Abstract
At present, there are 22 FDA-approved antiretroviral agents, which are categorised into four classes of drugs. Several others are in various stages of basic and clinical development. The authors of this paper review the general characteristics of each class of antiretrovirals, as well as individual investigational agents that are in advanced clinical development. A brief synopsis of US and WHO antiretroviral treatment guidelines is also provided.
Keywords::
- antiretrovirals
- antiretroviral therapy
- chemokine receptor antagonists
- entry inhibitors
- fusion inhibitors
- highly active antiretroviral therapy
- HIV
- integrase inhibitors
- maturation inhibitors
- non-nucleoside analogue reverse transcriptase inhibitors
- nucleoside analogue reverse transcriptase inhibitors
- protease inhibitors
- treatment guidelines
1. Introduction
The year 2006 marks the 10th anniversary of the introduction of highly active antiretroviral therapy (HAART), a treatment paradigm that has led to significant declines in HIV-associated morbidity and mortality Citation[1,2]. Currently, there are 22 FDA-approved individual antiretroviral drugs that are classified into four categories based on their mechanism of action (Box 1). Additional agents are in various stages of clinical and preclinical development. The authors provide an up-to-date overview of antiretrovirals that are currently used in the treatment of HIV infection, as well as newer antiretrovirals in advanced stages of clinical development. In addition, current guidelines for the use of antiretrovirals in the management of HIV infection will be briefly reviewed. Drug resistance and metabolic complications that are associated with antiretroviral therapy (ART) are beyond the aim and scope of this review and are not addressed.
2. The replication cycle of HIV
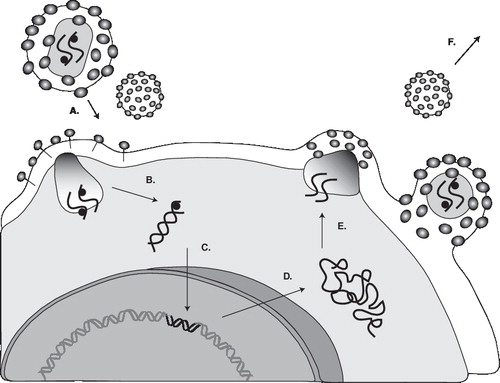
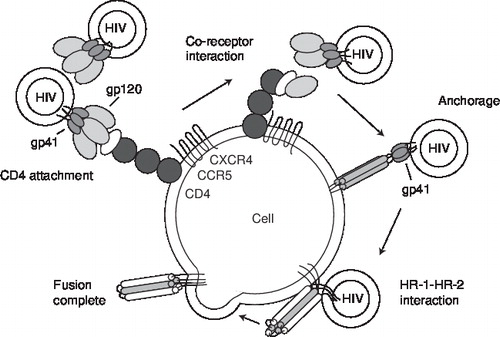
A working knowledge of the HIV replication cycle is essential for understanding the mechanism of action of antiretrovirals Citation[3]. depicts a simplified schema of the HIV-1 life cycle. The human immunodeficiency virus is an enveloped virus that contains two copies of viral genomic RNA in its core. In addition to the copies of RNA, the viral core also contains enzymes that are required for HIV replication: reverse transcriptase, integrase and protease. The first step in HIV replication cycle is the interaction between the envelope proteins of the virus and specific host-cell surface receptors (e.g., CD4 receptors). However, binding to the CD4 receptor by itself is not sufficient for entry of the virus into the cell. In addition to CD4, the human HIV requires a co-receptor for entry into target cells. The chemokine receptors CXCR4 and CCR5 have been identified as the principal co-receptors for entry of HIV into its target cells (). Chemokines (the abbreviation for chemoattractant cytokines) are a group of structurally related glycoproteins that play a role in activating the immune system’s response to infection. Their actions are mediated by a family of structurally related receptors. Binding of HIV to co-receptors causes conformational changes in the envelope proteins, ultimately resulting in the fusion of the viral envelope and the host cytoplasmic membrane. Fusion creates a pore through which the viral capsid enters the cell. Following entry into the cell, the viral reverse transcriptase enzyme catalyses the conversion of viral RNA into DNA. This viral DNA enters the nucleus and becomes inserted into the chromosomal DNA of the host cell (integration). Expression of the viral genes leads to production of precursor viral proteins. These proteins and viral RNA are assembled at the cell surface into new viral particles and leave the host cell by a process called budding. During the process of budding, they acquire the outer layer and envelope. At this stage, the protease enzyme cleaves the precursor viral proteins into their mature products. If this final phase of the replication cycle does not take place, the released viral particles are non-infectious and not competent to initiate the replication cycle in other susceptible cells.
There are four classes of antiretroviral drugs that have received FDA approval: nucleoside/nucleotide analogue reverse transcriptase inhibitors (NRTIs), non-nucleoside analogue reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs) and fusion inhibitors.
3. Nucleoside/nucleotide analogue reverse transcriptase inhibitors
NRTIs are structurally similar to the building blocks of nucleic acids (RNA, DNA) but differ from their natural analogues by the replacement of the hydroxy group in the 3′ position by another group that is unable to form the 5′ to 3′ phosphodiester linkage that is essential for DNA elongation. NRTIs block reverse transcriptase activity by competing with the natural substrates and incorporating into viral DNA and so to act as chain terminators in the synthesis of proviral DNA Citation[4]. To exert their antiviral activity, NRTIs must first be intracellularly phosphorylated to their active 5′ triphosphate forms by cellular kinases Citation[5]. Tenofovir is the only NRTI. As it already contains a phosphate molecule in its structure, it only requires phosphorylation by cellular enzymes to its diphosphate form for its antiviral activity.
The general characteristics of currently available NRTIs are shown in .
4. Non-nucleoside analogue reverse transcriptase inhibitors
NNRTIs bind directly and non-competitively to the enzyme reverse transcriptase Citation[6,7]. Although the drugs differ chemically from each other, they all bind to the same site: a site that is distinct from the substrate binding site. They block DNA polymerase activity by causing a conformational change and disrupting the catalytic site of the enzyme. Unlike nucleoside analogues, NNRTIs do not require phosphorylation to become active and are not incorporated into viral DNA. They also have no activity against HIV-2 Citation[8]. Nevirapine and efavirenz are inducers of the hepatic CYP system. Delavirdine, on the other hand, inhibits CYP Citation[9]. Through this interaction with the CYP enzyme system, NNRTIs may change the metabolism of, and thus lower (nevirapine, efavirenz) or increase (delavirdine), the plasma levels of coadministered drugs that are metabolised by the CYP system. Similarly, drugs that induce or inhibit CYP activity may have an effect on the plasma concentrations of NNRTIs. Delavirdin is rarely used in clinical practice because of its relatively low potency and large pill burden.
The most common side effect that is associated with all NNRTIs is rash, occurring in ≤ 35% patients who are receiving nevirapine, usually within the first 6 weeks of initiation of therapy Citation[10,11]. Nevirapine is additionally associated with hepatotoxicity, in particular in women with CD4+ counts > 250 cells/mm3 Citation[12,13]. The risk of hepatotoxicity is greatest in the first 6 weeks of therapy. However, it may occur at any time during treatment, and in some cases may not be reversible with discontinuation of therapy. Rash was observed in ∼ 50% of the patients with symptomatic hepatotoxicity.
Efavirenz on the other hand has been commonly associated with CNS side effects, ranging from dizziness to hallucinations, insomnia, nightmares and worsening of psychiatric illnesses [DuPont Pharma: efavirenz tablet, US prescribing information (1998)]. These CNS side effects are more common during the first 2 weeks of therapy and usually resolve by 24 weeks. Efavirenz also causes fetal malformations in pregnant monkeys, and has been associated with neural tube defects in children of pregnant women who had received this drug Citation[14,15]. The risk of hepatotoxicity is less with efavirenz compared with nevirapine. In the 2NN study, 4.5% of participants in the efavirenz arm had grade 3 or 4 liver abnormalities, compared with 8% for the nevirapine twice-daily and 135 for the nevirapine once-daily arm Citation[16].
Both nevirapine and efavirenz have been associated with dyslipidaemias. In a direct comparison of nevirapine and efavirenz in the 2NN study, nevirapine recipients had smaller increases in triglyceride levels, greater increases in high-density lipoprotein cholesterol levels, and larger decreases in the ratio of total cholesterol to high-density lipoprotein cholesterol than efavirenz recipients Citation[16].
The general characteristics of the three NNRTIs that are currently approved for clinical use are listed in .
5. Protease inhibitors
PIs exert their antiviral effect by inhibiting HIV-1 protease. HIV-1 protease is a complex enzyme that is composed of two identical halves (i.e., a symmetrical dimer) with an active site that is located at the base of the cleft Citation[17]. It is responsible for the cleavage of the large viral precursor polypeptide chains into smaller, functional proteins, thus allowing maturation of the HIV virion. This process takes place in the final stages of the HIV life cycle. Inhibition of the protease enzyme results in the release of structurally disorganised and non-infectious viral particles Citation[18]. PIs are metabolised by the CYP system and are themselves, to varying degrees, inhibitors of this system Citation[19]. This leads to a significant number of interactions with drugs that are inducers, inhibitors or substrates of this system. The general characteristics of the available PIs are listed in .
The inhibitory effect of PIs on each other drug’s metabolism has led to the evaluation of specific combinations that provide an advantageous pharmacokinetic profile, and which may delay or prevent the onset of resistance, allow for dose reductions, more convenient dosing regimens and less toxicity Citation[20,21]. All of the currently licensed PIs are commonly prescribed as boosted agents, with the exception of nelfinavir, which is not well or reliably augmented by ritonavir. In fact, two PIs, lopinavir and tipranavir, require coadministration with ritonavir to achieve effective serum concentrations.
6. Fusion inhibitors
Enfuvirtide (also known as T-20) is the first, and thus far the only, fusion inhibitor to be approved by the FDA. The HIV membrane consists of trimeric envelope structures consisting of two glycoproteins: gp120 and gp41 Citation[22,23]. In each envelope structure, gp120 molecules constitute the cap, and the stalk is formed by gp41 anchored in the viral lipid bilayer (). On the binding of gp120 to CD4 and chemokine receptors, a critical conformational change occurs in the structure of gp41. The two different peptide motifs of gp41, heptad repeat (HR) 1 and -2, unite to form a six-helix bundle hairpin structure, which then pulls the membranes of the virus and cell closer, resulting in membrane fusion. Enfuvirtide is a linear 36 amino acid peptide homologous to a segment of the HR2 region of gp41. It binds to the HR1 region of gp41 and blocks the formation of the six-helix bundle that is necessary for fusion. Enfuvirtide is indicated for the treatment of HIV-1 infection in treatment-experienced patients. It is not active against HIV-2. Analysis of results from the pivotal Phase III trials, TORO (T-20 versus Optimized Regimen Only)-I and -II, suggest that the optimal time to initiate enfuvirtide is when there are at least two drugs that retain partial antiviral activity; the CD4+ cell counts are > 100 × 106 cells/l; and plasma HIV-1 RNA are < 1 × 105 copies/ml Citation[24,25]. Injection site reactions are the most common adverse events that were reported with the use of enfuvirtide. Their manifestation includes erythema, induration, ecchymosis, nodules or cysts, and may present with symptoms of pruritus, pain or discomfort. A needle-free drug delivery system that may decrease the impact of injection site reactions is under investigation. Bacterial pneumonia (both Gram-positive and Gram-negative) has been observed in patients taking enfuvirtide at a higher rate than in those in the comparator study arms in the TORO studies. Rare cases of systemic hypersensitivity reactions to enfuvirtide have also been reported. These presented with rash, fever, nausea/vomiting, chills, rigors, hypotension and elevated liver enzymes Citation[26].
7. Antiretroviral agents in clinical development
Numerous antiretroviral candidate drugs are in various stages of preclinical and clinical development. Some are new agents within the existing drug classes, developed with a focus towards an improved pharmacokinetic and safety profile, higher genetic barriers to the development of resistance, and hopefully with retained or improved antiviral potency (). Others represent entirely new classes of agents that are directed against newer targets and with unique mechanisms of action ().
7.1 New non-nucleoside analogue reverse transcriptase inhibitors
Etravirine (TMC-125) is a new NNRTI that has potent in vitro anti-HIV activity against wild-type HIV-1 isolates, as well as isolates with key NNRTI resistance mutations. In a Phase IIb dose-ranging, randomised, partially blinded study, a mean change from baseline in viral load (VL) of -1.04 and -1.18 (etravirine 400- and 800 mg b.i.d., respectively) was observed at 24 weeks in HIV-1-infected patients with substantial treatment experience and documented evidence of NNRTI resistance and three or more PI mutations Citation[27]. Two ongoing, pivotal randomised, placebo-controlled Phase III clinical trials, DUET-I and -II, will evaluate etravirine in a larger cohort of treatment-experienced HIV-1 infected adults with documented NNRTI resistance and at least three primary PI mutations.
7.2 New protease inhibitors
Darunavir (TMC-114), a new PI, has shown potent in vitro activity against HIV-1 isolates with PI resistance mutations. POWER-1 and -2 were randomised Phase IIb trials of darunavir that are boosted with ritonavir (darunavir/r) in treatment-experienced patients Citation[28,29]. Both of the trials have identical protocols, and enrolled three-class-experienced patients with at least one primary PI mutation and with VL > 1000 copies/ml, who were then randomised to one of four darunavir/r doses (400/100 mg daily; 800/100 mg daily; 400/100 mg b.i.d.; 600/100 mg b.i.d.), or investigator-selected PIs. Patients received an optimised background regimen consisting of two or more NRTIs with or without enfuvirtide. POWER-1 was conducted in Europe, Brazil and Australia, and POWER-2 was conducted in the US. Data for the 24-week efficacy were recently reported for both trials. In POWER-1, the primary end point of > 1 log10 VL reduction from the baseline was achieved in 77% of darunavir/r patients versus 25% for control (p < 0.001). Mean VL decrease for darunavir/r was 2.03 versus 0.63 log10 copies/ml for control (p < 0.001). VL < 50 copies/ml occurred in 53% of darunavir/r patients versus 18% of controls (p < 0.001). In POWER-2, 62% of the patients achieved a reduction in VL of 1 log10 or more in the highest darunavir/r dose group (600/100 mg b.i.d.), compared with 14% in the control group. The percentage of patients reaching undetectable virus levels (< 50 copies/ml) was 39% in the highest darunavir/r dose group, compared with 7% in the control arm. Based on these results, the dose of darunavir/r 600/100 mg b.i.d. has been selected for further evaluation in Phase III trials. TMC-114, now called darunavir, was approved by the FDA in June 2006, for use in combination with other antiretroviral agents for the treatment of HIV infection in antiretroviral treatment-experienced adults.
7.3 Integrase inhibitors
The integration of HIV-1 proviral DNA into the host cell genome occurs in three steps Citation[30]. First, two nucleotides are removed from each 3′-end of the viral DNA, a process termed 3′-end processing. In the second step, DNA strand transfer, the processed viral DNA ends are inserted into the host DNA. The third step, whereby the two unpaired nucleotides at the 5′-ends of the viral DNA are removed, the single gaps are filled and ligated. HIV-1 integrase is a viral enzyme that catalyses the first two steps of integration; it is believed that the repair step is catalysed by cellular enzymes.
MK-0518 is a novel agent that inhibits the integrase enzyme at the level of the DNA strand transfer. It was evaluated in a Phase II multi-centre, randomised, double-blinded, dose-ranging, placebo-controlled study in 167 patients with three-class-resistant HIV, and VLs of > 5000 copies/ml and CD4+ counts > 50 cells/mm3 Citation[31]. Patients received either MK-0518 200,- 400-, or 600 mg or placebo, each dosed orally twice daily and in combination with optimised background treatment. At week 16, 64 – 84% of the patients in the MK-0518 arm achieved HIV RNA < 400 copies/ml, compared with 22% for the placebo arm. Of the patients in the MK-0518 arm, 56 – 72% achieved HIV RNA < 50 copies/ml, compared with 19% in the placebo arm. These results are quite exciting given that the study participants were highly treatment experienced. The pivotal Phase III studies of MK-0518, designated BENCHMRK-1 and -2, are expected to be initiated in the near future.
7.4 Inhibitors of viral entry
Three distinct classes of HIV entry inhibitors are in development: attachment inhibitors, chemokine co-receptor antagonists and fusion inhibitors Citation[32]. Individual agents that are in relatively advanced stages of clinical development are depicted in . Among the group, CCR5 receptor antagonists are the most advanced in their clinical development and are briefly discussed below.
Different HIV-1 isolates have distinct tropisms for various CD4+ human target cell types in vitro. Some replicate only in transformed T-cell lines and activated primary T cells (designated T tropic), whereas other HIV-1 strains (designated M tropic), replicate only in cells from the macrophage/monocyte lineage and activated primary T cells Citation[33,34]. Viral isolates obtained from HIV-1-infected persons in the early stage of infection are predominantly M tropic, whilst the presence of T-tropic virus is noted more commonly at lower CD4+ cell counts and higher VLs Citation[35]. The chemokine receptor CCR5 is the major co-receptor for M-tropic HIV-1 isolates. CXCR4 is the major co-receptor for T-tropic strains. The chemokine receptors CCR5 and CXCR4 are the major co-receptors, besides the cellular CD4 receptor, for entry of HIV-1 into its target cell.
A total of three CCR5 chemokine receptor antagonists had been in advanced clinical development. However, further development of two of the drugs has recently been halted. In the case of aplaviroc, all of the clinical trials were halted because of hepatotoxicity noted in a significant number of people participating in Phase IIb and Phase III trials Citation[36,101]. In the case of vicriviroc (SCH-417,690), vicriviroc plus combivir was found to be inferior in its antiviral efficacy, compared with a regimen of combivir plus efavirenz in a Phase II trial in treatment-naive patients. Thus, further development in treatment-naive patients has been halted Citation[102]. However, studies of vicriviroc in ART-experienced patients with more advanced HIV disease are continuing. Maraviroc (UK-427,857), the third CCR5 chemokine receptor antagonist, continues its clinical development in both treatment-naive and -experienced patients. In earlier multiple-dose Phase I/IIa studies, maraviroc was well tolerated at doses up to and including 300 mg b.i.d. Citation[37]. From an efficacy standpoint, 10-day monotherapy with maraviroc at doses of 300 mg/day and 300 mg b.i.d. in HIV-positive patients resulted in mean maximum HIV RNA reductions of 1.60 and 1.84 log10, respectively. It has received fast-track designation from the FDA and is undergoing Phase III clinical testing.
7.5 Maturation inhibitors
Bevirimat (PA-457) is the first drug in a new class of antiretrovirals: HIV-1 maturation inhibitors. Its mechanism of action occurs at a late stage in HIV-1 replication cycle, where it blocks conversion of the capsid precursor to mature capsid protein, resulting in the release of non-infectious virions Citation[38]. It has shown potent in vitro inhibition of HIV replication against both wild-type and resistant isolates, and adverse experiences seem to be mild-to-moderate. In a randomised, double-blind Phase IIa dose-ranging study, statistically significant reductions in HIV-1 RNA were achieved at the 100- and 200-mg doses compared with placebo (100 mg, -0.48 log10; and 200 mg, -1.03 log10) Citation[39].
8. Guidelines on the use of antiretroviral therapy for HIV infection
A panel of leading AIDS specialists, convened by the Department of Health and Human Services (DHHS) in collaboration with the Henry J Kaiser Family Foundation, has developed recommendations for the use of antiretroviral agents in HIV-infected adults and adolescents Citation[103]. A number of other organisations have developed similar guidelines Citation[40,104].
There is a general consensus for treating patients with symptoms ascribed to HIV infection. Thus, current DHHS guidelines recommend ART for all patients with a history of an AIDS-defining illness or severe symptoms of HIV infection, regardless of CD4+ T-cell count.
The recommendations on when to start ART in asymptomatic patients are more problematic and are based on the likelihood of the development of AIDS-defining illnesses or death. Few data from controlled trials are available for this purpose. However, data from observational cohorts of HIV-infected persons have provided some guidance to assist in the assessment of risk for disease progression. The CD4+ cell count appears to be a much more important prognostic indicator of disease progression than VL. However, an HIV-1 RNA (VL) value of > 100,000 copies/ml was also independently associated with a greater likelihood of disease progression. For example, the 3-year probability of disease progression for those patients with a CD4+ cell count of 100 – 199 cells/mm3 is 9.3%, compared with a probability of 3.4% for those with CD4+ cell counts > 350 cells/mm3. This risk increases to 12 and 4.4%, respectively, if the VL is > 100,000 copies/ml. Current DHHS guidelines recommend initiating ART in otherwise asymptomatic patients with < 200 CD4+ T cells/mm3. The evidence for initiating therapy in asymptomatic HIV-infected persons with a CD4+ T-cell count of 200 – 350 cells/mm3 is less strong. However, many HIV clinicians prescribe ART for such patients. The evidence for initiating ART for those that have CD4+ cell counts > 350 cells/mm3, but also have VLs > 100,000 copies/ml, is even less compelling. Many clinicians defer therapy in these patients. It should be emphasised that patients must actively participate in all of these decisions, understand the benefits and risks of treatment, and make an informed commitment to a complex long-term treatment.
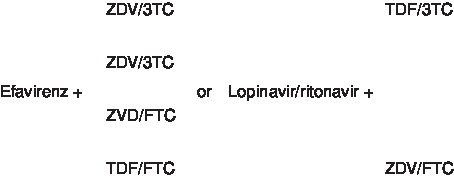
The latest (May 4, 2006) DHHS-recommended regimens for first-line ART are listed in . The preferred regimens are those that have clinical trial data support for their efficacy, durability of effect, tolerability and ease of use. Alternative regimens have exhibited some disadvantages in those factors, compared with the preferred regimens. A number of antiretroviral agents, either individually or in combination with certain other antiretroviral drugs, have been found to be inadequate for initial therapy due to either lack of efficacy, increased toxicity or significant inconvenience. These are listed in and . Two of these, triple nucleoside combination regimens and the nucleoside backbone of tenofovir and didanosine, warrant further discussion.
Triple NRTI combination regimens have attracted a lot of attention for a number of reasons including the availability of a fixed dose combination (Trizivir®; GlaxoSmithKline [lamivudine/zidovudine/abacavir]), a once-daily dosing option, as well as a potential for avoiding side effects that are seen with PIs and NNRTIs. However, several clinical trials have convincingly demonstrated that triple NRTI regimens are less potent than comparator NNRTI- or PI-based regimens. The combination of zidovudine/lamivudine/abacavir is the only triple NRTI combination that was shown to have equivalent virological outcome compared with a PI-based (indinavir) regimen in randomised, controlled trials Citation[41,42]. However, even in these studies, patients randomised to the triple NRTI arm who had high baseline plasma HIV-1 RNA > 100,000 copies/ml had a significantly inferior virological response than patients in the indinavir arm. Importantly, this same triple NRTI regimen has been found to be inferior to efavirenz-based regimens, regardless of whether the baseline HIV-1 RNA levels were greater than or less than 100,000 copies/ml. Based on these results, the current guidelines state that the triple NRTI regimen consisting of zidovudine/lamivudine/abacavir should only be used when a preferred or an alternative NNRTI- or a PI-based regimen cannot be used for reasons of toxicity, complexity or drug–drug interactions. Studies of other triple NRTI regimens, including stavudine/didanosine/lamivudine, stavudine/lamivudine/abacavir, didanosine/stavudine/abacavir, tenofovir/abacavir/lamivudine, and tenofovir/didanosine/lamivudine, have all shown inferior virological responses when compared with PI- or NNRTI-based regimens Citation[43-45]. These regimens are therefore not recommended for use.
The combination of tenofovir with didanosine presents the clinician with several challenges. First, tenofovir significantly increases didanosine exposure and thus increases the likelihood of didanosine-associated adverse events, including pancreatitis and neuropathy Citation[46]. The mechanism of this interaction is unknown. When coadministered with tenofovir, the didanosine dose should be reduced to 250 mg in adults weighing > 60 kg. Second, the combination of tenofovir and didanosine with either efavirenz or nevirapine as initial therapy has resulted in inferior virological response Citation[47-49]. The reason for this lack of response is not entirely clear. Third, the combination of tenofovir and didanosine has also been associated with a reduced CD4+ cell count response, despite VL suppression Citation[50-52]. Finally, didanosine use has been identified as one of the factors that is associated with renal impairment among patients receiving tenofovir Citation[53,54].
9. Antiretroviral therapy in resource limited settings
The AIDS epidemic affects a significant number of people throughout the world. According to ‘The AIDS Epidemic Update: December 2005’ released by the joint United Nations Programme on HIV/AIDS (UNAIDS) and the WHO, an estimated 40 million people are living with the HIV infection Citation[105]. There are > 90% of all of the people living with HIV who reside in developing countries, where resources for diagnosis, prevention and management of diseases are scarce. The WHO and the UNAIDS estimated that ∼ 6.5 million people worldwide are in immediate need of life-sustaining ART and launched an initiative called ‘3 by 5’ in 2003: an initiative that has the target of providing ART to 3 million people living with HIV/AIDS in low- and middle-income countries by the end of 2005 Citation[106]. According to the latest report released in March 2006, the number of people receiving combination ART in developing countries increased from 400,000 in December 2003 to 1.3 million in December 2005 Citation[107]. Thus, while the goal of the 3 by 5 initiative has not been met, there has been some progress in providing access to ART globally. A lot more still remains to be done.
The WHO has developed guidelines for the use of ART in resource-limited settings Citation[108]. Similar to the DHHS guidelines, the WHO guidelines address when ART should begin, which antiretroviral regimens should be selected and how treatment should be monitored. The WHO recommends that HIV-infected adults and adolescents should start ART therapy when there is WHO stage III or IV HIV disease, irrespective of the CD4+ cell count. For those with WHO stage I or II HIV disease, a CD4+ cell count of < 200/mm3 or a total lymphocyte count of ≤ 1200/mm3 should trigger initiation of ART. Because of its cost, complexity and unavailability, VL testing is not recommended for decision-making on initiation or regular monitoring of ART in resource-limited settings. The selection of first-line regimens has to take into consideration the issues of the availability of fixed-dose combinations, the lack of a requirement for refrigeration, drug availability and the cost, in addition to the usual criteria of efficacy, durability and tolerability. The preferred regimens for initiating ART are the combination of two nucleosides and one of the non-nucleoside drugs: stavudine or zidovudine plus lamivudine, plus nevirapine or efavirenz. PI-based regimens have been primarily reserved for second-line therapy because of their cost, relatively higher pill count, complexity, significant drug interactions and the requirement for cold storage for ritonavir-boosted agents. The most recent WHO clinical staging of HIV/AIDS is provided in .
10. Expert opinion
Considerable progress has been made in the treatment of HIV infection since the introduction of HAART in 1996. This progress has led to dramatic declines in HIV-associated sickness and death. However, significant challenges and a number of unresolved issues remain. Despite this decline in HIV-associated morbidity and mortality, new HIV-infection continues to be diagnosed at a rate of 5 million people globally in 2005, and ∼ 40,000 new HIV infections yearly in the US. There are ongoing controversies about the optimal time to start therapy, and the proper sequencing of antiretroviral regimens continues to be debated. Recent antiretroviral treatment outcomes are reported to be better than in the earlier years of HAART both in the clinical and clinical trial setting. However, a significant number of patients (in particular those patients that were initiated with ART prior to the HAART era and thus have been exposed to serial ineffective monotherapy regimens, as well as stepwise additions of single new agents to a failing antiretroviral regimen), are experiencing virological failure. The development of new agents with equal or increased potency, and the results from recently reported clinical trials of ART in heavily treatment-experienced patients, has generated some renewed optimism in the management of treatment failure.
With HIV-infected patients living for longer, long-term metabolic complications, such as disturbances of lipid and glucose metabolism, as well as lipid deposition disorders, have become important clinical issues. Additionally, the clinical consequences of HIV and hepatitis coinfection, including liver failure, as well as increased toxicity to antiretroviral agents, are increasingly being observed. The challenges in the developing world are somewhat different, although the challenges noted for the developed world are also applicable. The vast majority of people infected and affected by HIV live in the developing world and have barely begun to benefit from HAART. Current experience suggests that both the adherence rates and treatment responses to ART in resource-limited settings are comparable to those that have been observed in the developed world. Although it is disappointing that the goal of the 3 by 5 initiative has not been achieved, the mounting consensus to provide universal access to treatment to those who need it holds some hope for the future.
Box 1 . Current FDA-approved antiretrovirals.
| • | NRTI/NtRTIs: zidovudine, didanosine, zalcitabine*, stavudine, lamivudine, abacavir, enofovir, emtricitabine, combivir*, trizivir*, truvada*, epzicom* | ||||
| • | Fusion inhibitors: enfurvitide | ||||
| • | NNRTIs: nevirapine, delavirdine, efavirenz | ||||
| • | Protease inhibitors: saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, lopinavir/ritonavir, atazanavir, fosamprenavir, tipranavir, amprenavir, darunavir | ||||
Table 1. General characteristics of nucleoside analogue reverse inhibitors.
Table 2. General characteristics of the currently available non-nucleoside analogue reverse transcriptase inhibitors.
Table 3. General characteristics of currently available protease inhibitors
Table 4. Investigational drugs in existing classes of antiretrovirals in advanced clinical development.
Table 5. Investigational drugs in new classes of antiretrovirals in advanced clinical development.
Table 6. Antiretroviral drugs/components not recommended for initial therapy.
Table 7. Antiretroviral regimens or components that should not be offered at any time.
Table 8. Revised WHO clinical staging system for HIV/AIDS.
Notes
Zalcitabine is planned to be discontinued in 2006. Amprenavir has been discontinued by the manufacturer.
*Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of all nucleoside analogues and are believed to be caused by NRTI/NtRTI-induced mitochondrial damage. The deoxynucleoside analogue reverse transcriptase inhibitors (zalcitabine, stavudine, didanosine) have a higher potential to cause mitochondrial DNA depletion than other NRTI/NtRTIs.
NNRTI: Non-nucleoside analogue reverse transcriptase inhibitor; NRTI: Nucleoside/nucleotide analogue reverse transcriptase inhibitor; NtRTI: Nucleotide reverse transcriptase inhibitor.
Bibliography
- PALELLA FJ Jr, DELANEY KM, MOORMAN AC et al.: Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. (1998) 338:853-860.
- TORRES RA, BARR M: Impact of combination therapy for HIV infection on inpatient census. N. Engl. J. Med. (1997) 336:1531-1532.
- HASELTINE WA: Molecular biology of the human immunodeficiency virus type 1. FASEB J. (1991) 5(10):2349-2360.
- MITSUYA H, BRODER S: Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotropic virus III/ lymphadenopathy-associated virus (HTLV III/LAV) by 2′, 3′-dideoxy- nucleosides. Proc. Natl. Acad. Sci. USA (1986) 83:1911-1915.
- GAO WY, AGBARIA R, DRISCOLL JS, MITSUYA H: Divergent anti-human immunodeficiency virus activity and anabolic phosphorylation of 2′,3′-dideoxynucleoside analogs in resting and activated human cells. J. Biol. Chem. (1994) 269(17):12633-12638.
- GROB PM, WU JC, COHEN KA et al.: Nonnucleoside inhibitors of HIV-1 reverse-transcriptase: nevirapine as a prototype drug. AIDS Res. Hum. Retroviruses (1992) 8:145-152.
- MERLUZZI VJ, HARGRAVE KD, LABADIA M et al.: Inhibition of HIV-1 replication by a non-nucleoside reverse transcriptase inhibitor. Science (1990) 250:1411-1413.
- WITVROUW M, PANNECOUQUE C, SWITZER WM, FOLKS TM, DE CLERCQ E, HENEINE W: Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antiviral Therapy (2004) 9(1):57-65.
- VON MOLTKE LL, GREENBLATT DJ, GRANDA BW et al.: Inhibition of human cytochrome P450 isoforms by nonnucleoside reverse transcriptase inhibitors. J. Clin. Pharmacol. (2001) 41(1):85-91.
- POLLARD RB, ROBINSON P, DRANSFIELD K: Safety profile of nevirapine, a nonnucleoside reverse transcriptase inhibitor for the treatment of human immunodeficiency virus infection. Clin. Ther. (1998) 20:1071-1092.
- ANANWORANICH J, MOOR Z, SIANGPHOE U et al.: Incidence and risk factors for rash in Thai patients randomized to regimens with nevirapine, efavirenz or both drugs. AIDS (2005) 19(2):185-192.
- SULKOWSKI MS, THOMAS DL, MEHTA SH, CHAISSON RE, MOORE RD: Hepatotoxicity associated with nevirapine or efavirenz-containing antiretroviral therapy: role of hepatitis C and B infections. Hepatology (2002) 35(1):182-189.
- NUNEZ M, LANA R, MENDOZA JL, MARTIN-CARBONERO L, SORIANO V: Risk factors for severe hepatic injury after introduction of highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. (2001) 27(5):426-431.
- DE SANTIS M, CARDUCCI B, DE SANTIS L, CAVALIERE AF, STRAFACE G: Periconceptional exposure to efavirenz and neural tube defects. Arch. Intern. Med. (2002) 162(3):355.
- FUNDARO C, GENOVESE O, RENDELI C, TAMBURRINI E, SALVAGGIO E: Myelomeningocele in a child with intrauterine exposure to efavirenz. AIDS (2002) 16(2):299-300.
- VAN LETH F, PHANUPHAK P, RUXRUNGTHAM K et al.: 2NN Study team. Comparison of first-line antiretroviral therapy with regimens including nevirapine, efavirenz, or both drugs, plus stavudine and lamivudine: a randomised open-label trial, the 2NN Study. Lancet (2004) 363(9417):1253-1263.
- NAVIA MA, FITZGERALD PM, MCKEEVER BM et al.: Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature (1989) 159:87-94.
- PILLAY D, BRYANT M, GETMAN D et al.: HIV-1 protease inhibitors: their development, mechanism of action and clinical potential. Rev. Med. Virol. (1995) 5:23-33.
- KUMAR GN, RODRIGUES AD, BUKO AM et al.: Cytochrome P450- mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J. Pharmacol. Exp. Ther. (1996) 277:423-431.
- KEMPF DJ, MARSH KC, KUMAR G et al.: Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. (1997) 41:654-660.
- GERBER JG: Using pharmacokinetics to optimize antiretroviral drug–drug interactions in the treatment of human immunodeficiency virus infection. Clin. Infect. Dis. (2000) 30(Suppl. 2):S123-S129.
- ECKERT DM, KIM PS: Mechanism of viral membrane fusion and its inhibition. Ann. Rev. Biochem. (2001) 70:777-810.
- COOLEY LA, LEWIN SR: HIV-1 cell entry and advances in viral entry inhibitor therapy. J. Clin. Virol. (2003) 26:121-132.
- LALEZARI JP, HENRY K, O’HEARN M et al.: TORO 1 Study Group. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. (2003) 348(22):2175-2185.
- LAZZARIN A, CLOTET B, COOPER D et al.: TORO 2 Study Group. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. (2003) 348(22):2186-2195.
- TROTTIER B, WALMSLEY S, REYNES J et al.: Safety of enfuvirtide in combination with an optimized background of antiretrovirals in treatment-experienced HIV-1-infected adults over 48 weeks. J. Acquir. Immune Defic. Syndr. (2005) 40(4):413-421.
- GROSSMAN HA, HICKS C, NADLER J et al.: Efficacy and tolerability of TMC125 in HIV patients with NNRTI and PI resistance at 24 weeks: TMC125-c223. In: Program and Abstracts of the 45th Interscience Conference of Antimicrobial Agents and Chemotherapy, December 16-19 (2005) Washington, DC, USA. Abstract H-416c.
- KATLAMA C, CARVALHO MI, COOPER D et al.: TMC 114/r out performs investigator selected PI(s) in three class-experienced patients: week 24 primary analysis of POWER 1 (TMC 114-C213). In: Program and Abstracts of the 3rd IAS Conference on HIV Pathogenesis and Treatment, July 24-27 (2005), Rio de Janeiro, Brazil. Abstract WeOaLB0102.
- WILKIN T, HAUBRICH R, STEINHART CR et al.: TMC114/r superior to standard of care in 3-class-experienced patients: 24-wks primary analysis of the Power 2 Study (C202). In: Program and Abstracts of the 45th Interscience Conference of Antimicrobial Agents and Chemotherapy, December 16-19 (2005), Washington, DC, USA. Abstract H-413.
- CRAIGIE R: HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. (2001) 276(26):23213-23216.
- GRINSZTEJN B, NGUYEN BY, KATLAMA C et al.: Potent antiretroviral effect of MK-0518, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus. In: Program and Abstracts of the 13th Conference on Retroviruses and Opportunistic Infections; February 5-8, (2006) Denver, Colorado, USA. Abstract 159LB.
- MOORE JP, DOMS RW: The entry of entry inhibitors: a fusion of science and medicine. Proc. Natl. Acad. Sci. USA (2003) 100:10598-10602.
- LUCAS AD, GAUDIERI S, RAUCH A et al.: Cellular tropism of HIV-1 mediated and constrained by coreceptor dependencies. J. Viral Entry (2005) 1:17-27.
- PHILPOTT SM: HIV-1 coreceptor usage, transmission, and disease progression. Curr. HIV Res. (2003) 1:217-227.
- MOYLE GJ, WILDFIRE A, MANDALIA S et al.: Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis. (2005) 191(6):866-872.
- STEEL HM: Special presentation on aplaviroc-related hepatotoxicity. In: Program and Abstracts of the European AIDS Clinical Society 10th European AIDS Conference, November 17-20 (2005), Dublin, Ireland.
- MCHALE M, ABEL S, RUSSELL D et al.: Overview of Phase I and 2a safety and efficacy data of maraviroc (UK-427,857). In: Program and Abstracts of the 3rd International AIDS Society Conference on the HIV Pathogenesis and Treatment, July 24-27 (2005), Rio de Janeiro, Brazil. Abstract TuOa0204.
- LI F, GOILA-GAUR R, SALZWEDEL K et al.: PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. (2003) 100(23):13555-13560.
- BEATTY G, LALEZARI J, ERON J et al.: Safety and antiviral activity of PA-457, the first-in-class maturation inhibitor, in a 10-day monotherapy study in HIV-1 infected patients. In: Program and Abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy, December 16-19 (2005), Washington, DC, USA. Abstract H-416d.
- YENI PG, HAMMER SM, HIRSCH SM et al.: Treatment for adult HIV infection: 2004 recommendations of the International AIDS Society-USA Panel. JAMA (2004) 292:251-265.
- STASZEWSKI S, KEISER P, MONTANER JS et al.: Abacavir-lamivudine-zidovudine versus indinavir-lamivudine-zidovudine in antiretroviral-naïve HIV-infected adults: a randomized equivalence trial. JAMA (2001) 285(9):1155-1163.
- VIBHAGOOL A, CAHN P, SCHECHTER M et al.: Triple nucleoside treatment with abacavir plus the lamivudine/zidovudine combination tablet (COM) compared to indinavir/COM in antiretroviral therapy-naïve adults: results of a 48-weekopen-label, equivalence trial (CNA3014). Curr. Med. Res. Opin. (2004) 20(7):1103-1114.
- VAN LEEUWEN R, KATLAMA C, MURPHY RL et al.: A randomized trial to study first-line combination therapy with or without a protease inhibitor in HIV-1-infected patients. AIDS (2003) 17(7):987-999.
- BARTLETT JA, JOHNSON J, HERRERA G et al.: Abacavir/lamivudine (ABC/3TC) in combination with efavirenz (NNRTI), amprenavir/ritonavir (PI) or stavudine (NRTI): ESS4001(CLASS) preliminary 48 week results. In: XIV International AIDS Conference; July 7-12 (2002), Barcelona, Spain. Abstract TuOrB1189.
- GERSTOFT J, KIRK O, OBEL N et al.: Low efficacy and high frequencyof adverse events in a randomized trial of the triple nucleoside regimen abacavir, stavudine and didanosine. AIDS (2003) 17(14):2045-2052.
- PECORA FULCO P, KIRIAN MA: Effect of tenofovir on didanosine absorption in patients with HIV. Ann. Pharmacother. (2003) 37:1325-1328.
- JEMSEK J, HUTCHERSON P, HARPER E: Poor virologic responses and early emergence of resistance in treatment naïve, HIV-infected patients receiving a once daily triple nucleoside regimen of didanosine, lamivudine, and tenofovir DF. In: 11th Conference on Retroviruses and Opportunistic Infections; February (2004), San Francisco, CA, USA.
- LEON A, MARTINEZ E, MALLOLAS J et al.: Early virological failure in treatment-naive HIV-infected adults receiving didanosine and tenofovir plus efavirenz or nevirapine. AIDS (2005) 19:213-215.
- PODZAMCZER D, FERRER E, GATELL JM et al.: Early virological failure and occurrence of resistance in naive patients receiving tenofovir, didanosine and efavirenz. Antiviral Therapy (2004) 9:S172.
- BARRIOS A, RENDONA, NEGREDO E et al.: Paradoxical CD4+ T-cell decline in HIV-infected patients with complete virus suppression taking tenofovir and didanosine. AIDS (2005) 19(6):569-575.
- LACOMBE K, PACANOWSKI J, MEYNARD JL et al.: Risk factors for CD4 lymphopenia in patients treated with a tenofovir/didanosine high dose-containing highly active antiretroviral therapy regimen. AIDS (2005) 19(10):1107-1108.
- NEGREDO E, BONJOCH A, PAREDES R et al.: Compromised immunologic recovery in treatment-experienced patients with HIV infection receiving both tenofovir disoproxil fumarate and didanosine in the TORO studies. Clin. Infect. Dis. (2005) 41(6):901-905.
- COTE H, MAGIL A, HARRIGAN PR et al.: Kidney mitochondrial DNA levels in HIV patients with renal dysfunction on tenofovir-containing therapy are markedly reduced with concurrent didanosine therapy. In: Program and Abstracts of the 3rd IAS Conference on HIV Pathogenesis and Treatment, July 24-27, Rio de Janiero, Brazil (2005). Abstract TuPe2.3C19.
- LOUIE S, BALLARD C, BI L, BERINGER P: Factors increasing the risk of renal dysfunction with tenofovir difumarate (TDF). In: Program and Abstracts of the 3rd IAS Conference on HIV Pathogenesis and Treatment, July 24-27, Rio de Janiero, Brazil (2005). Abstract TuPe3.5B01.
Websites
- http://www.gsk.com/media/archive.htm Press release: GlaxoSmithKline terminates patient enrolment for Phase III studies of investigational HIV entry inhibitor APLAVIROC (GW873140). October 25 (2005), London, UK.
- http://www.scheringplough.com/schering_ plough/news/release.jsp?releaseID=774673 Press release: Schering-Plough discontinues Phase II study of vicriviroc in treatment-naive HIV patients, continues Phase II study in treatment-experienced HIV patients. October 27 (2005), Kenilworth, New Jersey, USA
- http:\\www:aidsinfo.nih.org Department of Health and Human Services (DHHS) panel on clinical practices for treatment of HIV infection: guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents (2005).
- http:\\www:bhiva.org Bhiva Writing Committee on behalf of the BHIVA Executive Committee: British HIV Association (BHIVA) guidelines for the treatment of HIV-infected adults with antiretroviral therapy (2005).
- http://www:unaids.org/epi/2005/doc/EPIupdate2005_pdf_en/epi-update2005_en.pdf. UNAIDS/WHO: AIDS epidemic update (2005).
- http://www:who.int/3by5/publications/ documents/en/3by5StrategyMakingIt Happen.pdf. The WHO strategy (2006).
- http://www:who.int/mediacentre/news/ releases/2006/pr13/en/index.html. WHO Press Release. Progress on global access to antiretroviral therapy (2006).
- http://www:who.int/hiv/pub/prev_care/en/ arvrevision2003en.pdf. WHO guidelines. Scaling up antiretroviral therapy in resource-limited settings: treatment guidelines for a public health approach (2003).