
Abstract
Introduction: In the era of multidrug-resistant, extensively drug-resistant (XDR) and even pandrug-resistant Gram-negative microorganisms, the medical community is facing the threat of untreatable infections particularly those caused by carbapenemase-producing bacteria, that is, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii. Therefore, all the presently available antibiotics, as well as for the near future compounds, are presented and discussed.
Areas covered: Current knowledge concerning mechanisms of action, in vitro activity and interactions, pharmacokinetic/pharmacodynamics, clinical efficacy and toxicity issues for revived and novel antimicrobial agents overcoming current resistance mechanisms, including colistin, tigecycline, fosfomycin, temocillin, carbapenems, and antibiotics still under development for the near future such as plazomicin, eravacycline and carbapenemase inhibitors is discussed.
Expert opinion: Colistin is active in vitro and effective in vivo against XDR carbapenemase-producing microorganisms in the critically ill host, whereas tigecycline, with the exception of P. aeruginosa, has a similar spectrum of activity. The efficacy of combination therapy in bacteremias and ventilator-associated pneumonia caused by K. pneumoniae carbapenemase producers seems to be obligatory, whereas in cases of P. aeruginosa and A. baumannii its efficacy is questionable. Fosfomycin, which is active against P. aeruginosa and K. pneumoniae, although promising, shares poor experience in XDR infections. The in vivo validity of the newer potent compounds still necessitates the evaluation of Phase III clinical trials particularly in XDR infections.
1. Introduction
Nosocomial infections caused by multidrug-resistant (MDR) and extensively drug-resistant (XDR) Gram-negative pathogens represent a major threat worldwide Citation[1]. The ESKAPE microorganisms, from the initials of the most frequently isolated MDR bacteria, that is, Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp., point out the ‘eskape’ effect from the action of antibacterial agents Citation[2]. Particularly, K. pneumoniae strains producing carbapenemases reach mortality rates ranging between 23 and 75%, which are mainly attributed to the lack of active antimicrobials Citation[3]. The last resort antibiotics, mostly prescribed off-label, are two revived antimicrobials of the 1970s to 1980s, colistin and fosfomycin, as well as tigecycline, which in combination with an aminoglycoside or with each other in case of in vitro activity have shown promising efficacy both in vitro and in vivo in the critically ill host Citation[4,5]. However, due to the increased use, particularly of colistin, resistance is rapidly increasing Citation[6-8]. Unfortunately, new therapeutic options, such as plazomicin and the extended spectrum β lactamase (ESBL)/carbapenem inhibitors, are still under development, whereas temocillin, an older antibiotic, which is active against ESBL-producing enterobacteriaceae, is available only in three European countries Citation[9-11]. In this review, the latest data regarding the in vitro activity, pharmacokinetic/pharmacodynamics (PK/PDs), clinical efficacy and safety tissues of the above antibiotics are reported and discussed.
2. Colistin
2.1 Mode of action – in vitro activity and interactions
Colistin is a cationic antimicrobial peptide discovered in 1947 from Bacillus polymyxa. It entered clinical use in 1958 but was abandoned in 1970s due to reported cases of nephrotoxicity and neurotoxicity. Reintroduction of the revived antibiotic was necessary due to the emerging increase of MDR Gram-negative pathogens in combination with the deficit of newer antimicrobial regimens Citation[1,5]. The target of antimicrobial action of colistin is based on the initial interaction of the cationic peptide and the negatively charged lipopolysaccharide of the bacterial cell membrane, leading to destabilization of the outer membrane by displacement of calcium and magnesium, enhancing the permeability of the cell envelope and eventually to cell death through leakage of cell contents Citation[5]. There are two forms of colistin commercially available: colistin sulfate for oral and topical use and colistin methanesulfonate (CMS) for parenteral administration Citation[5]. The parenteral products used in different global regions are standardized as: i) milligram colistin base activity (CBA) and ii) international units (IU), and 1 mg CBA is equivalent to 33.250 IU or 1 million IU is analogous to 30 mg CBA Citation[12]. Colistin is active against Enterobacteriaceae (Escherichia coli, Klebsiella spp., Enterobacter spp., Citrobacter spp., Salmonella spp. and Shigella spp., including ESBL, K. pneumoniae carbapenemase (KPC), VIM and New Delhi metallo (NDM)-1 producers), MDR and XDR P. aeruginosa and A. baumannii, Legionella pneumophila, Stenotrophomonas maltophilia and Aeromonas spp., where Proteus and Providencia spp. as well as Burkholderia spp., Serratia marcescens and Morganella morganii are inherently resistant. Gram-positive and most anaerobic strains are intrinsically resistant Citation[5]. The most recent susceptibility breakpoints are depicted in . In vitro interactions have led to synergistic results mainly with rifampicin and carbapenems against A. baumannii and P. aeruginosa. In 39 nosocomial A. baumannii strains, synergy between colistin and rifampicin was observed in 51.3 and 66.7% of isolates after 24 h for 1 × MIC and 4 × MIC, respectively Citation[13]. KPC-producing strains have also been studied and synergy has been found with previous antimicrobial regimens and with tigecycline. However, in the latter strains, combination of carbapenems and colistin has been reported to be antagonistic with colistin-resistant strains Citation[5]. Of great surprise are the synergistic results of colistin with antimicrobials with Gram-positive activity, that is, daptomycin and vancomycin against A. baumannii Citation[14].
Table 1. Susceptibility breakpoints (μg/ml) of colistin.
2.2 Pharmacokinetics/pharmacodynamics
CMS is an inactive prodrug that in vivo and in vitro is hydrolyzed to partially sulfomethylated derivatives and to colistin that exhibits antibacterial activity. CMS is eliminated mainly (∼ 70%) by the kidneys, whereas colistin undergoes extensive renal tubular reabsorption and predominately has a nonrenal route of elimination Citation[15]. Colistin possesses rapid concentration-dependent bacterial killing against susceptible strains and studies have demonstrated that AUC/MIC is the PK/PD index that mostly correlates with the antibacterial effect Citation[15]. The major issue associated with older colistin PK studies includes the scarcity of a reliable methodology because they were based on microbiological assays which are considered problematic due to incapability of distinguishing CMS from colistin. Nowadays, newer method such as liquid chromatography tandem mass spectrometry (LC/MS/MS) has been developed and is being considered as the only valid approach for quantifying colistin levels with precision Citation[16]. The first major PK study in critically ill patients using LC/MS/MS for determination of CMS and colistin was conducted by Plachouras et al. Eighteen patients (age range 40 – 83 years) were enrolled with moderate-to-good renal function (creatinine clearance [CrCL]: 41–126 ml/min/1.73 m2) and an intravenous (i.v.) dose of 3 million IU (MU) every 8 h was administrated. The half-time of CMS disposition was 2.3 h, whereas the half-time of colistin was determined at 14.4 h. The predicted Cmax of colistin was 0.6 mg/l after the first dose and 2.3 mg/l at steady state. The latter results indicated insufficient colistin concentrations for the first 48 h of treatment with the risk of therapeutic failures and resistance development () Citation[16]. The same group of investigators evaluated the application of a loading dose of 6 MU CMS in 10 critically ill patients (6 male; mean age, 54 years; mean CrCL 82 ml/min/1.73 m2) and in 19 critically ill patients (9 male; mean age, 56 years; median CrCL 87 ml/min/1.73 m2) a loading dose of 9MU with a maintenance dose of 4.5 MU every 12 h commenced after 24 h was evaluated. The Cmax of colistin after the loading dose was found to be 1.34 and 2.5 mg/l, respectively () Citation[17,18]. Recently, a multicenter, multinational study on PK/PD of colistin in critically ill patients was completed with a recruitment of 228 patients, including various categories of patients with renal insufficiently Citation[19,20]. Analyses of data have been published up to date for 162 patients not on renal replacement therapy with a median CrCL of 63 ml/min/1.73 m2, for 12 patients on intermittent hemodialysis and for 4 patients on continuous renal replacement therapy. Model-fitted parameter were utilized for the proposal of algorithms on loading dose and maintenance dose of colistin taking into consideration CrCL, body weight (lower of either actual or ideal) and targeted steady-state colistin concentration levels. It is of great significance to be emphasized that the first maintenance dose should be given after 24 h and maximum loading dose should not exceed 10 MU (300 mg CBA) based on current upper limit of production information in combination with potential increase in nephrotoxicity with higher dosages. A substantial finding in this study was that patients with high CrCL, ≥ 80 ml/min/1.73 m2, had decreased ability of achieving steady-state colistin concentrations > 2 mg/l with currently recommended dosage. Therefore, in these patients, highly active combination therapy should be employed in particular when treating MDR Gram-negative with MIC ≥ 1 μg/ml Citation[19,20]. It has been also shown that CMS and colistin are efficiently cleared by hemodialysis and during continuous renal replacement therapy Citation[19,21]. Dosages of various categories of patients are illustrated in Citation[19,20,22].
Figure 1. Graphical representation of CMS and colistin concentrations observed at a dose 3 MU q8 h and with a loading dose of 6 MU and 9 MU (infusion over 0.5 h and 1 h).
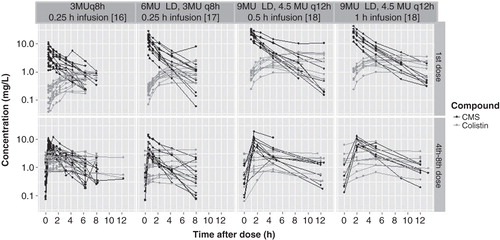
Table 2. Suggested loading and maintenance dosage of colistin methanesulfonate in million international unit in various patient categories.
2.3 Clinical studies
There have been a large number of clinical reports on efficacy of i.v. CMS in patients with pneumonia without cystic fibrosis. Since 1999, 12 single-arm studies and 6 comparator studies with 316 and 396 patients retrospectively have been published with a clinical cure of 25 – 71% and a mortality rate ranging between 29 and 64% Citation[5,23,24]. It should be pointed out that clinical efficacy in nosocomial pneumonia exceeded 55% and was comparable to comparators (carbapenems, piperacillin/tazobactam and ampicillin/sulbactam) Citation[24]. However, all studies shared similar limitations mainly concerning retrospective design, heterogeneity of doses and simultaneous administration of other antibiotics Citation[5,23,24]. In addition, in the large randomized trial with in which serious infections caused by XDR A. baumannii were included, the efficacy of the addition of rifampicin was as follows: 210 patients mainly with ventilator-associated pneumonia (VAP) (69.8%) and bloodstream infections (20.1%) were randomized (1:1) for i.v. receiving colistin alone at a dose of 2 MU every 8 h (n = 105) or colistin in combination with rifampicin 600 mg every 12 h (n = 105). The addition of rifampicin had no impact on reduction of 30-day mortality, infection-related death and length of hospitalization. However, an increase rate of A. baumannii eradication was observed in the combination group, whereas higher levels of hepatotoxicity were also depicted Citation[25]. In a recent literature review, including 298 patients infected with carbapenemase-producing K. pneumoniae strains and suffering mostly from bacteremia, combination treatment with two active drugs, that is, colistin, tigecycline, gentamicin or carbapenems (when MIC ≤ 4 μg/ml) was found superior compared to monotherapy, whereas mortality rates with colistin monotherapy in these cases had similar rates with inappropriate treatment Citation[3]. However, it seems that in case of P. aeruginosa or A. baumannii infections, monotherapy should be adequate Citation[26,27]. Recent studies implementing higher doses of colistin have been very promising. In a recent prospective study on 28 patients with severe sepsis (57.1%) and septic shock (42.1%) due to Gram-negative bacteria sensitive only to colistin, a loading dose of 9 MU with a maintenance dose of 4.5 MU twice daily was administrated Citation[28]. The main type of infection were bloodstream infection (64.3%) and VAP (35.7%) caused by A. baumannii (46.4%), K. pneumoniae (46.4%) and P. aeruginosa (7.2%). In 14 patients, CMS was administrated as monotherapy and clinical cure was found to be 82.1% (23/28) with acute kidney failure of 17.8% Citation[28]. In another recent study, higher colistin doses were evaluated retrospectively in 76 patients suffering from Gram-negative bacteremia treated with colistin for at least 3 days. Median colistin doses were significantly higher in patients who attained microbiological success (2.9 vs 1.5 mg/kg/day) as well as for survivors at day 7 (2.9 vs 1.5 mg/kg/day). However, no statistical difference was found in mortality at day 28 and incidence of nephrotoxicity was increased in patients who were given 3.8 mg/kg/day compared to those administered with 1.6 mg/kg/day Citation[29]. The unorthodox combination of colistin with a glycopeptide has been evaluated in two recent studies with equivocal results and the effect has been assumed to be associated with the interaction of colistin with the outer membrane, facilitating the entry of glycopeptide to the cell wall targets from which they are excluded Citation[30,31]. In a multicenter retrospective study on 166 patients with infections caused by Gram-negative bacteria, a combination of colistin (median dose 6 MU/day) with glycopeptide were administrated in 68 patients (62 patients received vancomycin and 6 patients received teicoplanin with a median dose of 2 g and 400 mg/day accordingly). The most common infection was VAP (64.5%) followed by bloodstream infections (19.9%) and the main implicated pathogens were MDR A. baumannii, MDR P. aeruginosa and carbapenem-resistant K. pneumoniae in 59.6, 18.7 and 14.5% of patients, respectively. No difference regarding 30-day mortality was documented (33.8 vs 29.6%) and nephrotoxicity rates were similar. However, regarding mortality, in multivariate analysis, combination therapy for ≥ 5 days was a protective factor Citation[30]. In a second retrospective study with 57 patients, 29 patients were treated with colistin 3 MU every 8 h plus vancomycin 1 g every 12 h and 28 patients were treated with colistin alone for MDR A. baumannii infections for at least 5 days. Clinical cure (55.2 vs 67.9%), microbiological eradication (54.2 vs 65.2%) and mortality rates (48.3 vs 50%) were similar in both groups; however, rates of nephrotoxicity differed and were increased in the combination group (55.2 vs 28.6%, p = 0.04) Citation[31].
Adjunctive inhalation therapy has been implied in cases of VAP and tracheobronchitis. High lung concentration levels without considerable systemic absorption and toxicity are considered major benefits of this practice. It is of great significance to emphasize that the use of a vibrating mesh nebulizer is preferred as around 60% of the inhaled dose reaches the respiratory tract, whereas only 10 – 15% reaches the respiratory tract when utilizing conservative nebulizers Citation[32]. In a recent systemic review and meta-regression with 14 single-arm studies and 6 controlled studies (including 11 studies with i.v. colistin, 5 with aerolized colistin monotherapy and 4 studies with combination of i.v. and aerolized colistin), a overall favorable clinical response rate of 72% was observed, whereas no difference regarding mortality, toxicity or clinical achievement was described between colistin and control groups Citation[24]. The largest retrospective matched case-control study evaluating the efficacy and safety of i.v. colistin against i.v. plus aerolized colistin for microbiologically documented VAP caused by MDR Gram-negative pathogens included 121 patients in the first arm and 104 in the later arm. Daily dose of i.v. colistin was 7 ± 2.5 MU, whereas inhaled colistin was administrated at a dose of 1 MU three times a day with an ultrasonic nebulizer Citation[33]. Authors reported that combination treatment was associated with higher rates of clinical cure (69.2 vs 54.8%, p = 0.03) and significantly less days of mechanical ventilation (12 vs 8 days, p = 0.03). The daily dose suggested for inhaled colistin is 1 MU every 8 h. However, a recent PK study in 20 patients with ventilator-associated tracheobronchitis where the previous dose of inhaled colistin was administrated as monotherapy showed that the median levels of colistin concentrations in epithelial fluid were 6.7, 3.9 and 2 μg.ml at 1, 4 and 8 h, respectively Citation[34]. Based on the latter findings, it is assumed that the latter dosage might not be adequate due to subtherapeutic concentrations at the end of the dosing interval. Higher doses of aerolized colistin up to 5 MU every 8 h have been suggested in clinical practice for the treatment of nosocomial VAP Citation[32].
Of particular interest are the impressive results of intraventricular or intrathecal colistin in MDR-XDR A. baumannii ventriculitis or meningitis in the neurosurgery setting. In a recent literature review of 81 patients treated with intraventricular or intrathecal colistin at a median dose of 125,000 IU (range 20,000 – 500,000 IU) for a median period of 18.5 days, the rate of successful outcome was shown to be 89% and toxicity was mainly manifested as reversible chemical ventriculitis or meningitis in nine (11%) cases Citation[35].
Unfortunately, the excessive use of colistin has been associated with the emergence of MDR Gram-negatives particularly in carbapenemase-producing K. pneumoniae (CPKP). This phenomenon has led to colonization and subsequent infections with colistin-resistant K. pneumoniae strains, increasing up to 30% Citation[6,7]. In the multivariable model, the use of colistin for > 14 days was determined as the only independent risk factor related to resistance development to colistin in K. pneumoniae, A. baumannii and P. aeruginosa Citation[8].
2.4 Adverse events
The most common adverse effect of colistin is nephrotoxicity Citation[36]. A discrepancy between old and recent reports on renal dysfunction exists; in general, lower incidences of renal toxicity are reported in the modern era. Major factors associated with this disparity are the use of more purified colistin, the administration of colistimethate instead of colistin sulfate and close monitoring of patients receiving colistin. Nephrotoxicity rates vary widely and depend on definitions applied. Recently, the RIFLE criteria, a validated tool for evaluation of acute kidney injury was introduced and has been utilized in newer publications, with renal toxicity calculated between 18 and 53.5% Citation[36-38]. Risk factors are hypoalbuminemia, receipt of ≥ 3 concomitant nephrotoxins, diabetes mellitus, obesity, total cumulative dose and duration of CMS therapy Citation[36,38]. It is of great significance to mention that higher daily doses are associated with renal toxicity in a dose-dependent fashion. In particular, in a large academic health system, > 30% of patients receiving colistin dose between 3 – 4.9 mg/kg/day reported nephrotoxicity and this number scaled up to 69% when doses administered surpassed ≥ 5 mg/kg/day, indicating a major risk using dosage above upper limit label product labeling (300 mg CBA) Citation[38]. In general, CMS-induced renal toxicity is mostly mild and reversible with renal replacement therapy occasionally required and permanent renal damage rarely observed Citation[36]. The incidence of neurotoxicity in earlier studies has been reported at approximately 7% mainly documented as peripheral or orofacial paresthesias, vertigo, visual disturbances, confusion, seizures and the detrimental event of neuromuscular blockade leading to respiratory muscle paralysis and apnea. However, in more recent studies neurotoxic phenomena are rarely reported Citation[36].
3. Tigecycline
3.1 In vitro activity and interactions
Tigecycline, a glycylcycline, is a bacteriostatic derivative of minocycline with the ability to overcome the active efflux and the ribosomal protein resistance mechanisms, which inactivate older tetracyclines Citation[39]. It was approved by the FDA and the European Medicines Agency (EMA) in 2005 and 2006, respectively, for the treatment of complicated intra-abdominal infections (cIAIs) and complicated skin and skin structure infections, and the FDA in 2009 added community-acquired pneumonia to the list. However, nowadays tigecycline is frequently administered off-label for treating XDR infections Citation[5,40]. Tigecycline antimicrobial spectrum includes ESBL-producing enterobacteriaceae, MDR and XDR A. baumannii and CPKP Citation[41,42]. In the Tigecycline Evaluation and Surveillance Trial study between 2005 and 2011 among MDR A. baumannii and ESBL-positive E. coli and K. pneumoniae strains, MIC50/MIC90 were 0.5/1 and 0.25/0.5 μg/ml, respectively Citation[43], whereas in a resistant surveillance, including 22,005 unique clinical isolates collected worldwide in 2011, tigecycline susceptibility against meropenem non-susceptible K. pneumoniae and Acinetobacter spp. were found to be between 94.3 and 100% and 83.8 and 93.9% Citation[44]. However, Pseudomonas, Proteus, Providencia and M. morganii are inherently resistant to tigecycline Citation[39]. The breakpoint for enterobacteriaceae and Acinetobacter spp. according to FDA and European Committee on Antimicrobial Susceptibility Testing (EUCAST) is defined as 2 and 1 μg/ml, respectively Citation[45,46]. The in vitro interactions of tigecycline with other antimicrobials is usually indifferent; however, when combined with colistin against metallo-β-lactamase (MBL) and ESBL-producing K. pneumoniae synergy has been shown Citation[5,47]. Unfortunately, due to extensive use of tigecycline as an off-label antibiotic in XDR infections in endemic regions for CPKP infections, resistance to tigecycline is increasing Citation[48].
3.2 Pharmacokinetics/pharmacodynamics
Tigecycline is available only as an i.v. formulation and the standard regimen after a loading dose of 100 mg is 50 mg every 12 h. The drug has a protein binding of 78% and is primarily excreted in the bile (59%), a 50% reduction of the maintenance dose suggested severe liver insufficiency, while the dose should not be changed in renal failure or hemodialysis Citation[49]. Kinetic parameters are shown in Citation[49], whereas the drug creates advantageous levels in the bile, gallbladder wall as well as in the colon wall and blood neutrophils Citation[49]. The area under the unbound drug concentration–time curve (fAUC)/MIC is the PK/PD index best correlated with the in vivo activity of the drug, and higher response is observed in nosocomial pneumonia whenever fAUC/MIC approaches 1 Citation[39]. However, because of tigecycline's high volume of distribution, the drug is rapidly accumulated in the various tissue compartments resulting in low drug levels in blood, epithelial lining fluid (ELF) and the urinary tract, where only 15 – 22% of tigecycline is eliminated. The latter findings offer a plausible explanation to the reported failures in bloodstream infections and VAP, indicating that in similar situations combination therapy should be a priority Citation[50-52].
Table 3. Main plasma pharmacokinetic parameters of tigecycline in humans.
3.3 Clinical studies
Several studies on MDR Gram-negative infections have been published, most of them being retrospective and non-comparative, in which other antibiotics were combined with tigecycline rendering the elucidation of the true efficacy of tigecycline in the world of MDR-XDR pathogens rather obscure Citation[3,53-55]. In a Greek study, in which criteria for definition of resistance pathogens were based on MICs, tigecycline as monotherapy or presumed active monotherapy was given at the standard low dose for A. baumannii and K. pneumoniae infections with an MDR/XDR profile Citation[56]. Overall successful clinical response increased to 80%, including 17 with septic shock. However, 13 episodes of breakthrough infections and superinfections were observed in 10 patients with Gram-negative pathogens inherently resistant to tigecycline, that is, Proteus spp. and P. aeruginosa. In another prospective, double-blind, randomized trial, tigecycline at the standard dose compared to imipenem were given in 511 patients with health care-associated pneumonia/VAP Citation[57]. Tigecycline compared to imipenem did not reach ‘noninferiority’, and the disappointing result observed in A. baumannii infections led to a negative approval by the FDA, attributed to the low tigecycline serum levels (≤ 0.6 μg/ml). Subsequently, the results of a recent study in patients with hospital-acquired pneumonia (HAP) where tigecycline was prescribed at two high-dosage regimens (150 mg followed by 75 mg every 12 h in 23 patients or 200 mg followed by 100 mg every 12 h in 20 patients) versus imipenem at a dose of 1 g every 8 h in 24 patients, despite the rather low numbers of recruited patients, supported the latter consideration Citation[58]. A numerically higher clinical response of 85% was observed with the higher dose of tigecycline versus 69.6% in the lower dose versus 75% in patients given imipenem. Therefore, it seems that a MIC of tigecycline of 0.5 or 1 μg/ml requires higher dosage schedule (75 mg or 100 mg every 12 h following a loading dose of 150 or 200 mg respectively), whereas for MICs < 0.5 μg/ml, 50 mg every 12 h should be adequate. Although further studies in a large number of patients are necessary, the reported results support the consideration that the obtained higher AUC/MIC after the higher prescribed dose of tigecycline was connected with the improved efficacy. The necessity and the validity of tigecycline combinations were evaluated from a recent review in 77 severe infections caused by CPKP derived from 34 already published studies, where failure rates with tigecycline monotherapy increased to 46% ranging, however, from 0 to 15% whenever colistin or an aminoglycoside or meropenem (with an MIC ≤ 4 μg/ml) were given simultaneously, thus depicting the necessity of the combinations at least in case of infections caused by XDR-CPKP Citation[3]. Recently, in a retrospective study, the outcome of tigecycline monotherapy or in combination with ceftazidime, ceftriaxone, piperacillin/tazobactam or a carbapenem in the treatment of healthcare-associated MDR A. baumannii infections in 266 patients versus 120 patients in the non-tigecycline group, who were treated with imipenem plus sulbactam, were reported Citation[59]. No significant difference in survival rates at 30 days was observed, as well as in the length of hospital or intensive care unit (ICU) stay between the two groups (36.1 vs 38.3%); however, the rate of unfavorable outcome was significantly lower (50 vs 69.2%, p < 0.05) in the group of patients who received tigecycline. In case of empirical therapy in the critically ill, the lack of activity of tigecycline against P. aeruginosa mandates the addition of an antipseudomonal antibiotic.
3.4 Adverse events
A significant number of patients recruited in Phase II studies complained of nausea, vomiting and diarrhea, whereas few cases of pancreatitis have been also reported. Also the possibility of decreased fibrinogen levels is of concern (H. Giamarellou, personal communication). Two similar FDA warnings in 2010 and 2013 are of importance because it indicated that the drug was associated with increased risk of death compared to other antibiotics used for treating similar infections Citation[60,61]. Overall, death occurred in 3.9 versus 2.9% respectively, whereas for approved indications death rate was 2.5 versus 1.8% (p = 0.09), with the reported difference attributed mainly to VAP and baseline bacteremia suffering patients Citation[62]. However, a meta-analysis including 14 randomized trials, comprising about 7400 patients, showed that despite the all-cause mortality being numerically higher in the tigecycline than in the comparator groups, the difference was not statistically significant Citation[63]. In Europe (as of November 2012), the benefit/risk evaluation of the EMA's Committee of Medicinal Products for Human Use concluded that ‘the benefits of tigecycline continue to outweigh its risks’, but it also recommended changes to the product information to ensure that it is used appropriately by making prescribers aware that the medicine had been associated with an increased mortality in clinical studies Citation[64].
4. Fosfomycin
4.1 Mode of action – in vitro activity and interactions
Fosfomycin, a revived antibiotic, discovered in Spain in 1969, belongs to the class of phosphonic compounds Citation[65]. Fosfomycin inhibits phosphoenolpyruvate transferase, the first enzyme involved in the synthesis of peptidoglycan, inhibiting cell-wall synthesis Citation[66]. It is an advantageous molecule because, among all known antibiotics, it has the smallest molecular mass (138 Da), ensuring extensive diffusibility Citation[67]. Fosfomycin tromethamine, a soluble salt of fosfomycin, is licensed in several parts of the world to be given as single-dose oral therapy for uncomplicated urinary tract infections (UTIs) in women caused by E. coli and Enterococcus faecalis Citation[68]. The relevant available formulation for i.v. administration is fosfomycin disodium.
Fosfomycin is active against a broad spectrum of Gram-positive and Gram-negative bacteria, possessing a low potential for cross resistance with other classes of antibiotics Citation[65,66]. In a review of 11 studies, among 5057 isolates of Enterobacteriaceae, including E. coli (2205 strains) and K. pneumoniae (764 strains), 88% of which produced ESBL, 91.3% were found susceptible to fosfomycin Citation[69]. Recently, fosfomycin was evaluated against 542 consecutive non-duplicate urine isolates of ESBL-producing E. coli and K. pneumoniae versus non-ESBL-producing strains Citation[70]. Susceptibilities of E. coli were 86 versus 97% for ESBL- and non-ESBL-producing isolates and 62 versus 78% for the relevant K. pneumoniae strains, with only amikacin and imipenem being more active. Fosfomycin activity against carbapenem-resistant enterobacteriaceae and preferably K. pneumoniae strains has been also shown Citation[71,72] with susceptibilities increasing between 93 to 99.04% and 95% for serine and metallo-β-lactamase-producing strains Citation[71-73]. Regarding the non-fermenters, Acinetobacter spp. is inherently resistant to fosfomycin, whereas P. aeruginosa, including MDR strains, are mostly sensitive Citation[74]. In a systematic review of 23 microbiological studies out of 1743 MDR P. aeruginosa, ≥ 90% of the isolates were found to be susceptible to fosfomycin. Regarding the interpretation of susceptibilities, either the older Clinical and Laboratory Standards Institute criteria used for E. coli in UTIs (i.e., susceptible at MICs of ≤ 64 μg/ml or with zones of ≥ 16 mm) or the newer EUCAST and lower clinical breakpoints for enterobacteriaceae (i.e., susceptible at MIC of ≤ 32 μg/ml) should be applied Citation[72,75]. As it concerns the in vitro interactions of fosfomycin with other antibiotics against MDR Gram-negatives, out of several studies, the following can be concluded Citation[76-78]: i) against KPC(+) strains, synergy was mostly observed with carbapenems in 30 – 78% of the tested isolates, as well as with colistin or tigecycline in ∼ 35%, whereas results were unpredictable with aminoglycosides, cephalosporins and ciprofloxacin combinations. Resistance development to fosfomycin was shown in 100% of the control strains, which however was decreased to 53.8, 69.2 and 81.8% after fosfomycin is combined with meropenem, colistin or gentamicin, respectively Citation[78]; and ii) against K. pneumoniae OXA-48 producers, combination of fosfomycin with imipenem, meropenem and tigecycline expressed synergy in 42, 33 and 33% of the strains, respectively, whereas the combination of fosfomycin with colistin was antagonistic against all tested isolates Citation[79]. Regarding P. aeruginosa, synergy was observed with ciprofloxacin in 27 – 78.4% of strains, with amikacin in 18.8 – 100%, with imipenem in 0 – 73.3% and with ceftazidime in 0 – 70% Citation[76]. Recently the combinations of fosfomycin with minocycline and polymyxin B against pandrug-resistant (PDR) A. baumannii demonstrated either synergistic or additive results Citation[80].
4.2 Pharmacokinetics/pharmacodynamics
In serious systemic infections, fosfomycin is usually prescribed at a i.v. dose of 4 – 8 g , 2 h infusion every 8 h Citation[65-67]. In uncomplicated UTIs, 3 g per os [p.o.] as a single dose is adequate, whereas in complicated UTIs 3 g p.o. every 2 – 3 days up to 21 days on an empty stomach should be given Citation[68]. After 4 g i.v. dose, Cmax ranges between 105 and 120 μg/ml, whereas after doubling the dose to 8 g, 260 – 442 μg/ml have been detected with a half-life of 3.7 ± 2.2 h Citation[67,81]. Fosfomycin being hydrophilic is exclusively eliminated via glomerular filtration, with its clearance correlated with glomerular filtration rate Citation[82]. In healthy volunteers, volume distribution (Vd) of 20.6 L comparable with β-lactams and the aminoglycosides has been observed Citation[83]; however, in the critically ill patients, as a result of changes in the vascular endothelium causing increased capillary permeability Citation[67], Vd is increased as much as 50% resulting in lower Cmax reaching 35%. Fosfomycin is not metabolized and is not protein-bound, which is an advantage for noninflamed tissues Citation[84]. It is totally removed by hemodialysis and therefore re-dosing at the end of the session is necessary, whereas in critically ill patients undergoing continuous veno-venous hemofiltration, > 75% of fosfomycin is removed, not necessitating dosage adjustment Citation[81]. The presence of hepatic insufficiency does not require any dosage modification Citation[81]. Fosfomycin is bactericidal with a post-antibiotic effect of 3.4 – 4.7 h for enterobacteriaceae and an AUC of 929 ± 280 mgh/l Citation[82,84]. As with the β-lactams, the main PD parameter is %T>MIC, which should be ≥ 40% Citation[67]. Therefore, an MIC of 8 μg/ml succeeds the target attainment at 98%, an MIC of 16 μg/ml at 92% and an MIC of 32 μg/ml at 61%, indicating that if MICs are ≥ 32 μg/ml, the highest doses should be given Citation[81]. As depicted in the low fosfomycin molecular weight contributes to its high penetration ability into a variety of human tissues and compartments Citation[67,81,84-86].
Table 4. Pharmacokinetics of parenteral fosfomycin in various tissue and sites.
4.3 Clinical studies
Unfortunately, and despite fosfomycin's promising activity in non-MDR infections Citation[87], experience with XDR pathogens is still limited. In a recent French prospective cohort study, the efficacy of parenteral fosfomycin at a i.v. dose of 4 g every 8 h, mostly against MDR and XDR bacterial infections was analyzed in 116 adult and pediatric patients Citation[88]. The main indications for use were bacteremia, osteomyelitis, lung infection and UTI. Bacteria most frequently involved were P. aeruginosa and methicillin-resistant S. aureus. MDR microorganisms were isolated in 71.5% of cases, especially MDR P. aeruginosa (n = 28), among which 24 strains were XDR. Critical situations were common, with 44% of patients hospitalized in the ICU and 22.4% presented with septic shock. The overall outcome was favorable in 76.8% of cases. However, fosfomycin was given in combination with other antibiotics, to which susceptibilities of the pathogen were not clarified, thus hindering the real evaluation of the drug. The efficacy of oral fosfomycin in 41 hospitalized patients with MDR pathogens, including KPC(+)strains isolated in urine, who received an average of 2.9 doses of 3 g/day fosfomycin tromethamine, were reported in a retrospective study Citation[89]. Microbiologically, 59% of patients were cured, with most failures encountered in solid organ transplants associated also with the presence of a ureteral stent. In another retrospective study, fosfomycin was given at an i.v. dose of 2 g every 8 h in combination with either high dose 4 h infusion of 1 g doripenem or colistin (2.5 mg/kg every 12 h) in 49 patients with VAP or HAP caused by P. aeruginosa with meropenem and imipenem median MIC > 32 μg/ml and doripenem 4 μg/ml Citation[90]. No differences either in clinical cure (60 vs 58%) or bacterial eradication (72 vs 75%) and all-cause mortality (40 vs 42%) were observed. The most extensive study in 48 critically ill ICU patients treated with fosfomycin for infections due to PDR and XDR carbapenemase-producing Gram-negative bacteria was recently published Citation[91]. The study was multicentered, observational and prospective, and fosfomycin-treated patients suffered from XDR or PDR fosfomycin-susceptible, microbiologically documented infections, including mainly primary bacteremia (37.5%), catheter-related bacteremia (14.6%) and VAP (29.2%). Thirty patients (65.2%) were in severe sepsis/septic shock, whereas 85.4% had already unsuccessfully received antimicrobials with anti-Gram-negative activity (colistin 64.6%, tigecycline 35.4%, any carbapenem 27%, any aminoglycoside 27.1%, piperacillin/tazobactam 8.3%). KPC-2 predominated in 41 patients exhibiting an XDR profile in 26 patients and a PDR profile in 15 patients. In 17 patients, VIM-2 producing P. aeruginosa was isolated with an XDR profile in 16 of them. On admission to the study 83.3, 45.8 and 12.5% of patients were in respiratory, cardiovascular and renal failure, respectively, with a mean acute physiology and chronic health evaluation II score of 20.5 ± 7.6 and a median ICU length of stay of 34 (23 – 51) days. Fosfomycin was given i.v. at a median dose of 24 g/day (8 g every 8 h) for a median of 14 days, mainly in combination with colistin (66.7%) or tigecycline (39.6%) or gentamicin (31.3%). Overall clinical outcome at day 14 was successful in 54.2%, with failure in 33.3%, microbiological eradication in 56.3%, superinfection in 6.3% and resistance development to fosfomycin in 3 cases with crude mortality of 37.5% at day 28. In the reported study, there were two subsets of patients who served as probable surrogate markers of fosfomycin real efficacy. The first consisted of a subgroup of 15 patients with PDR-CPKP, in whom 5 were in septic shock and 4 in severe sepsis, for whom fosfomycin was the only active in vitro antibiotic. The reported 60% successful clinical and microbiological outcome should be considered as a proof of the real effectiveness of fosfomycin, the in vivo efficacy of which was equal to monotherapy, since it is very probable that without fosfomycin most patients should have died. In the second subgroup, 12 patients who did not respond to previous in vitro active antibiotics, had a successful therapeutic result of 50% with the addition of fosfomycin. Based on the latter reported results, there is no doubt that fosfomycin deserves further clinical evaluation in patients with XDR/PDR infections. On the other hand, development of resistance to fosfomycin monotherapy in vivo, as it is observed in vitro, is a major issue which still requires clarification. Mechanisms of resistance have been attributed either to mutations in the chromosomally encoded transport systems or to fosfomycin-modifying enzymes Citation[92]. However, resistant mutants, because of the biological cost they carry, exhibit decreased growth rate and low adherence to epithelial cells and their in vivo significance necessitating further studies. However, for the time being, it should be prudent, at least in the critically ill patients, to avoid monotherapy with fosfomycin.
4.4 Adverse events
Fosfomycin, in general, is a safe antibiotic with limited adverse events Citation[93]. The most significant is hypokalemia, observed in 20 – 25% of patients, attributed to a tubular effect on the kidney. On the other hand, the high sodium intake (1 g of i.v. fosfomycin possesses 0.33 g of sodium) could be a limitation in patients with heart or renal failure. Strongly, despite the frequent coadministration of aminoglycosides, the incidence of nephrotoxicity decreases, an event attributed to fosfomycin's protective effect on the lysosomal membrane integrity Citation[94].
5. Carbapenems
5.1 In vitro and in vivo activity against carbapenemase-producing strains
Until August 2012, > 120 carbapenemases have been described, hydrolyzing all carbapenems and almost all cephalosporins and β-lactams, including the inhibitors Citation[40]. The most important ones are the IMP types from P. aeruginosa, the KPC types mainly from K. pneumoniae, the NDM types from enterobacteriaceae, and the OXA types mostly from A. baumannii strains Citation[3]. Based on the in vitro observation that both VIM- and KPC-producing K. pneumoniae could posses low MICs to carbapenems (0.12 – 32 μg/ml), it was reported in a Greek prospective study on 162 consecutive patients with K. pneumoniae bacteremia that mortality rates in VIM-positive strains with meropenem MIC ≤ 4 μg/ml after the combination of two active in vitro antibiotics, one of which was meropenem, as 8.3 versus 27 and 27.8% whenever one active in vitro antibiotic or inappropriate therapy were given respectively Citation[4]. Similar results were also obtained in 298 patients compiled from 34 studies, in whom combination of a carbapenem with MIC ≤ 4 μg/ml with an active in vitro aminoglycoside or colistin or tigecycline were administered, provided that the carbapenem, depending on the time that blood levels are sustained above the MIC (T>MIC = 40 – 50%), is given in the highest dose and in prolonged infusions (3 h meropenem, 4 h doripenem) Citation[3,95]. However, in case of a carbapenem MIC > 4 μg/ml, the combination of two active in vitro antibiotics, excluding carbapenems, was superior in vivo to any active monotherapy. In two recent studies, the latter results were verified, indicating also that even for strains with carbapenem MICs 8 – 16 μg/ml, the combinations are advantageous, pointing out also the need of triple combination Citation[96,97].
5.2 Combination of two carbapenems
Recently the Bulik and Nicolau revolutionary successful approach of the combination of two carbapenems (double carbapenems [DC]) in the in vitro chemostat model, as well as in the in vivo thigh model, was applied for the first time in three patients Citation[98,99]. Two of them were septic, whereas in all three patients MICs to all carbapenems were high (> 32 μg/ml) as well as to all available antibiotics. The regimen included the administration of ertapenem, based on its increased affinity for KPCs, hindering subsequently doripenem or meropenem degradation in the environment of the targeted CPKP strains. Two patients suffered from PDR-KPC-2 K. pneumoniae bacteremia and one patient suffered from UTI. All responded successfully to the administration of 1 g ertapenem given every 24 h, followed after 1 h by 2 g meropenem every 8 h in 3 h infusion, without relapse at the follow up. Subsequently, 26 septic patients with XDR or PDR-CPKP bacteremia (17 patients), and UTIs (9 patients) were treated with the DC regimen with clinical success in 21 patients (80.7%), microbiological eradication in 25 patients (96%) and relapse in two patients Citation[100,101]. The obtained results render DC regimen a probable candidate therapeutic approach in XDR and PDR-CPKP infections that in the era of diminishing effective antimicrobials deserves further evaluation in well-controlled clinical studies in order to establish the real efficacy of the DC regimen.
6. Temocillin
6.1 In vitro activity
Temocillin is a β-a-methoxy-derivative of ticarcillin which was marketed by Beecham Pharmaceuticals in the UK in the 1980s Citation[102], and because of its interesting in vitro spectrum of activity, it was recently relaunched in the UK by Eumedica and is also available in Belgium and Luxembourg. The chemical modification of ticarcillin to temocillin increased its stability to β-lactamases in which ESBLs and the AmpC were included, with temocillin being very active against enterobacteriaceae but inactive against P. aeruginosa, Acinetobacter spp. and anaerobes. Against a collection of ESBL-producing E. coli harvested over a 4-year period harboring CTX-M+TEM (35%), TEM alone (44%), CTM-M alone (6%) and CTX-M+SHV (2%), the proportion of susceptible isolates to temocillin was 92% with MIC50/MIC90 of 8/32 μg/ml Citation[10]. Subsequently, in a vitro study against 652 clinical isolates of enterobacteriaceae collected prospectively from ICU patients in 2005, temocillin was active against almost all ESBL- and AmpC-producing enterobacteriaceae Citation[103]. Similar were the in vitro results from a collection of 846 survey isolates from England, including strains with ESBL and AmpC phenotypes, as well as Klebsiella oxytoca hyperproducers of K1 enzyme Citation[104]. The modal MIC values of temocillin were 8 μg/ml, whereas > 88% of the AmpC-and ESBL-producing strains were susceptible to < 16 μg/ml and 99% of the AmpC-and ESBL-producing strains were susceptible to ≤ 32 μg/ml. The activity of temocillin was also investigated against KPC-producing K. pneumoniae and E. coli strains with MICs ranging between 8 and 64 μg/ml, MIC50/MIC90 being both 32 μg/ml, whereas the frequencies of mutants were 1 × 10−10 at both 1 × MIC and at 2 × MICs Citation[105]. However OXA-48 and the metalloenzymes IMP, NDM and VIM conferred clear resistance to temocillin Citation[106]. Regarding the interpretation of susceptibilities to temocillin the British Society for Antimicrobial Chemotherapy defined MIC breakpoints for enterobacteriaceae as ≤ 8 and ≤ 32 μg/ml in systemic and UTIs, respectively, whereas, regarding zone diameters for a 30 μg temocillin disc, corresponding zone breakpoints were 20 and 12 mm, respectively Citation[106], the latter results of which was confirmed by the EUCAST methodology for disc diffusion susceptibility testing Citation[107].
6.2 Pharmacokinetics/pharmacodynamics
Temocillin PK/PD studies in ICU patients with various infections after an i.v. dose of 2 g every 12 h have determined the Cmax to be 147 ± 12 (85 – 223) μg/ml, a half-life to be 4.3 ± 0.3 (3.8 – 5.3) h, serum protein binding to be 85% and AUC24 (mg·h/l) to be 1856 ± 282 with renal clearance of 40.7 ± 6.5 Citation[108]. The Monte-Carlo simulation for target attainment at the twice-daily administration schedule proved that a fT > MIC of ≥ 40% for a MIC slightly above 8 μg/ml will be reached with a probability of 95% Citation[108].
6.3 Clinical studies
Despite being in the market for more than 20 years, clinical experience with temocillin is still very limited. Early clinical studies in 2006 and 2008 reported the efficacy of temocillin in Burkholderia cepacia infections in cystic fibrosis patients Citation[109]. Currently registered indications of temocillin in Belgium and the UK include UTIs, sepsis and lower respiratory tract infections due to enterobacteriaceae, mostly those producing ESBLs and derepressed AmpC cephalosporinases as an alternative to carbapenems. The largest experience on the efficacy of temocillin refers to a retrospective study in 92 patients in whom the drug was given for UTIs, bacteremia and HAP caused by enterobacteriaceae producing ESBL and/or derepressed AmpC β-lactamases Citation[110]. Clinical and bacteriological efficacy increased to 86 and 84%, respectively, thus supporting the possibility of carbapenem-sparing alternatives. However, the significance of an optimal therapeutic regimen of 2 g every 12 h or renally adjusted equivalent when compared with a suboptimal dosage was pointed out since clinical efficacy of 91% dropped to 73% and microbiological eradication dropped from 92 to 63%. Unfortunately, to our knowledge, no clinical experience with temocillin in the treatment of KPC-producing enterobacteriaceae has been reported. Based on temocillin in vitro activity, it is evident that the latter revived β-lactam deserves further clinical experience in a variety of serious MDR infections. Probably a higher dose would permit treatment of isolates with MICs of 16 ×μg/ml, whereas, in the critically ill patient, the addition of an antipseudomonal agent should be considered.
7. Newer drugs
7.1 Ceftolozane/tazobactam
Ceftolozane is a novel cephalosporin which is currently combined with the β-lactamase inhibitor tazobactam in a fixed 2:1 ratio. Ceftolozane inhibits the penicillin-binding proteins exhibiting greater affinity to the essential ones Citation[111]. Ceftolozane has demonstrated increased stability to AmpC β-lactamases and potent activity against P. aeruginosa with MICs at 4- to 16-fold dilutions below the comparative MICs for ceftazidime, whereas it has been shown to be unaffected by upregulation of efflux pumps or loss of porin channels Citation[111]. However, it is not active against carbapenemase-producing Gram-negative bacteria. The addition of tazobactam broadens the spectrum of ceftolozane, including ESBL producers and anaerobic species Citation[112]. In healthy adults, Cmax and plasma half-life for a dosage of 1000/500 mg and 2000/1000 mg infused over 60 min every 8 h were 74.4 mg/l and 3.12 h and 117 mg/l and 2.67 h, respectively. Ceftolozane is primarily eliminated via urinary excretion (≥ 92%), and dose adjustments is required in patients with a CrCL < 50 ml/min. Ceftolozane/tazobactam exhibits excellent lung penetration and could be considered a potential candidate for nosocomial pneumonia Citation[111,113]. The drug has completed Phase III trial (). The most common adverse events reported are gastrointestinal and sleep disorders, headache and infusion-site reactions Citation[111,113].
Table 5. Phase II and Phase III of newer antibiotics.
7.2 Plazomicin
Plazomicin is a semisynthetic derivative of sisomicin with significantly improved activity against strains that are amikacin- or gentamicin-resistant Citation[9]. Plazomicin also manifests bactericidal activity against AmpC cephalosporinases and ESBL-producing pathogens, including fluoroquinolone, aminoglycoside-resistant and carbapenemases-producing Gram-negative bacteria with the exception of Proteus species. However, strains with the presence of either an ArmA or RmtC 16S rRNA methyltransferase, often apparent in isolates carrying the NDM-1 carbapenemase, are resistant. Plazomicin is not hydrolyzed by any known aminoglycoside-modifying enzymes apart from N-acetlyltransferases AAC (2′)-Ia, -Ib and -Ic (only found in Providencia spp.) Citation[9]. Plazomicin exhibits in vitro synergy against P. aeruginosa when combined with cefepime, doripenem, imipenem or piperacillin/tazobactam. After i.v. administration to humans at a dose of 15 mg/kg, the Cmax was 113 μg/ml, with the AUC0 – 24 of 235 h·μg/ml, the half-life of 4 h and the steady-state Vd of 0.248 L/kg with lung penetration (calculated as the ratio of ELF-to-plasma AUC) of approximately 13% Citation[9,114]. Plazomicin is eliminated renally (87%); thus, dose adjustments are required for moderate and severe renal dysfunction Citation[9,114,115]. The drug has completed four Phase I trials and one Phase II trial for the treatment of complicated UTIs and is entering a Phase III trial (). Surprisingly, no evidence of nephrotoxicity has been described in all trials and most adverse events reported are mild to moderate (i.e., tinnitus, nausea, dizziness and hypertension) Citation[115].
7.3 Eravacycline
Eravacycline is a novel fully synthetic tetracycline antibiotic with potent antibacterial activity spectrum, including enterobacteriaceae-producing ESBL, MDR A. baumannii, carbapenemase-producing isolates with the exception of P. aeruginosa and B. cepacia Citation[116]. The antibacterial activity of eravacycline has been shown to be minimally affected by expression of tetracycline-specific efflux and ribosomal protection mechanisms in clinical isolates and has demonstrated a greater than twofold better potency over tigecycline against significant number of isolates Citation[116]. In healthy volunteers, plasma PK was dose-dependent and linear, illustrating a relative increase with dose, reaching steady-state values of AUC of around 8000 ng·h/ml when given at 1.5 mg/kg/day and 12,688 ng·h/ml when given 1 mg/kg twice daily with a mean half-life of 35.5 h and renal clearance of 15.5% Citation[117]. Eravacycline concentrations in the ELF and alveolar macrophages achieved greater levels than plasma by 6- and 50-fold, respectively, supporting further study in patients with respiratory infections Citation[118]. Eravacycline has completed a Phase II study and has commenced a Phase III study on cIAIs (). The most common adverse events were gastrointestinal, administration site and vascular disorders. Interestingly, eravacycline is also available in an oral formula Citation[119].
7.4 Avibactam
Avibactam is a novel synthetic, β-lactamase inhibitor that hinders the activities of several β-lactam hydrolyzing enzymes and has been combined with ceftazidime currently in Phase III clinical trials Citation[11]. It is active against strains producing ESBL, AmpC and KPC enzymes, including carbapenem-resistant isolates due to porin loss. However, metallo-β-lactamases, that is, VIM, IMP, NDM, are resistant. In healthy volunteers, avibactam has a Vd of 20 – 24 l, a half-life of 1.5 – 2.7 h, an average clearance of 10.4 – 13.8 l/h and Cmax of 100 mg/l Citation[11]. Elimination of avibactam is predominantly by renal excretion of unchanged drug, and thus dosage adjustment is required in renal impairment. Clinical trials to date suggest that ceftazidime-avibactam is as effective as standard carbapenem therapy in cIAIs and cUTIs, including those caused by ceftazidime-resistant Gram-negative bacilli () Citation[120,121]. The most common reported adverse events were nausea, vomiting, abdominal pain, pyrexia and elevations in liver enzymes Citation[120,121].
8. Expert opinion
Infections due to MDR-XDR-PDR Gram-negatives, particularly in the critically ill ICU patients, nowadays represent a reality as well as a threat worldwide. K. pneumoniae, P. aeruginosa and A. baumannii producing MBL, KPC, OXA and NDM carbapenemases are the commonest implicated microorganisms causing mostly bacteremia and VAP, followed by mortality exceeding 50% in most series, attributed both to the virulence as well as to the lack of appropriate antimicrobial therapy. Therefore, it was necessary that the older antibiotics of the 1970s sustaining their in vitro activity against the former microorganisms be revived, whereas newer antibiotics overcoming resistance mechanisms be developed.
Colistin is the major representative of revived antibiotics with the broadest spectrum of activity. A wide range of efficacy in nosocomial MDR and XDR infections has been reported ranging between 25 and 71%; however, conducted trials are mostly retrospective, nonrandomized, with dosage heterogeneity and simultaneous administration of other active in vitro antibiotics, rendering therefore inconclusive the efficacy of monotherapy with colistin. It should be also pointed out that due to the advancements in methodological assays a tremendous progress in PK/PDs of colistin has been achieved, permitting in vivo application of more accurate dosage schedules. However, in bacteremias and in VAP caused by carbapenemase-producing enterobacteriaceae, the combination with another or even two active in vitro antibiotics (e.g., tigecycline or gentamicin or meropenem/doripenem [with MIC ≤ 8 μg/ml]) seems to be prioritized. On the other hand, the role of inhaled colistin in VAP based on therapeutic discrepancies is still obscure, necessitating well-controlled studies and reliable nebulizers. As it concerns safety issues, nephrotoxicity, the major adverse effect in the past, is nowadays lower, whereas neurotoxicity is rather negligible. Unfortunately, prolonged and unjustified therapy with colistin was followed by resistance development, thus mandating appropriate duration of therapy as well as rapid de-escalation when appropriate.
Tigecycline, a modified minocycline that overcomes resistance mechanisms that are encountered with the older tetracyclines, is potent in vitro against MDR-XDR K. pneumoniae and A. baumannii. The in vivo activity of tigecycline, which represent an off-label indication, is rather promising in the critically ill host but combination therapy is a priority. The major problem with tigecycline is the low dosage schedule which creates low-blood levels, frequently not exceeding the MICs of the incriminated pathogens, leading to failure in case of bacteremias and HAP. Therefore, clinical trials are required to define the most appropriate therapeutic schedules. On the other hand, the two FDA warnings in 2010 and 2013, which connect tigecycline with increased death rates, are of concern. Probably the latter serious notification necessitates further clarification.
Fosfomycin, another revived antibiotic of the 1970s, based on its mode of action, is advantageous because it does not share cross resistance with other classes of antibiotics, being active in vitro against MDR-XDR enterobacteriaceae and P. aeruginosa. Unfortunately, fosfomycin is still an almost ‘unknown’ antibiotic since clinical experience is mostly based on UTIs and gastrointestinal infections, with limited experience in MDR-XDR pathogens. Its PK/PD necessitates further exploration in order to determine the appropriate therapeutic regimen, whereas the possibility of monotherapy to induce resistance in vivo requires careful clinical studies.
Temocillin, a modified ticarcillin of the 1980s, based on its activity in vitro against ESBL-producing enterobacteriaceae, including KPC producers, is currently relaunched in some European countries. Despite promising clinical efficacy, mostly in UTIs, the exact dosage schedule requires remodeling, since by increasing the dose, even bacteremias with MIC above the sensitivity breakpoints could be captured. Unfortunately, temocillin limited distribution requires availability at least in countries with high prevalence of MDR pathogens.
Among the newer antimicrobial agents, still under Phase III trial, two of them appear as very promising, namely, plazomicin and avibactam. Plazomicin, a non-nephrotoxic aminoglycoside, is a semisystemic derivative of sisomicin. In vitro it is active against ESBL and carbapenemase-producing Gram-negatives; however, in case of 16SrRNA methyltransferase production, the drug is inactivated. Therefore, before broader application of plazomicin, epidemiological studies to determine the latter enzyme worldwide distribution are required. In vivo plazomicin activity is proved up to now only in UTIs, whereas results in bacteremias and HAP in seriously ill patients with carbapenemase-producing pathogens in comparison to colistin are pending. On the other hand, avibactam, a β-lactamase inhibitor inactivating the ESBLs, AmpC, KPC, OXA-48 carbapenemase, and ceftazidime, is under Phase III evaluation to meropenem or doripenem in HAP, cUTIs, as well as in cIAI.
It is evident that there is still a long route before newer compounds active against XDR and PDR strains become available in the market. In the meantime, how should the clinicians confront their critically ill XDR-PDR infected patients? A new revolutionary approach based on the combination of two carbapenems (ertapenem plus doripenem or meropenem), which at the molecular level binds KPC carbapenemases, seems to be promising. On the other hand, clinical trials in off-label indications of the above-discussed antibiotics, following fast procedures, are urgently required.
There is no doubt that it is time for the clinicians, when facing the critical shortage of new active antibiotics, to react by applying a multifaceted interventional approach based mainly on antimicrobial stewardship programs, which every physician is called to apply and proclaim and to follow strictly as well.
Article highlights.
Resistance to carbapenems in Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa, mainly determined by the production of carbapenem-hydrolyzing β-lactamases, have emerged worldwide with a mortality exceeding 50%, which is attributed mainly to the lack of effective antimicrobial regimens.
Colistin, a polypeptide antibiotic of 1950s, has reentered clinical practice due to emergence of resistance. It is systemically administrated in the form of colistin methanesulfonate (CMS) which is hydrolyzed to colistin that exhibits antibacterial activity. With the introduction of new assays that distinguish CMS from colistin, more accurate dosage schedules have been implied with the application of a loading dose and maintenance dose with longer intervals. Clinical efficacy varies but is based mainly on heterogeneous retrospective studies. Therefore, randomized prospective clinical studies are warranted.
Tigecycline, a modified minocycline derivative is potent in vitro against multidrug-resistant (MDR)–extensively drug-resistant (XDR) K. pneumoniae and A. baumannii and has been utilized off-label in critically ill patients. However, due to disadvantageous PK, combination treatment and higher doses are suggested.
Fosfomycin, a revived antibiotic of 1970s, is active in vitro against MDR – XDR enterobacteriaceae and P. aeruginosa has been used in the past for urinary tract infections (UTIs) and gastrointestinal infections. Due to increasing antimicrobial resistance rates, high dose intravenous fosfomycin is timidly administrated in critically ills patient with XDR strains with promising results.
Temocillin, active against extended spectrum β lactamase (ESBL)-producing enterobacteriaceae and K. pneumoniae carbapenemase (KPC) producers, is currently relaunched in some European countries. Clinical studies demonstrating efficacy are mainly associated with UTIs, bacteremia and hospital-acquired pneumonia.
Newer drugs in Phase III studies for the combat of MDR and XDR Gram-negative strains include ceftolozane, a novel cephalosporin, active against AmpC β-lactamases and P. aeruginosa unaffected by efflux pump or loss of porin channels, combined with tazobactam. Plazomicin is a semisynthetic derivative of sisomycin, active against ESBL, carbapenemase producers with the exception of strains with the presence of either ArmA or RmtC 16srRNA methyltransferase. Eravacycline, a fully synthetic tetracycline antibiotic is active against ESBL, MDR A. baumannii, carbapenemase-producing strains with the exception of P. aeruginosa and Burkholderia cepacia. Avibactam, a synthetic non-β-lactam, β-lactamase inhibitor, when combined with ceftazidime is active against ESBL, AmpC and KPC producers, but not metallo β-lactamases, that is, VIM, IMP and NDM-1.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
Notes
This box summarizes key points contained in the article.
Bibliography
- Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012;18:268-81
- Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 2009;48:1-12
- Tzouvelekis LS, Markogiannakis A, Psichogiou M, et al. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 2012;25:682-707
- Daikos GL, Petrikkos P, Psichogiou M, et al. Prospective observational study of the impact of VIM-1 metallo-beta-lactamase on the outcome of patients with Klebsiella pneumoniae bloodstream infections. Antimicrob Agents Chemother 2009;53:1868-73
- Giamarellou H, Poulakou G. Multidrug-resistant Gram-negative infections: what are the treatment options? Drugs 2009;69:1879-901
- Antoniadou A, Kontopidou F, Poulakou G, et al. Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: first report of a multiclonal cluster. J Antimicrob Chemother 2007;59:786-90
- Marchaim D, Chopra T, Pogue JM, et al. Outbreak of colistin-resistant, carbapenem-resistant Klebsiella pneumoniae in metropolitan Detroit, Michigan. Antimicrob Agents Chemother 2011;55:593-9
- Kontopidou F, Plachouras D, Papadomichelakis E, et al. Colonization and infection by colistin-resistant Gram-negative bacteria in a cohort of critically ill patients. Clin Microbiol Infect 2011;17:E9-E11
- Galani I. Plazomicin. Drugs Future 2014;39:25-35
- Rodriguez-Villalobos H, Malaviolle V, Frankard J, et al. In vitro activity of temocillin against extended spectrum beta-lactamase-producing Escherichia coli. J Antimicrob Chemother 2006;57:771-4
- Zhanel GG, Lawson CD, Adam H, et al. Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination. Drugs 2013;73:159-77
- Nation RL, Li J, Turnidge JD. The urgent need for clear and accurate information on the polymyxins. Clin Infect Dis 2013;57:1656-7
- Giamarellos-Bourboulis EJ, Xirouchaki E, Giamarellou H. Interactions of colistin and rifampin on multidrug-resistant Acinetobacter baumannii. Diagn Microbiol Infect Dis 2001;40:117-20
- Galani I, Orlandou K, Moraitou H, et al. Colistin/daptomycin: an unconventional antimicrobial combination synergistic in vitro against multidrug-resistant Acinetobacter baumannii. Int J Antimicrob Agents 2014; published online 4 February 2014, doi: 10.1016/j.ijantimicag.2013.12.010
- Bergen PJ, Landersdorfer CB, Zhang J, et al. Pharmacokinetics and pharmacodynamics of ‘old' polymyxins: what is new? Diagn Microbiol Infect Dis 2012;74:213-23
- Plachouras D, Karvanen M, Friberg LE, et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by gram-negative bacteria. Antimicrob Agents Chemother 2009;53:3430-6
- Mohamed AF, Karaiskos I, Plachouras D, et al. Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother 2012;56:4241-9
- Karaiskos I, Eriberg LE, Pontikis K, et al. Colistin pharmacokinetics after application of a loading dose of 9MU colistin methanesulfonate (CMS) in critically ill patients. Poster A1054b. 53rd Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), 10 – 13 September 2013; Denver, Co
- Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011;55:3284-94
- Garonzik S, Forrest A, Li J, et al. Population pharmacokinetic (popPK)-model of colistin (C) & colistin methanesulfonate (CMS) in critically-ill patients not on renal replacement therapy (RRT). O576, 23rd European Society of Clinical Microbiology and Infectious Diseases (ECCMID); 27 – 30 April 2013, Berlin, Germany
- Karvanen M, Plachouras D, Friberg LE, et al. Colistin methanesulfonate and colistin pharmacokinetics in critically ill patients receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother 2013;57:668-71
- Koomanachai P, Landersdorfer CB, Chen G, et al. Pharmacokinetics of colistin methanesulfonate and formed colistin in end-stage renal disease patients receiving continuous ambulatory peritoneal dialysis. Antimicrob Agents Chemother 2014;58:440-6
- Molina J, Cordero E, Pachón J. New information about the polymyxin/colistin class of antibiotics. Expert Opin Pharmacother 2009;10:2811-28
- Florescu DF, Qiu F, McCartan MA, et al. What is the efficacy and safety of colistin for the treatment of ventilator-associated pneumonia? A systematic review and meta-regression. Clin Infect Dis 2012;54:670-80
- Durante-Mangoni E, Signoriello G, Andini R, et al. Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: a multicenter, randomized clinical trial. Clin Infect Dis 2013;57(3):349-58
- Falagas ME, Rafailidis PI, Ioannidou E, et al. Colistin therapy for microbiologically documented multidrug-resistant Gram-negative bacterial infections: a retrospective cohort study of 258 patients. Int J Antimicrob Agents 2010;35:194-9
- Hu Y, Li L, Li W, et al. Combination antibiotic therapy versus monotherapy for Pseudomonas aeruginosa bacteraemia: a meta-analysis of retrospective and prospective studies. Int J Antimicrob Agents 2013;42:492-6
- Dalfino L, Puntillo F, Mosca A, et al. High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 2012;54:1720-6
- Vicari G, Bauer SR, Neuner EA, Lam SW. Association between colistin dose and microbiologic outcomes in patients with multidrug-resistant gram-negative bacteremia. Clin Infect Dis 2013;56:398-404
- Petrosillo N, Giannella M, Antonelli M, et al. Clinical experience of colistin-glycopeptide combination in critically ill patients infected with Gram-negative bacteria. Antimicrob Agents Chemother 2014;58:851-8
- Garnacho-Montero J, Amaya-Villar R, Gutiérrez-Pizarraya A, et al. Clinical efficacy and safety of the combination of colistin plus vancomycin for the treatment of severe infections caused by carbapenem-resistant Acinetobacter baumannii. Chemotherapy 2013;59:225-31
- Lu Q, Luo R, Bodin L, et al. Efficacy of high-dose nebulized colistin in ventilator-associated pneumonia caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Anesthsiology 2012;117:1335-47
- Tumbarello M, De Pascale G, Trecarichi EM, et al. Effect of aerosolized colistin as adjunctive treatment on the outcomes of microbiologically documented ventilator-associated pneumonia caused by colistin-only susceptible gram-negative bacteria. Chest 2013;144:1768-75
- Athanassa ZE, Markantonis SL, Fousteri MZ, et al. Pharmacokinetics of inhaled colistimethate sodium (CMS) in mechanically ventilated critically ill patients. Intensive Care Med 2012;38:1779-86
- Karaiskos I, Galani L, Baziaka F, Giamarellou H. Intraventricular and intrathecal colistin as the last therapeutic resort for the treatment of multidrug-resistant and extensively drug-resistant Acinetobacter baumannii ventriculitis and meningitis: a literature review. Int J Antimicrob Agents 2013;41:499-508
- Spapen H, Jacobs R, Van Gorp V, et al. Renal and neurological side effects of colistin in critically ill patients. Ann Intensive Care 2011;1:14
- Bellomo R, Ronco C, Kellum JA, et al. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004;8:R204-12
- Pogue JM, Lee J, Marchaim D, et al. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011;53:879-84
- Stein GE, Babinchak T. Tigecycline: an update. Diagn Microbiol Infect Dis 2013;75:331-6
- Hanberger H, Giske CG, Giamarellou H. When and how to cover for resistant gram-negative bacilli in severe sepsis and septic shock. Curr Infect Dis Rep 2011;13:416-25
- Castanheira M, Sader HS, Deshpande LM, et al. Antimicrobial activities of tigecycline and other broad-spectrum antimicrobials tested against serine carbapenemase-and metallo-beta-lactamase-producing Enterobacteriaceae: report from the SENTRY Antimicrobial Surveillance Program. Antimicrob Agents Chemother 2008;52:570-3
- Zarkotou O, Pournaras S, Altouvas G, et al. Comparative evaluation of tigecycline susceptibility testing methods for expanded-spectrum cephalosporin- and carbapenem-resistant gram-negative pathogens. J Clin Microbiol 2012;50:3747-50
- Denys GA, Callister SM, Dowzicky MJ. Antimicrobial susceptibility among gram-negative isolates collected in the USA between 2005 and 2011 as part of the Tigecycline Evaluation and Surveillance Trial (T.E.S.T.). Ann Clin Microbiol Antimicrob 2013;12-12
- Sader HS, Flamm RK, Jones RN. Tigecycline activity tested against antimicrobial resistant surveillance subsets of clinical bacteria collected worldwide (2011). Diagn Microbiol Infect Dis 2013;76:217-21
- Clinical Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. 23rd Informational Supplement M100-S23. CLSI; Wayne, PA, USA; 2013
- The European Committee on Antimicrobial Susceptibility Testing-EUCAST. Clinical Breakpoints. Available from: http://www.eucast.org/ [Accessed 1 January 2014]
- Vouillamoz J, Moreillon P, Giddey M, Entenza JM. In vitro activities of tigecycline combined with other antimicrobials against multiresistant gram-positive and gram-negative pathogens. J Antimicrob Chemother 2008;61:371-4
- Nigo M, Cevallos CS, Woods K, et al. Nested case-control study of the emergence of tigecycline resistance in multidrug-resistant Klebsiella pneumoniae. Antimicrob Agents and Chemother 2013;57:5743-6
- Giamarellou H, Poulakou G. Pharmacokinetic and pharmacodynamic evaluation of tigecycline. Expert Opin Drug Metab Toxicol 2011;7:1459-70
- Barbour A, Schmidt S, Ma B, et al. Clinical pharmacokinetics and pharmacodynamics of tigecycline. Clin Pharmacokinet 2009;48:575-84
- Xie J, Wang T, Sun J, et al. Optimal tigecycline dosage regimen is urgently needed: results from a pharmacokinetic/pharmacodynamic analysis of tigecycline by Monte Carlo simulation. Int J Infect Dis 2014;18:62-7
- Zavascki AP, Bulitta JB, Landersdorfer CB. Combination therapy for carbapenem-resistant Gram-negative bacteria. Expert Rev Anti Infect Ther 2013;11:1333-53
- Anthony KB, Fishman NO, Linkin DR, et al. Clinical and microbiological outcomes of serious infections with multidrug-resistant gram-negative organisms treated with tigecycline. Clin Infect Dis 2008;46:567-70
- Curcio D, Fernández F, Vergara J, et al. Late onset ventilator-associated pneumonia due to multidrug-resistant Acinetobacter spp.: experience with tigecycline. J Chemother 2009;21:58-62
- Kontopidou F, Giamarellou H, Katerelos P, et al. Infections caused by carbapenem-resistant Klebsiella pneumoniae among patients in intensive care units in Greece: a multi-centre study on clinical outcome and therapeutic options. Clin Microbiol Infect 2014;20:O117-23
- Poulakou G, Kontopidou FV, Paramythiotou E, et al. Tigecycline in the treatment of infections from multi-drug resistant gram-negative pathogens. J Infect 2009;58:273-84
- Freire AT, Melnyk V, Kim MJ, et al. Comparison of tigecycline with imipenem/cilastatin for the treatment of hospital-acquired pneumonia. Diagn Microbiol Infect Dis 2010;68:140-51
- Ramirez J, Dartois N, Gandjini H, et al. Randomized phase 2 trial to evaluate the clinical efficacy of two high-dosage tigecycline regimens versus imipenem-cilastatin for treatment of hospital-acquired pneumonia. Antimicrob Agents Chemother 2013;57:1756-62
- Lee YT, Tsao SM, Hsueh PR. Clinical outcomes of tigecycline alone or in combination with other antimicrobial agents for the treatment of patients with healthcare-associated multidrug-resistant Acinetobacter baumannii infections. Eur J Clin Microbiol Infect Dis 2013;32:1211-20
- FDA. A drug safety communication: increased risk of death with Tygacil (tigecycline) compared to other antibiotics used to treat similar infections. 2010. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm224370.htm [Accessed 23 October 2012]
- FDA. A drug safety communication: FDA warns of increased risk of death with IV antibacterial Tygacil (tigecycline) and approves new Boxed Warning. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm369580.htm [Accessed 27 September 2013]
- McGovern PC, Wible M, El-Tahtawy A, et al. All-cause mortality imbalance in the tigecycline phase 3 and 4 clinical trials. Int J Antimicrob Agents 2013;41:463-7
- Tasina E, Haidich AB, Kokkali S, Arvanitidou M. Efficacy and safety of tigecycline for the treatment of infectious diseases: a meta-analysis. Lancet Infect Dis 2011;11:834-44
- EMA. Questions and answers on the review of Tygacil (tigecycline). Outcome of a renewal procedure CMA/121925/2011, EMA/H/C/000644/R/54. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/ human/000644/WC500102228.pdf [Accessed 17 February 2011]
- Raz R. Fosfomycin: an old–new antibiotic. Clin Microbiol Infect 2012;18:4-7
- Popovic M, Steinort D, Pillai S, Joukhadar C. Fosfomycin: an old, new friend? Eur J Clin Microbiol Infect Dis 2010;29:127-42
- Parker S, Lipman J, Koulenti D, et al. What is the relevance of fosfomycin pharmacokinetics in the treatment of serious infections in critically ill patients? A systematic review. Int J Antimicrob Agents 2013;42:289-93
- Keating GM. Fosfomycin trometamol: a review of its use as a single-dose oral treatment for patients with acute lower urinary tract infections and pregnant women with asymptomatic bacteriuria. Drugs 2013;73:1951-66
- Falagas ME, Kastoris AC, Kapaskelis AM, Karageorgopoulos DE. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum beta-lactamase producing, Enterobacteriaceae infections: a systematic review. Lancet Infect Dis 2010;10:43-50
- Araj GF, Jaber FA. In vitro activity of fosfomycin and other antimicrobials against uropathogenic Escherichia coli and Klebsiella pneumoniae at a tertiary care center in Lebanon. J Med Liban 2012;60:142-7
- Falagas ME, Maraki S, Karageorgopoulos DE, et al. Antimicrobial susceptibility of multidrug-resistant (MDR) and extensively drug-resistant (XDR) Enterobacteriaceae isolates to fosfomycin. Int J Antimicrob Agents 2010;35:240-3
- Endimiani A, Patel G, Hujer KM, et al. In vitro activity of fosfomycin against blaKPC-containing Klebsiella pneumoniae isolates, including those nonsusceptible to tigecycline and/or colistin. Antimicrob Agents Chemother 2010;54:526-9
- Tuon FF, Rocha JL, Formighieri MS, et al. Fosfomycin susceptibility of isolates with blaKPC-2 from Brazil. J Infect 2013;67:247-9
- Falagas ME, Kastoris AC, Karageorgopoulos DE, Rafailidis PI. Fosfomycin for the treatment of infections caused by multidrug-resistant non-fermenting Gram-negative bacilli: a systematic review of microbiological, animal and clinical studies. Int J Antimicrob Agents 2009;34:111-20
- Clinical Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing; 19th informational supplement. CLSI document M100-S19. Clinical and Laboratory Standards Institute. CLSI; Wayne, PA, USA; 2009
- Kastoris AC, Rafailidis PI, Vouloumanou EK, et al. Synergy of fosfomycin with other antibiotics for Gram-positive and Gram-negative bacteria. Eur J Clin Pharmacol 2010;66:359-68
- Samonis G, Maraki S, Karageorgopoulos DE, et al. Synergy of fosfomycin with carbapenems, colistin, netilmicin, and tigecycline against multidrug-resistant Klebsiella pneumoniae, Escherichia coli, and Pseudomonas aeruginosa clinical isolates. Eur J Clin Microbiol Infect Dis 2012;31:695-701
- Souli M, Galani I, Boukovalas S, et al. In vitro interactions of antimicrobial combinations with fosfomycin against KPC-2-producing Klebsiella pneumoniae and protection of resistance development. Antimicrob Agents Chemother 2011;55:2395-7
- Evren E, Azap OK, Çolakoğlu Ş, Arslan H. In vitro activity of fosfomycin in combination with imipenem, meropenem, colistin and tigecycline against OXA 48-positive Klebsiella pneumoniae strains. Diagn Microbiol Infect Dis 2013;76:335-8
- Zhang Y, Chen F, Sun E, et al. In vitro antibacterial activity of combinations of fosfomycin, minocycline and polymyxin B on pan-drug-resistant Acinetobacter baumannii. Exp Ther Med 2013;5:1737-9
- Roussos N, Karageorgopoulos DE, Samonis G, Falagas ME. Clinical significance of the pharmacokinetic and pharmacodynamic characteristics of fosfomycin for the treatment of patients with systemic infections. Int J Antimicrob Agents 2009;34:506-15
- Bergan T, Thorsteinsson SB, Albini E. Pharmacokinetic profile of fosfomycin trometamol. Chemotherapy 1993;39:297-301
- Frossard M, Joukhadar C, Erovic BM, et al. Distribution and antimicrobial activity of fosfomycin in the interstitial fluid of human soft tissues. Antimicrob Agents Chemother 2000;44:2728-32
- Sauermann R, Karch R, Langenberger H, et al. Antibiotic abscess penetration: fosfomycin levels measured in pus and simulated concentration-time profiles. Antimicrob Agents Chemother 2005;49:4448-54
- Pfausler B, Spiss H, Dittrich P, et al. Concentrations of fosfomycin in the cerebrospinal fluid of neurointensive care patients with ventriculostomy-associated ventriculitis. J Antimicrob Chemother 2004;53:848-52
- Gardiner BJ, Mahony AA, Ellis AG, et al. Is fosfomycin a potential treatment alternative for multidrug-resistant gram-negative prostatitis? Clin Infect Dis 2014;58:e101-5
- Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Fosfomycin: use beyond urinary tract and gastrointestinal infections. Clin Infect Dis 2008;46:1069-77
- Dinh A, Salomon J, Bru JP, Bernard L. Fosfomycin: efficacy against infections caused by multidrug-resistant bacteria. Scand J Infect Dis 2012;44:182-9
- Neuner EA, Sekeres J, Hall GS, van Duin D. Experience with fosfomycin for treatment of urinary tract infections due to multidrug-resistant organisms. Antimicrob Agents Chemother 2012;56:5744-8
- Apisarnthanarak A, Mundy LM. Carbapenem-resistant Pseudomonas aeruginosa pneumonia with intermediate minimum inhibitory concentrations to doripenem: combination therapy with high-dose, 4-h infusion of doripenem plus fosfomycin versus intravenous colistin plus fosfomycin. Int J Antimicrob Agents 2012;39:271-2
- Pontikis K, Karaiskos I, Bastani S, et al. Outcomes of critically ill intensive care unit patients treated with fosfomycin for infections due to pandrug-resistant and extensively drug-resistant carbapenemase-producing Gram-negative bacteria. Int J Antimicrob Agents 2014;43:52-9
- Karageorgopoulos DE, Wang R, Yu XH, Falagas ME. Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J Antimicrob Chemother 2012;67:255-68
- Florent A, Chichmanian RM, Cua E, Pulcini C. Adverse events associated with intravenous fosfomycin. Int J Antimicrob Agents 2011;37:82-3
- Morin JP, Olier B, Viotte G, Fillastre JP. Can fosfomycin reduce the nephrotoxicity of aminoglycosides? Pathol Biol (Paris) 1984;32:338-42
- Daikos GL, Markogiannakis A. Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 2011;17:1135-41
- Qureshi ZA, Paterson DL, Potoski BA, et al. Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 2012;56:2108-13
- Tumbarello M, Viale P, Viscoli C, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 2012;55:943-50
- Wiskirchen DE, Crandon JL, Nicolau DP. Impact of various conditions on the efficacy of dual carbapenem therapy against KPC-producing Klebsiella pneumoniae. Int J Antimicrob Agents 2013;41:582-5
- Giamarellou H, Galani L, Baziaka F, Karaiskos I. Effectiveness of a double-carbapenem regimen for infections in humans due to carbapenemase-producing pandrug-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 2013;57:2388-90
- Karaiskos I, Masgala A, Galani L, et al. Double carbapenem regimens for infections in humans due to carbapenemase-producing Klebsiella pneumoniae (CPKP). Poster K-186. 53rd Interscience Conference in Antimicrobial Agents and Chemotherapy (ICAAC); 10 – 13 September 2013; Denver, Co
- Nogid B, Venugopolan V, Bias T, et al. Double carbapenem therapy for bacteremia due to carbapenem resistant Klebsiella pneumoniae: from test tube to clinical practice. Poster 2110. 23rd European Congress of Clinical Microbiology and Infectious Diseases (ECCMID); 27 – 30 April 2013; Berlin, Germany
- Slocombe B, Basker MJ, Bentley PH, et al. BRL 17421, a novel beta-lactam antibiotic, highly resistant to beta-lactamases, giving high and prolonged serum levels in humans. Antimicrob Agents Chemother 1981;20:38-46
- Glupczynski Y, Huang TD, Berhin C, et al. In vitro activity of temocillin against prevalent extended-spectrum beta-lactamases producing Enterobacteriaceae from Belgian intensive care units. Eur J Clin Microbiol Infect Dis 2007;26:777-83
- Livermore DM, Hope R, Fagan EJ, et al. Activity of temocillin against prevalent ESBL-and AmpC-producing Enterobacteriaceae from south-east England. J Antimicrob Chemother 2006;57:1012-14
- Adams-Haduch JM, Potoski BA, Sidjabat HE, et al. Activity of temocillin against KPC-producing Klebsiella pneumoniae and Escherichia coli. Antimicrob Agents Chemother 2009;53:2700-1
- Woodford N, Pike R, Meunier D, et al. In vitro activity of temocillin againstmultidrug-resistant clinical isolates of Escherichia coli, Klebsiella spp. and Enterobacter spp., and evaluation of high-level temocillin resistance as a diagnostic marker for OXA-48 carbapenemase. J Antimicrob Chemother 2014;69:564-7
- Vanstone GL, Dilley R, Schwenk S, et al. Temocillin disc diffusion susceptibility testing by EUCAST methodology. J Antimicrob Chemother 2013;68:2688-9
- De Jongh R, Hens R, Basma V, et al. Continuous versus intermittent infusion of temocillin, a directed spectrum penicillin for intensive care patients with nosocomial pneumonia: stability, compatibility, population pharmacokinetic studies and breakpoint selection. J Antimicrob Chemother 2008;61:382-8
- Lekkas A, Gyi KM, Hodson ME. Temocillin in the treatment of Burkholderia cepacia infection in cystic fibrosis. J Cyst Fibros 2006;5:121-4
- Balakrishnan I, Awad-El-Kariem FM, Aali A, et al. Temocillin use in England: clinical and microbiological efficacies in infections caused by extended-spectrum and/or derepressed AmpC beta-lactamase-producing Enterobacteriaceae. J Antimicrob Chemother 2011;66:2628-31
- Zhanel GG, Chung P, Adam H, et al. Ceftolozane/tazobactam: a novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 2014;74:31-51
- Hong MC, Hsu DI, Bounthavong M. Ceftolozane/tazobactam: a novel antipseudomonal cephalosporin and β-lactamase-inhibitor combination. Infect Drug Resist 2013;29:215-23
- Miller B, Hershberger E, Benziger D, et al. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob Agents Chemother 2012;56:3086-91
- Zhanel GG, Lawson CD, Zelenitsky S, et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther 2012;10:459-73
- Riddle V, Cebrik D, Armstrong E, et al. Plazomicin safety and efficacy in patients with complicated urinary tract infection (cUTI) or acute pyelonephritis (AP) [abstract L2-2118a]. 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); 9 - 12 September 2012; San Francisco, USA
- Sutcliffe JA, O'Brien W, Fyfe C, Grossman TH. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob Agents Chemother 2013;57:5548-58
- Sutcliffe JA. Antibiotics in development targeting protein synthesis. Ann NY Acad Sci 2011;1241:122-5
- Connors KP, Housman ST, Pope JS, et al.A Phase I. Open-Label, Safety and Pharmacokinetic Study to Assess Bronchopulmonary Disposition of Intravenous Eravacycline in Healthy Men and Women. Antimicrob Agents Chemother 2014; published online 27 January 2014, doi: 10.1128/AAC.02036-13
- Solomkin JS, Ramesh MK, Cesnauskas G, et al. Efficacy and safety of two dose regimens of eravacycline versus ertapenem in adult community-acquired complicated intra-abdominal infections: results of a phase 2, randomized, double-blind study. Antimicrob Agents Chemother 2013; published online 16 December 2013, doi: 10.1128/AAC.01614-13
- Lucasti C, Popescu I, Ramesh MK, et al. Comparative study of the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infections in hospitalized adults: results of a randomized, double-blind, Phase II trial. J Antimicrob Chemother 2013;68:1183-92
- Vazquez JA, González Patzán LD, Stricklin D, et al. Efficacy and safety of ceftazidime-avibactam versus imipenem-cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: results of a prospective, investigator-blinded, randomized study. Curr Med Res Opin 2012;28:1921-31