
Abstract
Standard therapy for phenylketonuria (PKU), the most common inherited disorder in amino acid metabolism, is an onerous phenylalanine-restricted diet. Adherence to this stringent diet regimen decreases as patients get older, and this lack of adherence is directly associated with cognitive and executive dysfunction and psychiatric issues. These factors emphasize the need for alternative pharmacological therapies to help treat patients with PKU. Sapropterin dihydrochloride is a synthetic form of tetrahydrobiopterin, the cofactor of phenylalanine hydroxylase that in pharmacological doses can stabilize and increase residual enzyme activity in some patients with PKU. About one-third of all patients with PKU respond to oral sapropterin. Phenylalanine ammonia lyase (PAL) is a prokaryotic enzyme that converts phenylalanine to ammonia and trans-cinnamic acid. Phase I and II trials have shown that injectable recombinant Anabaena variabilis PAL produced in Escherichia coli conjugated with PEG can reduce phenylalanine levels in subjects with PKU. The most frequently reported adverse events were injection-site reactions, dizziness and immune reactions. Additionally, oral administration of PAL and delivery of enzyme substitution therapies by encapsulation in erythrocytes are being investigated. Novel therapies for patients with PKU appear to be options to reduce phenylalanine levels, and may reduce the deleterious effects of this disorder.
1. Introduction
Phenylketonuria (PKU; OMIM 261600) is a metabolic disorder caused by inherited deficiency of phenylalanine hydroxylase (PAH; EC 1.14.16.1), the enzyme that converts phenylalanine to tyrosine (), which leads to accumulation of phenylalanine and subsequent neurocognitive dysfunction if untreated Citation[1]. Early detection by newborn screening can prevent intellectual disability by initiation of a phenylalanine-restricted diet soon after birth. However, on average, patients with PKU have lower intelligence quotient scores than matched individuals without PKU and can show deficits in multiple other aspects of neuropsychological function, including cognitive and executive function, working memory and emotional well-being Citation[2,3]. Adherence to the phenylalanine-restricted diet regimen is difficult, and often becomes poor as patients reach adolescence. Thus, despite the success of newborn screening and the phenylalanine-restricted diet in preventing severe mental disability, substantial unmet need remains for patients with PKU, and only limited therapeutic options exist. Although administration of large-neutral amino acids mixtures Citation[4] and new chemical chaperons Citation[5] is also the subject of investigations, we discuss the two most important alternative treatments for patients with PKU: sapropterin dihydrochloride and pegylated phenylalanine ammonia lyase (PAL).
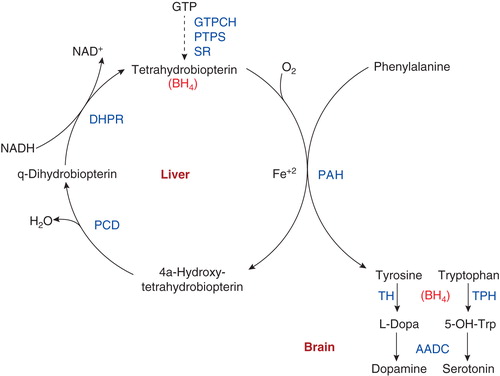
Figure 1. The phenylalanine hydroxylating system in the liver and the function of tetrahydrobiopterin in the brain. BH4 is synthesized from GTP by the enzymes GTPCH, PTPS, and SR. Hydroxylation of phenylalanine to tyrosine by PAH – and of tyrosine to l-DOPA by TH, and Trp to 5-OH-Trp by TPH – results in oxidation of BH4 to a carbinolamine intermediate, which is then reduced back to BH4 by PCD and DHPR. Dopamine and serotonin are produced by decarboxylation of their respective precursors by AADC.
2. Sapropterin dihydrochloride for tetrahydrobiopterin-responsive PKU
Tetrahydrobiopterin (BH4) is an essential cofactor of the PAH enzyme (). For a subset of patients with BH4-responsive PKU, the activity of their variant PAH enzymes can be increased by additional BH4; for these patients, treatment with exogenous BH4 results in reduced blood phenylalanine levels and improved tolerance for dietary phenylalanine. While a reduction of 30% or more in blood phenylalanine levels during a BH4 loading test is often considered to be the threshold for classification as ‘BH4 responsive,’ the BH4 loading test has not been standardized and the 30% threshold is arbitrary Citation[6,7]. It is estimated that 20 – 60% of patients with PKU have BH4-responsive PKU, and patients with less-severe PKU are more likely to be BH4 responsive Citation[2,3,6,8,9]. Although there is a general relationship between the particular variants in the PAH gene and patient’s BH4 responsiveness, the PAH genotype is not always predictive of phenotype Citation[2,7-9]. Data from the locus-specific database PAHdb, with 859 variants, and the genotypes database BIOPKU, with 7552 patients (both on www.biopku.org), allowed prediction of BH4 responsiveness from a patient genotype in 70% of all patients with PKU Citation[9]. The primary mechanisms of BH4 action are an increase in residual PAH enzyme activity and/or the multifactorial stabilization of the PAH enzyme tetramer Citation[7,10,11].
Sapropterin dihydrochloride (Kuvan® [BioMarin, Novato, California, USA]) is a synthetic form of BH4 that is approved by the US FDA for the treatment of BH4-responsive PKU (in combination with a phenylalanine-restricted diet) and by the European Medicines Agency for BH4-responsive PKU and BH4 deficiency. Multiple clinical trials in patients with BH4-responsive PKU have shown that sapropterin treatment is well tolerated and associated with reductions in blood phenylalanine levels in both pediatric and adult patients () Citation[6], and is also well tolerated in patients with cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy Citation[12]. A recent long-term retrospective follow-up study reported outcomes in 147 European patients with BH4-responsive PKU Citation[13]. These patients had been treated with BH4/sapropterin for at least 6 months, with follow-up periods of up to 12 years. Sixty-four percent of patients were treated with sapropterin alone and 36% of patients were treated with sapropterin in combination with a phenylalanine-restricted diet; of the latter group, 70% were able to transition to treatment with sapropterin alone. Sapropterin treatment was well tolerated. Blood phenylalanine levels were stable and within the therapeutic range for all patients, and for 38 patients with more severe PKU, sapropterin treatment was associated with a significant increase in median dietary phenylalanine tolerance (from 500 to 2500 mg/d). Quality of life was improved for 50% of patients, and dietary compliance was improved for 47%.
Table 1. Summary of clinical trials and selected other studies assessing treatment with sapropterin dihydrochloride in patients with elevated blood phenylalanine concentrations.
Two recent prospective long-term follow-up studies examined sapropterin treatment outcomes in pediatric patients. A study in 43 Japanese patients with BH4-responsive PKU compared sapropterin treatment outcomes between patients who were under (n = 21) or over (n = 22) 4 years of age Citation[14]. In each age group, most patients were treated for between 1 and 3 years. Sapropterin treatment reduced mean blood phenylalanine levels to within the recommended optimal control range in both age groups; however, patients who initiated treatment under 4 years of age had superior control of blood phenylalanine levels. Mean blood phenylalanine levels were similar in patients treated with sapropterin alone and those treated with sapropterin and diet. All patients who initiated sapropterin prior to 4 years of age developed normally. A different long-term follow-up study examined outcomes after 2 years of treatment in 55 Canadian and US patients who initiated sapropterin treatment between birth and the age of 6 years Citation[15]. After 2 years of sapropterin treatment, patients had mean blood phenylalanine levels that remained below baseline, and their mean prescribed phenylalanine intake increased. Importantly, neurocognitive function and development were maintained within the normative range, and no children appeared to be at risk for developmental delays. In both these pediatric studies, sapropterin was well tolerated Citation[14,15].
3. Phenylalanine ammonia lyase
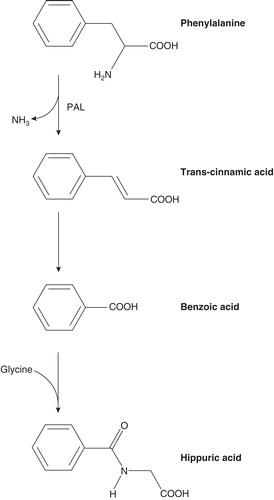
PAL is an enzyme not present in mammals that converts phenylalanine to ammonia and trans-cinnamic acid (). As a potential enzyme substitution therapy in patients with PKU, PAL can be given orally to decrease phenylalanine content in the gut or injected to reduce blood phenylalanine levels. Initial trials of oral PAL in humans yielded a modest decrease in phenylalanine levels Citation[16]. Subsequent studies in animal models led to development of a recombinant Anabaena variabilis PAL (genetically modified to improve protease resistance) produced in Escherichia coli conjugated with PEG to reduce immunogenicity (rAvPAL–PEG) Citation[17]. In a murine PKU model deficient in PAH activity (BTBRPahenu2 [ENU2]), blood phenylalanine levels were greatly lowered by weekly subcutaneous administration of rAvPAL–PEG over 3 months Citation[17] and, to a lesser extent, by oral administration of rAvPAL–PEG Citation[18].
Figure 2. Degradation of phenylalanine to trans-cinnamic acid and ammonia by the enzyme PAL. Trans-cinnamic acid is converted to benzoic acid by a mechanism that is not yet well described; benzoic acid is conjugated with glycine to form hippuric acid and excreted in urine.
In humans, Phase I studies demonstrated that subcutaneous administration of rAvPAL–PEG in a single dose of 0.1 mg/kg was safe and well tolerated in adult patients with PKU and led to significant reductions in blood phenylalanine levels Citation[19]. Adverse events included injection-site reactions, dizziness and rashes (local or generalized). No significant changes in laboratory test results were observed, but all patients developed antidrug antibodies. Blood PAL levels peaked about 5 days after drug administration and caused a mean 54% reduction in blood phenylalanine levels, with a nadir ∼ 6 days after injection. There was an inverse correlation between drug and phenylalanine concentrations in plasma. Phenylalanine returned to near-baseline concentrations ∼ 21 days after injection.
Limited data are currently available concerning repeated administration of rAvPAL–PEG to subjects with PKU (). Fifty-six patients with PKU enrolled into four Phase II studies (165-205, PAL-002, PAL-003, PAL-004). Patients in these studies were asked to maintain consistent diets and had mean baseline blood phenylalanine levels of 1360 μmol/l. In the 25 patients who were treated for at least 1 year, blood phenylalanine levels reduced by an average of 68% from baseline. Twenty of the 56 patients self-injected at least some of the doses. The most common adverse events are hypersensitivity-type reactions consisting of injection-site reactions, disseminated skin reactions or joint pains. While some of these reactions were observed in nearly all patients during initial drug administration, the reactions were generally mild to moderate and self-limited, and patients with these reactions were all successfully re-treated. During long-term exposure to rAvPAL–PEG, injection-site and hypersensitivity reaction rates decreased and there was no laboratory evidence of liver or kidney injury. Not only did rAvPAL–PEG reduce blood phenylalanine levels to the therapeutic range for PKU (120 – 360 µmol/l), but 17 patients had at least 1 day during which rAvPAL–PEG reduced phenylalanine levels below the normal range (≤ 30 µmol/l). The median duration of consecutive days with blood phenylalanine ≤ 30 µmol/l was 8, with one patient achieving these low levels for 135 consecutive days with no side effects. Two Phase III studies to evaluate efficacy and safety of self-administered injections of rAvPAL–PEG by adults with PKU were recently initiated (165 – 301 and 165 – 302).
Table 2. Summary of clinical trials for rAvPAL–PEG (from Clinicaltrials.gov).
An alternative approach for the delivery of potentially immunogenic prokaryotic enzymes has been tested in mice: infusion of red blood cells (RBCs) loaded with recombinant Chromobacterium violaceum PAH Citation[20] or rAvPAL enzyme Citation[21]. Administration of RBCs loaded with PAH, together with BH4, reduced blood phenylalanine levels in wild-type mice but not in the ENU2 PKU mouse model Citation[20]. In contrast, repeated administration of RBCs loaded with rAvPAL, at 9 – 10-day intervals for 10 weeks, provided a persistent reduction in blood phenylalanine levels in the ENU2 PKU mouse model that was not affected by the generation of antidrug antibodies Citation[21].
4. Expert opinion
The combination of newborn screening and the phenylalanine-restricted diet has been successful in the prevention of the most severe manifestations of PKU. However, lifetime adherence to the restricted diet is onerous and substantial unmet medical needs remain, particularly but not only for patients with the neurocognitive manifestations of PKU. This situation has led to the development of alternative therapeutic options for patients with PKU, and the two novel therapies discussed here both have the potential to improve treatment outcomes for these patients.
Treatment with BH4/sapropterin is an approved therapy that is well tolerated and effective in reducing blood phenylalanine levels in patients with BH4-responsive PKU. In addition, new data from long-term follow-up studies show that treatment with sapropterin allows some patients to discontinue the phenylalanine-restricted diet Citation[13] and raise the possibility that early treatment with sapropterin may ameliorate some of the neurocognitive manifestations of PKU Citation[14,15]. Unfortunately, the BH4 loading test that assesses patients’ BH4 responsiveness has not been standardized and the responsiveness threshold is arbitrary, suggesting that there are likely to be patients who are not classified as BH4 responsive who might clinically benefit from treatment with sapropterin. Therefore, we suggest that the long-term efficiency testing (of several weeks or months) should follow the positive initial short-term BH4 challenge (typically 48 h). Enzyme substitution therapy holds great promise for all patients with PKU. Clinical development of rAvPAL–PEG is now well advanced, with Phase II and Phase III studies currently in progress. In the human trials to date, injection of rAvPAL–PEG has been fairly safe and well tolerated and has shown initial efficacy, although the relationship between repeated dosing, immunogenicity, and treatment efficacy will need to be more fully evaluated Citation[19]. Additionally, studies in mice are investigating the delivery of enzyme substitution therapies by encapsulation in RBCs Citation[20,21], an approach that may limit any potential loss of treatment efficacy from neutralizing antidrug antibodies. An efficacious and safe oral enzyme substitution therapy would be a great advance for patients with PKU.
As lifelong control of blood phenylalanine concentration by diet alone is difficult, substantial unmet needs remain for patients with PKU, and only limited therapeutic options exist. However, novel therapies currently in development may help to address these unmet medical needs.
Declaration of interest
N Longo receives clinical trial grant support from BioMarin Pharmaceutical Inc. and is an Advisor for BioMarin and Codexis, Inc. USA and N Blau receive grant support for clinical research from and act as advisors for Merck KGaA, Germany and Codexis, Inc., USA. Writing assistance was utilized in the preparation of this paper, it was carried out by Karl Zawadzki, PhD (Health Interactions, San Francisco), and supported by BioMarin Pharmaceutical, Inc., Novato, USA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
Bibliography
- Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat 2007;28:831-45
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010;376:1417-27
- Feillet F, van Spronsen FJ, MacDonald A, et al. Challenges and pitfalls in the management of phenylketonuria. Pediatrics 2010;126:333-41
- van Spronsen FJ, de Groot MJ, Hoeksma M, et al. Large neutral amino acids in the treatment of PKU: from theory to practice. J Inherit Metab Dis 2010;33:671-6
- Santos-Sierra S, Kirchmair J, Perna AM, et al. Novel pharmacological chaperones that correct phenylketonuria in mice. Hum Mol Genet 2012;21:1877-87
- Blau N. Sapropterin dihydrochloride for phenylketonuria and tetrahydrobiopterin deficiency. Expert Rev Endocrinol Metabol 2010;5:483-94
- Heintz C, Cotton RG, Blau N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Hum Mutat 2013;34:927-36
- Blau N, Shen N, Carducci C. Molecular genetics and diagnosis of phenylketonuria: state of the art. Expert Rev Mol Diagn 2014;14:655-71
- Wettstein S, Underhaug J, Perez B, et al. Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur J Hum Genet 2014; Epub ahead of print
- Pey AL, Stricher F, Serrano L, et al. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet 2007;81:1006-24
- Staudigl M, Gersting SW, Danecka MK, et al. The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum Mol Genet 2011;20:2628-41
- De Maria R, Campolo J, Frontali M, et al. Effects of sapropterin on endothelium-dependent vasodilation in patients with CADASIL: a randomized controlled trial. Stroke 2014;45:2959-66
- Keil S, Anjema K, van Spronsen FJ, et al. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics 2013;131:e1881-8
- Shintaku H, Ohura T. Sapropterin is safe and effective in patients less than 4-years-old with BH-responsive phenylalanine hydroxylase deficiency. J Pediatr 2014;165:1241-4
- Longo N, Siriwardena K, Feigenbaum A, et al. Long-term developmental progression in infants and young children taking sapropterin for phenylketonuria: a two-year analysis of safety and efficacy. Genet Med 2014; Epub ahead of print
- Hoskins JA, Jack G, Wade HE, et al. Enzymatic control of phenylalanine intake in phenylketonuria. Lancet 1980;1:392-4
- Sarkissian CN, Gamez A, Wang L, et al. Preclinical evaluation of multiple species of PEGylated recombinant phenylalanine ammonia lyase for the treatment of phenylketonuria. Proc Natl Acad Sci USA 2008;105:20894-9
- Sarkissian CN, Kang TS, Gamez A, et al. Evaluation of orally administered PEGylated phenylalanine ammonia lyase in mice for the treatment of Phenylketonuria. Mol Genet Metab 2011;104:249-54
- Longo N, Harding CO, Burton BK, et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicentre, phase 1 dose-escalation trial. Lancet 2014;384:37-44
- Yew NS, Dufour E, Przybylska M, et al. Erythrocytes encapsulated with phenylalanine hydroxylase exhibit improved pharmacokinetics and lowered plasma phenylalanine levels in normal mice. Mol Genet Metab 2013;109:339-44
- Rossi L, Pierige F, Carducci C, et al. Erythrocyte-mediated delivery of phenylalanine ammonia lyase for the treatment of phenylketonuria in BTBR-Pah mice. J Control Release 2014;194C:37-44
- Levy HL, Milanowski A, Chakrapani A, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 2007;370:504-10
- Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 2007;30:700-7
- Wasserstein M, Burton B, Cederbaum S. Results of a Phase II, multicenter, open-label study of sapropterin dihydrochloride in subjects with hyperphenylalaninemia related to primary BH4 deficiency. Presented at the Annual Meeting of the American Society for Human Genetics (ASHG), 2008; 11–15 November 2008; Philadelphia, PA, USA
- Lee P, Treacy EP, Crombez E, et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet A 2008;146A:2851-9
- Trefz FK, Burton BK, Longo N, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 2009;154:700-7
- Burton BK, Nowacka M, Hennermann JB, et al. Safety of extended treatment with sapropterin dihydrochloride in patients with phenylketonuria: results of a phase 3b study. Mol Genet Metab 2011;103:315-22
- Fernhoff PM, Burton B, Nowacka M, et al. PKU 008: a long-term, open-label study of sapropterin dihydrochloride (Kuvan®) in PKU subjects. Presented at the American College of Medical Genetics Annual Meeting; 25–29 March 2009; FL USA
- Douglas TD, Ramakrishnan U, Kable JA, et al. Longitudinal quality of life analysis in a phenylketonuria cohort provided sapropterin dihydrochloride. Health Qual Life Outcomes 2013;11:218
- Grange DK, Hillman RE, Burton BK, et al. Sapropterin dihydrochloride use in pregnant women with phenylketonuria: an interim report of the PKU MOMS sub-registry. Mol Genet Metab 2014;112:9-16
- Available from: www.Clinicaltrials.gov [Accessed 6 December 2014]
- BioMarin. Press Release 26 Sep 2012 Available from: http://investors.bmrn.com/releasedetail.cfm?ReleaseID=716661
- BioMarin. Press Release 5 June 2013 Available from: http://investors.bmrn.com/releasedetail.cfm?ReleaseID=769256