Abstract
Prostate cancer (PCa) is the second most common type of cancer among men in Western societies. Once in the castrate-resistant metastatic setting therapeutic options are limited. The importance of angiogenesis in the progression of PCa has been widely reported providing a rationale to test anti-angiogenic compounds for PCa treatment in clinical trials (CTs). However, in spite of the promising results shown in preclinical models and some anti-tumor activity observed in CTs, to date, no angiogenic inhibitor has been approved for use in PCa. This editorial outlines the latest clinical evidence regarding anti-angiogenic therapies in PCa treatment.
Expert opinion
Numerous evidences support the role of angiogenesis in the progression of PCa, providing a rationale for the use of anti-angiogenic compounds in PCa clinical trials. However, while anti-angiogenic therapies have shown promising results in preclinical models, they have not shown an overall survival (OS) benefit in PCa patients. Moreover, several challenges including intrinsic or acquired resistance, enhanced metastasis during treatment, relevant side-effects, and the lack of biomarkers for monitoring response to treatment must be solved before introducing them into clinical practice. As a result, to date there is no anti-angiogenic therapy approved by the FDA for PCa treatment. Further research will lead to a better understanding of the anti-angiogenic cascade, which will allow for additional molecular targets that, together with the results of ongoing clinical trials will help to clarify the usefulness of anti-angiogenic therapies in the treatment of PCa. Until then, it is unlikely that anti-angiogenic therapies will be used for the treatment of PCa in the clinical setting.
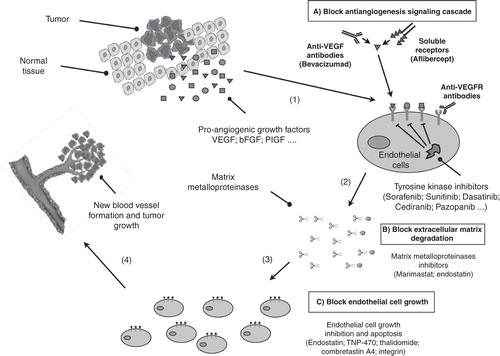
Prostate cancer (PCa) ranks second to skin cancer as the most common cancer among men in Western societies and is the second cause of death due to cancer after lung cancer. Since the availability and increased use of prostate specific antigen (PSA) as a screening tool, PCa is diagnosed earlier and most of the new diagnosed cases are confined to the prostate while the diagnosis of locally advanced and metastatic disease has decreased. However, patients with aggressive localized and locally advanced disease have an increased risk of developing metastasis Citation[1]. At the metastatic stage, patients may respond to androgen deprivation therapy but eventually the tumor will become independent of androgens to grow, resulting in tumor progression and death of the patient. Since 2004, the only Food and Drug Administration (FDA)-approved therapies for the treatment of metastatic castration-resistant prostate cancer (mCRPC) were docetaxel and prednisone. There was, therefore, an increasing interest in developing new drugs for mCRPC treatment and from 2010 on, new additional treatments have been approved by the FDA Citation[2]. Experimental evidences support a potential role of angiogenesis in the progression of PCa, providing a rationale to use anti-angiogenic compounds in CTs Citation[3,4]. This editorial summarizes the latest clinical evidence regarding anti-angiogenic therapies in the treatment of PCa ().
Figure 1. Main anti-angiogenic strategies for prostate cancer treatment. Pro-angiogenic factors (VEGF, bFGF …) are synthesized inside tumor cells and secreted into the surrounding tissue where they bind to specific receptors located on the outer surface of the endothelial cells (1). The binding of pro-angiogenic factors to its specific receptor, activate endothelial cells, which produce matrix metalloproteinases, a class of enzymes that break down the extracellular matrix allowing the migration of endothelial cells (2). As they migrate into the surrounding tissue, activated endothelial cells begin to divide (3) and organize into a network of new blood vessels (4). Angiogenesis plays a role in prostate cancer progression providing a rationale to test anti-angiogenesis inhibitors in prostate cancer patients. These inhibitors fall into different categories, depending on their mechanism of action. Some inhibit the anti-angiogenic signaling cascade (A), others block the ability of endothelial cells to break down the extracellular matrix, whereas others inhibit endothelial cell growth directly (C).
The process of forming new blood vessels from pre-existing vessels is called angiogenesis. One of the first observations made in the field of angiogenesis was that tumor growth is frequently accompanied by an increase in the number of blood vessels in the tumor area. This observation, together with the discovery that tumors secrete factors that are involved in the formation of new blood vessels, led to the hypothesis that tumor growth is highly dependent on the development of new blood vessels that supply nutrients and oxygen (and eliminate tumor metabolic waste) to the tumor cells resulting in tumor growth. In the early stages of the tumorigenic process, when tumors are smaller than 2 mm in diameter, they are able to obtain nutrients and oxygen from nearby capillaries. However, when the limit of 2 mm is reached, the nutritional needs of tumors increase significantly, and they cannot continue growing without extra nutrients and oxygen supply. The mechanism of angiogenesis is then activated to form new blood vessels that provide the necessary nutrients and oxygen for tumor growth. Angiogenesis contributes to tumor growth and proliferation and also plays a decisive role in the metastatic process helping tumor cells not only to invade tissues adjacent to the primary tumor, but also spreading of cells that are released from the primary tumor to colonize distant tissues, seriously threatening the patient's life Citation[5].
In 1971, Dr. Judah Folkman launched the hypothesis that the inhibition of angiogenesis could be an effective strategy in the treatment of cancer, and thereafter various pro-angiogenic and anti-angiogenic regulatory elements of angiogenesis have been identified, some of them representing therapeutic targets Citation[5,6].
Angiogenesis depends on the balance between pro-angiogenic and anti-angiogenic factors. Normally, anti-angiogenic factors predominate, blocking growth. However, in the tumoral process, since tumors secrete large amounts of pro-angiogenic factors, an imbalance in favor of these factors occurs overcoming the anti-angiogenic factors and leading to the formation of new blood vessels around the tumor, in a process termed as the “angiogenic switch” Citation[7].
Hypoxia represents the major trigger for angiogenesis. As the tumor grows, enhanced nutritional and oxygen requirements produce a hypoxic tumor micro-environment that leads to the expression of hypoxia-inducible factor 1 alpha (HIF1α), which activates the expression of pro-angiogenic genes, including, among others, the vascular endothelial growth factor (VEGF), the basic fibroblast growth factor (bFGF), the platelet-derived growth factor beta (PDGFβ), and the tumor necrosis factor alpha (TNF-α). Several naturally occurring anti-angiogenic factors have been described, including angiostatin, endostatin, thrombospondin, and cytokines such as IL-12 Citation[8].
The VEGF plays a central role in the process of angiogenesis. It comprises seven members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, PlGF (placenta growth factor) and Trimeresurus flavoviridis svVEGF. It is synthesized inside tumor cells and secreted into circulation where it binds to and activates a family of specific receptors, the vascular endothelial growth factor receptors (VEGFR), which are located in the membrane of vascular endothelial cells. This process stimulates the proliferation and migration of vascular endothelial cells leading to blood vessel basement membrane and the surrounding extracellular matrix degradation, allowing the endothelial cell precursors to migrate toward the angiogenic stimulus to form new capillary tubes with the cooperation of surrounding periendothelial and vascular smooth muscle cells, ultimately leading to tumor angiogenesis Citation[9].
Due to the many pro-angiogenic factors discovered, it was initially thought that inhibiting only one of them would have no effect on tumor growth. However, experiments in animal and cell culture models showed that blocking the VEGF alone substantially inhibited the angiogenic process and tumor growth. Accordingly, two main strategies were designed to inhibit the VEGF pathway; i) inhibition of VEGF ligands and ii) inhibition of the receptors Citation[10,11].
The importance of angiogenesis in the progression of PCa has been widely documented. Microvessel density is significantly greater in patients with metastatic PCa than in patients with localized disease. Both VEGF-A and VEGF-B have been detected in prostate tumor cells and in tumor blood vessels, and the plasma levels of the VEGF in patients with metastatic PCa are significantly higher than in those with localized disease. It has been reported than blood and urine VEGF levels correlate with PCa patient outcomes Citation[3]. In addition, an association between PSA progression and markers of angiogenesis has been described Citation[12]. Anti-angiogenic agents have received considerable attention and are currently under evaluation in ongoing CTs. Anti-angiogenic therapies are currently approved for the treatment of several types of cancers including glioblastoma, colorectal cancer, liver cancer, non-small cell lung cancer, chronic myeloid leukemia, and gastrointestinal stromal tumors Citation[3,4,12,13]. However, anti-angiogenic therapies have not shown, so far, an OS benefit in PCa patients.
Bevacizumab, a humanized variant of an anti-VEGF monoclonal antibody targeting VEGF-A, has shown anti-tumor activity in several cancers including renal cell carcinoma, colorectal cancer, glioblastoma, and non-small cell lung cancer all in the metastatic setting. The action mechanism of bevacizumab is partly due to the inhibition of the VEGF pathway through blockage of the VEGFR producing a normalization of the tumor vascularity, reduced tumor permeability and improved access of chemotherapy drugs to the tumor Citation[10,11]. Bevacizumab was first assessed as a single agent in PCa treatment and in a Phase II clinical trial no effect on objective or PSA response was reported. Bevacizumab was then used, in a Phase II clinical trial, in combination with docetaxel in 20 pre-treated patients with mCRPC. The study showed a PSA partial response rate of 55%, a partial objective response of 38%, and the treatment was generally well tolerated by patients. In a Phase II clinical trial (CALGB 90006), 79 chemotherapy-naïve patients with mCRPC were treated with bevacizumab combined with docetaxel and estramustine. Although the primary endpoint of progression-free survival (PFS) was not reached, increased antitumor activity and OS were observed. In view of these promising results, the drug was evaluated in a multicenter Phase III trial (CALGB 90401), comparing docetaxel and prednisone with or without bevacizumab in 1050 chemotherapy-naïve patients with mCRPC. Although an improvement in PFS, objective response rates and PSA decline were observed in the bevacizumab-treated group, the addition of bevacizumab to docetaxel showed no improved OS and it was associated with greater morbidity and mortality Citation[12,13].
An alternative strategy to block the VEGF is the use of soluble receptors, called VEGF-traps. Aflibercept blocks circulating VEGF-A, VEGF-B, and PIGF, limiting the amount of the VEGF available for binding to the cell receptors. Aflibercept did not extend OS in combination with docetaxel and prednisone or prednisolone in the randomized Phase III VENICE trial Citation[13].
Additionally, a second mechanism to inhibit the VEGF pathway involves blocking VEGFRs. A variety of small molecule receptor tyrosine kinase inhibitors (TKI) targeting the VEGFR has been developed and is currently being tested in CTs for PCa treatment. Sorafenib is a TKI that targets growth signaling by blocking the Raf kinase, a critical component of the RAF/MEK/ERK signaling pathways that controls cell division and proliferation. It also blocks angiogenesis by inhibiting the VEGFR-2/platelet-derived growth factor receptor beta (PDGFRβ) signaling cascade. Sorafenib showed minimal activity in monotherapy regimes in CRPC with or without metastases. CTs evaluating sorafenib in early PCa and in combination with chemotherapy and hormone therapy are currently ongoing, although only modest antitumor activity and no robust PSA declines have been reported.
Sunitinib is a TKI that blocks VEGFR2, PDGFRβ, and c-kit, thereby inhibiting angiogenesis and cell proliferation. Based on PSA decline ≥ 50% and radiographic and histological evidence of regression, sunitinib does not seem to be significantly active in PCa Citation[3,4].
Several evidences support the role of PDGF in PCa growth and provide a rationale for PDGF-targeted therapies in PCa. However, so far, these treatments have proved disappointing. Imatinib inhibits TK encoded by the bcr-abl, c-kit, and PDGF oncogenes. Three Phase II studies showed no significant decrease in PSA and significant toxicity after imatinib treatment. It was also evaluated in combination with docetaxel in a Phase II study in mCRPC. PSA decline ≥ 50%, PFS and OS were not significantly different and considerable toxicity in the imatinib group was reported Citation[3].
Other TKI such as Dasatinib, Cediranib, and Pazopanib are at different stages of clinical development Citation[3,4,9].
Extracellular matrix inhibitors are being evaluated in the treatment of PCa. After angiogenesis activation, endothelial cells produce matrix metalloproteinases (MMPs) that are released into the surrounding tissue. Metalloproteinases break down the extracellular matrix allowing the migration of endothelial cells, which begin to divide and integrate in a network of blood vessels. Overexpression of MMPs has been associated with tumor progression in PCa. Therefore, MMP inhibitors are potential therapies for PCa treatment. Marimastat has biological effect and may delay PCa progression although dose-limiting toxicity has been described Citation[9].
Other substances that modulate angiogenesis through endothelial cell inhibition include endostatin, TNP-470, and thalidomide. Preclinical studies suggest that endostatin may prevent metastasis in early-stage mCRPC. On the other hand, Phase I studies with TNP-470 and have yielded disappointing results Citation[9]. An alternative mechanism is used by cilengitide, which interacts with integrin inhibiting endothelial cell–cell interactions, endothelial cell–matrix interactions, and angiogenesis. Once again, Phase II CTs did not show significant clinical activity Citation[3,4,12,13].
Thalidomide has shown antitumor activity in PCa. Although the exact mechanism by which thalidomide inhibits angiogenesis is not fully understood, it has been suggested that it may control the expression of several MMPs and inhibits VEGF and bFGF. Thalidomide has been evaluated in combination with docetaxel in a Phase II trial in 75 chemotherapy-naïve mCRPC patients. Addition of thalidomide showed a higher percentage of patients with a greater than or equal decline in PSA (53% vs. 37%), PFS (5.9 vs. 3.7 months), and OS at 18 months (68.2 vs. 42.9%). In a Phase II study, thalidomide was combined with bevacizumab, docetaxel, and prednisone in 60 chemotherapy-naïve mCRPC patients. A PSA decline was observed in 88% of patients and the median OS was 28.2 months in the combination group. Given these promising results lenalidomide, a less toxic immunomodulatory derivative of thalidomide is being tested combined with docetaxel, prednisone, and bevacizumab in patients with mCRPC in ongoing Phase I/II and III CTs Citation[3,4,12,13].
In conclusion, since angiogenesis inhibitors have demonstrated therapeutic value in other types of cancers, they have potential to enhance the therapeutic arsenal for patients with PCa. However, while the results of anti-angiogenic therapy in preclinical models have provided promising results, there is some discrepancy between these data and those obtained in CTs, and to date, there is no anti-angiogenic treatment approved by the FDA for the treatment of PCa. Anti-angiogenic treatments face several major challenges including intrinsic or acquired resistance, enhanced metastasis during treatment, relevant side-effects, and the lack of validated biomarkers for monitoring response to therapy that must be solved before introducing them into clinical practice Citation[3,10,14,15]. As we continue to learn more about the angiogenesis process, therapies with new molecular targets will emerge, and together with the numerous ongoing and future CTs they will help to clarify the usefulness of anti-angiogenic treatments in PCa. Until then, it is unlikely that anti-angiogenic therapies will be used for the treatment of PCa in the clinical setting.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
- Siegel R, Naishadham D, Jemal A. Cancer Statistics 2012. CA Cancer J Clin 2012;62:10-29
- Attard G, de Bono JS. Translating scientific advancement into clinical benefit for castration-resistant prostate cancer patients. Clin Cancer Res 2011;17:3867-75
- Hwang C, Heath EI. Angiogenesis inhibitors in the treatment of prostate cancer. J Hematol Oncol 2010;3:26
- Aragon-Ching JB, Madan RA, Dahit WL. Angiogenesis inhibition in prostate cancer: current uses and future promises. J Oncol 2010;Article ID 361836:7 pages
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005;438:967-74
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971;285:1182-6
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996;86:353-64
- Chung A, Lee J, Ferrara N. Targeting the tumor vasculature: insights from physiological angiogenesis. Nat Rev Cancer 2010;10:505-14
- Jimenez JA, Kao C, Raikwar S, Gardner TA. Current status of anti-angiogenesis therapy for prostate cancer. Urol Oncol 2006;24:260-8
- Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett 2012;320:130-7
- Ferrara N. From the discovery of vascular endothelial growth factor to the introduction of avastin in clinical trials - an interview with Napoleone Ferrara by Domenico Ribatti. Int J Dev Biol 2011;55:383-8
- Bianchini D, Zivi A, Sandhu A, de Bono JS. Horizon scanning for novel therapeutics for the treatment of prostate cancer. Expert Opin Investig Drugs 2010;19:1487-502
- Osanto S, Van Poppel H. Emerging new therapies for advanced prostate cancer. Ther Adv Urol 2012;4:3-12
- Roukos DH, Tzakos A, Zografos G. Current concerns and challenges regarding tailored anti-angiogenic therapy in cancer. Expert Rev Anticancer Ther 2009;9:1413-16
- Ribatti D. Antiangiogenic therapy accelerates tumor metastasis. Leuk Res 2011;35:24-6