZLI will play a major role in the chronic suppression of P. aeruginosa in CF patients Citation[2,8].
Study design for antibiotic studies in patients with CF has become complex. Since a chronic suppressive strategy of patients cycling ‘on’ and ‘off’ treatment with TIS is standard of care, it makes prospective studies difficult. The study design has to consider when a new investigative therapy is being commenced with regard to the antibiotic cycling and whether the study subject has grown to expect antibiotic relief with some regularity. Study design is further compounded by lack of sufficiently sensitive, meaningful, primary outcome measures. Measurements of airway physiology such as FEV1 do not change sufficiently quickly to allow for studies much less than 6 months in duration. Event-driven studies such as those in which the occurrence of a pulmonary exacerbation or a hospitalization are the end point are more challenging, since improving care of patients with CF has led to a decrease in the total numbers of these events. In addition, the definition of pulmonary exacerbation, physician treatment preferences, and patient social situations and expectations can heavily influence the results of an event-driven study. Despite these concerns, AIR-CF2 showed a positive delay in need for additional antibiotics. Improvement of symptoms as captured by patient-reported outcome (PRO) measurements is clearly of importance to patients and is increasingly attractive as a clinically meaningful primary outcome measure in antibiotic studies in CF. Although the PRO CFQ-R has been used in more than 50 studies, AIR-CF1 is the first pivotal study of a new therapy to use it as a primary outcome measure. Therefore, while AZLI had positive benefit on a number of outcome measures, the significant and clinically meaningful improvement in patients' symptoms is noteworthy.
AZLI has clearly been shown to improve key outcomes that are important for an antibiotic therapy in CF: i) it has an antimicrobial effect; ii) it improves a measure of lung function, FEV1; iii) it reduces patients' symptoms; and iv) it reduces the frequency of pulmonary exacerbations experienced by patients. These results were seen in CF patients with both heavy use of existing therapies (AIR-CF2) as well as those with less use of existing therapies (AIR-CF1). While these were shorter studies, the long-term, open-label, follow-on trial AIR-CF3 showed continued efficacy in patients with repeated use. AZLI will be particularly valuable in patients who can tolerate neither TIS nor colistin due to airway hypersensitivity and is likely to become the primary treatment for such individuals. For individuals with CF who cannot tolerate the month-off cycle of TIS because of the severity of their disease, the use of AZLI in the ‘off’ month is an attractive option. Although the current studies were not designed to determine how AZLI might be used when combined with inhaled aminoglycosides, the run-in phase of AIR-CF2 showed incremental benefit when AZLI was added immediately following a course of TIS. Also of interest, during AIR-CF3 some patients used TIS or other inhaled antibiotic therapy during the ‘off’ month, yet still experienced a decline in FEV1 and/or CFQ-R Respiratory Symptoms scores during the off-AZLI periods. More exploration is needed to understand better the role of AZLI in combination with other inhaled antibiotics.
While AZLI is likely, initially, to be used to complement the current treatments, it may also have potential as part of the treatment strategy of eradication Citation[16-19]. No studies have been reported using AZLI for the treatment of new isolates of P. aeruginosa. The strategy of aggressive therapy for new isolates is now well established, and efficacy has been shown for colomycin and ciprofloxacin in combination and for TIS used as a single agent. The use of AZLI in this context will be important to establish, as this initial treatment often sets the scene for long-term treatment in those individuals where first-line eradication treatment has failed. There is an attractive possibility that AZLI may also be effective in colistin and ciprofloxacin or TIS failures. Additional studies on this issue will be very helpful.
While the CF medical community has largely accepted the need and utility of frequent antibiotic use, there is concern that inhaled antibiotics, especially when used repeatedly, may contribute to driving up bacterial resistance to antibiotics. Inhaled tobramycin has been on the market in the USA and in Europe for approximately 10 years, and the rates of tobramycin resistance in P. aeruginosa isolates from demographically similar patients with CF have increased from 5.4% in 1998 to 20.0% in 2008 Citation[70]. Although in vitro breakpoints cannot be extrapolated to sputum, AZLI is shown to have clinical benefit even when the MIC exceeds the parenteral breakpoint. It is hoped that the availability of an antibiotic class in addition to the aminoglycosides and colistin may decrease selection pressure that contributes to antibiotic resistance, while providing CF patients beneficial antibiotic therapy.
There is now a range of other inhaled antibiotics under investigation, including ciprofloxacin, levofloxacin and amikacin Citation[71]. Ciprofloxacin, colistin and tobramycin are being developed as dry powder inhalers, which would be attractive as it would negate the need to use a nebulizer. Each inhaled antibiotic will have a unique profile with respect to mechanism of action, tolerability profile and ease of use, which will help inform how it will be best used in the treatment of CF. How these different antibiotics might be used with current treatments remains to be seen, but it is likely that the future of antibiotic therapy will be in cycling different classes of antibiotics in patients with CF who have chronic infection with P. aeruginosa.
Declaration of interest
The paper was funded by Gilead Sciences, Inc. and Queens University Belfast. JS Elborn has received research grants from NIG, NIRDO and the CF Trust. NR Henig is an employee of Gilead Sciences, Inc.
Acknowledgements
Kate Loughney provided medical writing assistance under the sponsorship of Gilead Sciences. Crystal Anglen provided publication management assistance.
Notes
Bibliography
- Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996;154:1229-56
- Cystic Fibrosis Foundation Patient Registry. 2008 Annual Data Report to the Center Directors. Bethesda, Maryland: Cystic Fibrosis Foundation; 2007
- Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 1992;12:158-61
- Pamukcu A, Bush A, Buchdahl R. Effects of Pseudomonas aeruginosa colonization on lung function and anthropometric variables in children with cystic fibrosis. Pediatr Pulmonol 1995;19:10-5
- Corey M, Edwards L, Levison H, Knowles M. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr 1997;131:809-14
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918-51
- Fogarty A, Hubbard R, Britton J. International comparison of median age at death from cystic fibrosis. Chest 2000;117:1656-60
- Cystic Fibrosis Trust. UK CF Registry Annual Data Report 2008. Available from: http://www.cftrust.org.uk/aboutcf/publications/cfregistryreports/UK_CF_Registry-Annual_ Data_Report_2008.pdf [Last accessed 27 January 2010]
- Rosenfeld M, Emerson J, Williams-Warren J, Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr 2001;139:359-65
- Amadori A, Antonelli A, Balteri I, Recurrent exacerbations affect FEV1 decline in adult patients with cystic fibrosis. Respir Med 2009;103:407-13
- Hansen CR, Pressler T, Høiby N. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J Cyst Fibros 2008;7:523-30
- Foweraker J. Recent advances in the microbiology of respiratory tract infection in cystic fibrosis. Br Med Bull 2009;89:93-110
- Tunney MM, Field TR, Moriarty TF, Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 2008;177:995-1001
- Courtney JM, Bradley J, Mccaughan J, Predictors of mortality in adults with cystic fibrosis. Pediatr Pulmonol 2007;42:525-32
- Bittar F, Richet H, Dubus JC, Molecular detection of multiple emerging pathogens in sputa from cystic fibrosis patients. PLoS One 2008;3:e2908
- Lee TW, Brownlee KG, Denton M, Reduction in prevalence of chronic Pseudomonas aeruginosa infection at a regional pediatric cystic fibrosis center. Pediatr Pulmonol 2004;37:104-10
- Taccetti G, Campana S, Neri AS, Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis. J Chemother 2008;20:166-9
- Ho SA, Lee TW, Denton M, Regimens for eradicating early Pseudomonas aeruginosa infection in children do not promote antibiotic resistance in this organism. J Cyst Fibros 2009;8:43-6
- Douglas TA, Brennan S, Gard S, Acquisition and eradication of P. aeruginosa in young children with cystic fibrosis. Eur Respir J 2009;33:305-11
- Flume PA, O'sullivan BP, Robinson KA, Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176:957-69
- Heijermann H, Westerman E, Conway S, Inhaled medication and inhalation devices for lung disease in patients with cystic fibrosis: a European consensus. J Cyst Fibros 2009;8:295-315
- Smyth A, Elborn JS. Exacerbations in cystic fibrosis: 3 – Management. Thorax 2008;63:180-4
- Flume PA, Mogayzel PJ Jr, Robinson KA, Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 2009;180:802-8
- Ramsey BW, Pepe MW, Quan JM, Cystic fibrosis inhaled tobramycin study group. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Eng J Med 1999;340:23-30
- US prescribing information, TOBI®, tobramycin inhalation solution, USP. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/tobi.pdf [Last accessed 27 January 2010]
- Hodson ME, Gallagher CG. New clinical evidence from the European tobramycin trial in cystic fibrosis. J Cyst Fibros 2002;1(Suppl 2):199-202
- Summary of Product Characteristics (SPC), TOBI® 300°mg/5°ml Nebuliser Solution Website of UK licensed medicines. Available from: http://emc.medicines.org.uk/ [Last accessed 27 January 2010]
- Summary of Product Characteristics (SPC), Promixin® 1 MIU Powder for Nebuliser Solution. Website of UK licensed medicines. Available from: http://emc.medicines.org.uk/ [Last accessed 27 January 2010]
- Coulthard K. Maximizing the efficacy and safety of colistimethate therapy [abstract S19.2]. Pediatr Pulmonol 2008;43(Suppl 31):193-5
- Li J, Nation RL, Turnidge JD, Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet 2006;6:589-601
- Kuhn RJ. Formulation of aerosolized therapeutics. Chest 2001;120:94S-8S
- European Medicines Agency. Summary of Product Characteristics (SPC), Cayston® 75 mg powder and solvent for nebuliser solution. Available from: http://www.emea.europa.eu/humandocs/PDFs/EPAR/cayston/emea-combined-h996en.pdf [Last accessed 27 January 2010]
- Heath Canada. Product Monograph: Cayston® (Aztreonam for Inhalation Solution 75 mg aztreonam/vial antibiotic. Available from: http://www.hc-sc.gc.ca/index-eng.php [Last accessed 27 January 2010]
- FDA. Prescribing Information: Cayston® (aztreonam for inhalation solution). Available from: http://www.cayston.com/media/CAYSTON_prescribe_info.pdf [Last accessed 26 February 2010]
- Prescribing information: AZACTAM® (aztreonam) for injection, USP. Available from: http://www.elan.com/ [Last accessed 27 January 2010]
- In: Parfitt K, editor, Aztreonam. Martindale: the complete drug reference. 32nd edition. Pharmaceutical Press, London; 1999. p. 156-7
- Dietzsch HJ, Gottschalk B, Heyne K, Cystic fibrosis: comparison of two mucolytic drugs for inhalation treatment (acetylcysteine and arginine hydrochloride). Pediatrics 1975;55:96-100
- Georgopapadakou NH, Smith SA, Sykes RB. Mode of action of aztreonam. Antimicrob Agents Chemother 1982;21:950-6
- Johnson DH, Cunha BA. Aztreonam. Med Clin North Am 1995;79:733-43
- Craig WA. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect Dis Clin North Am 2003;17:479-501
- Sykes RB, Bonner DP. Aztreonam: the first monobactam. Am J Med 1985;78:2-10
- Brogden RN, Heel RC. Aztreonam. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 1986;31:96-130
- Adkinson NF Jr. Immunogenicity and cross-allergenicity of aztreonam. Am J Med 1990;88(3C):12S-5S, discussion 38S-42S
- Geller DE. The science of aerosol delivery in cystic fibrosis. Pediatr Pulmonol 2008;43(Suppl 9):5-17
- Gibson RL, Retsch-Bogart GZ, Oermann C, Microbiology, safety and pharmacokinetics of aztreonam lysinate for inhalation in patients with cystic fibrosis. Pediatr Pulmonol 2006;41:656-65
- Retsch-Bogart GZ, Quittner AL, Gibson RL, Efficacy and safety of inhaled aztreonam lysine for airway Pseudomonas in cystic fibrosis. Chest 2009;135:1223-32
- McCoy KS, Quittner AL, Oermann CM, Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med 2008;178:921-8
- Hofmann T, Otto K, Kirihara J, Safety and tolerability study of aztreonam for inhalation (AI) in healthy volunteers [abstract 195]. Pediatr Pulmonol 2003;36(Suppl 25):251
- Retsch-Bogart GZ, Burns JL, Otto KL, A phase 2 study of aztreonam lysine for inhalation to treat patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatr Pulmonol 2008;43:47-58
- Oermann CM, Retsch-Bogart GZ, McCoy KS, Effect of multiple courses of aztreonam for inhalation solution (AZLI) on disease-related endpoints and safety in patients with CF: final analysis of 18-month data [abstract 241]. Pediatr Pulmonol 2009;44(Suppl 32):296
- Oermann CS, McCoy KS, Retsch-Bogart GZ, Effect of repeated exposure to aztreonam for inhalation solution (AZLI) therapy on cystic fibrosis respiratory pathogens [abstract 353]. Pediatr Pulmonol 2009;44(Suppl 32):335-6
- Oermann CS, McCoy KS, Retsch-Bogart GZ, Antibiotic susceptibility in Pseudomonas aeruginosa (PA) isolates following repeated exposure to aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis [abstract 278]. Pediatr Pulmonol 2009;44(Suppl 32):309
- Oermann CM, Retsch-Bogart GZ, Quittner AL, An 18-month study, AIR-CF3, of the safety and improvement in pulmonary function and respiratory symptoms with repeated courses of aztreonam for inhalation solution in patients with cystic fibrosis and airway Pseudomonas aeruginosa. Pediatr Pulmonol 2010; in press
- Quittner AL, Sweeny S, Watrous M, Translation and linguistic validation of a disease-specific quality of life measure for cystic fibrosis. J Pediatr Psychol 2000;25:403-14
- Modi AC, Quittner AL. Validation of a disease-specific measure of health-related quality of life for children with cystic fibrosis. J Pediatric Psychol 2003;28:535-46
- Quittner AL, Buu A, Messer MA, Development and validation of the cystic fibrosis questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest 2005;128:2347-54
- Abbott J. Health-related quality of life measurement in cystic fibrosis: advances and limitations. Chron Respir Dis 2009;6:31-41
- US FDA Guidance for Industry. Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. Available from: http://www.fda.gov/downloads/Drugs/GuidanceCompliance RegulatoryInformation/Guidances/UCM193282.pdf [Last accessed 27 January 2010]
- Guyatt GH, Kirshner B, Jaeschke R. Measuring health status: what are the necessary measurement properties? J Clin Epidemiol 1992;45:1341-45
- Juniper EF, Guyatt GH, Willan A, Griffith LE. Determining a minimal important change in a disease-specific quality of life questionnaire. J Clin Epidemiol 1994;47:81-7
- Testa MA. Interpretation of quality-of-life outcomes: issues that affect magnitude and meaning. Med Care 2000;38:166-74
- Guyatt GH. Making sense of quality-of-life data. Med Care 2000;38:175-9
- Terwee CB, Dekker FW, Wiersinga WM, On assessing responsiveness of health-related quality of life instruments: guidelines for instrument evaluation. Qual Life Res 2003;12:349-62
- Goss CH, Quittner AL. Patient-reported outcomes in cystic fibrosis. Proc Am Thorac Soc 2007;4:378-86
- Quittner AL, Modi A, Wainwright C, Determination of the minimal clinically important difference (MCID) scores for the Cystic Fibrosis Questionnaire-Revised (CFQ-R) Respiratory Symptom scale in two populations of patients with CF and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135:1610-18
- Montgomery AB, S Lewis M, K Higuchi M, Hospitalization risk of current standard of care (SOC) vs. aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis (CF) [abstract]. Am J Respir Crit Care Med 2009;179:A1188
- Braff M, Retsch-Bogart G, McCoy K, Effect of 28-day aztreonam for inhalation solution (AZLI) therapy on cystic fibrosis (CF) respiratory pathogens [abstract]. Am J Respir Crit Care Med 2009;179:A5944
- McCoy KS, Retsch-Bogart G, Gibson R, Relevance of established susceptibility breakpoints to clinical efficacy of inhaled antibiotic therapies in cystic fibrosis [abstract 418]. Pediatr Pulmonol 2008:43(Suppl 31):351
- Elborn JS, Hodson M, Bertram C. Implementation of European standards of care for cystic fibrosis – control and treatment of infection. J Cyst Fibros 2009;8:211-17
- Burns JL, Emerson J, McNamara S, Antibiotic resistance in cystic fibrosis sputum isolates [abstract 373]. Pediatr Pulmonol 2008;43(Suppl 31):334
- Jones AM, Helm JM. Emerging treatments in cystic fibrosis. Drugs 2009;69:1903-10