Abstract
Rapamycin might have beneficial effects, some of them acting via autophagy, in several cellular, fly and mouse models of degenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) and dominant spinocerebellar atrophy (SCA). Other more sophisticated mechanisms have been described such as intervention in cell signaling and cell transcription particularly related to PD. Yet AD, PD, HD and SCA3 are chronic degenerative diseases, and chronic administration of rapamycin at advanced clinical stages may result in deleterious systemic effects due to chronic inhibition of mammalian target of rapamycin (mTOR). Studies are needed to uncover more specific inhibitors of particular mTOR signaling pathways, and to establish realistic guidelines for treatment at early stages of neurodegenerative processes.
Keywords::
Rapamycin is a natural antibiotic and immunosuppressant drug which is widely used to prevent rejection in organ transplantation. Moreover, due to its antiproliferative properties, rapamycin has also been approved for use in coronary-artery eluting stents and has become a promising anti-cancer agent. During the last decade, a growing number of studies have pointed toward rapamycin as a potential therapeutic agent against neurodegenerative diseases. The present paper first summarizes the molecular mechanisms of rapamycin and then reviews the main experimental studies dealing with a potential role for rapamycin in neurodegenerative diseases.
1. The mTOR signaling pathway
Rapamycin or sirolimus, a macrolide produced by Streptomyces hygroscopicus found in the soil of Rapa Nui Island, influences several essential cellular processes, including cell growth and proliferation, energetic metabolism, protein synthesis and autophagy, by inhibiting the activity of mammalian target of rapamycin (mTOR) Citation[1]. mTOR is a ubiquitous serine/threonine kinase member of the phosphoinositide 3-kinase-related protein kinase family that forms two functionally different enzymatic complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The two complexes share the catalytic subunit (mTOR) and two regulators (positive MLST8 and negative DEPTOR regulators) but differ in the composition of their scaffolding proteins Raptor (for mTORC1) and Rictor (for mTORC2), which give mTOR complexes the specificity for different substrates and regulators. Moreover, other unique components include the negative regulator PRAS40 of mTORC1, and PROTOR and mSIN1 proteins of mTORC2, which participate in the complex assembly and in targeting mTORC2 to membranes, respectively Citation[1,2]. Rapamycin does not impair mTOR kinase activity per se but rather disrupts the assembly of mTOR complexes by specifically binding FKBP12 protein which, in turn, inhibits Raptor-bound, but not Rictor-bound, mTOR. Thus, rapamycin's cellular effects are mainly due to its ability to disrupt mTORC1 activity rather than that of mTORC2, although prolonged treatment with high doses of rapamycin can also inhibit mTORC2 activity in specific tissues and cell lines.
mTORC1 is a key integrator of signaling produced by several nutrients, stressors, growth factors and energy signals. Therefore, mTORC1 activity is crucial in regulating cellular responses to external stimuli. Signal integration occurs at the level of tuberous sclerosis 1 (TSC1)-TSC2 complex, which exerts an inhibitory effect on mTORC1 signaling through Rheb acting as a mediator. PI3K is an important upstream mediator of amino acid responses in this pathway. Depending on the effect of nutrients, growth factors and stressors on TSC1-TSC2, mTORC1 can promote protein synthesis, mitochondrial and lipid metabolism and autophagy (). Signal integration also occurs at the lysosomes, where mTORC1 can become directly activated when it is recruited by RAG GTPases to the lysosomal surface in response to amino acids, resulting in the inhibition of autophagy, a molecular process which is aimed at degrading intracellular components for the recycling of molecules necessary to rebuild cellular components. It is worth noting that rapamycin promotes autophagy not only by blocking the inhibitory effect of mTORC1 but also by directly inducing the formation of autophagosome. The best characterized substrates of mTORC1 are ribosomal p70 S6 protein kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), with which mTORC1 is able to regulate the protein translation essential in synaptic plasticity Citation[1,2]. Moreover, mTORC1 is able to stimulate lipid synthesis, and mitochondrial proliferation and function, by regulating the expression of key genes in response to growth factors and nutrients, resulting in cell growth and accumulation of energy stores, and anticipating responses in the face of nutrient starvation Citation[1,2].
Figure 1. The mTOR-signaling pathway. Rapamycin bound to FKBP12 intracellular receptor influences several essential cellular processes, including cell survival, metabolism and mitochondrial function, protein synthesis and autophagy, by inhibiting the mTOR activity. mTOR is a serine/threonine kinase that forms two functional complexes, mTORC1 and mTORC2, which share the catalytic subunit (mTOR) and two regulators (MLST8 and DEPTOR) but differ in other components (See text for details). The TSC1-TSC2 complex integrates the signaling produced by several nutrients, stressors and growth factors and depending on external stimuli its activity may result in direct inhibition of mTORC1 and activation of mTORC2. mTORC1 promotes protein and lipid synthesis, as well as modulating mitochondrial function, by regulating gene expression and S6K and 4E-BP1 activity. In response to amino acids, mTORC1 is recruited by RAG GTPases to lysosomal membranes, where it is activated and inhibits autophagy. mTORC2 is activated in response to growth factors and promotes the AKT pathway, which is involved in cell survival, metabolism and cytoskeletal organization.
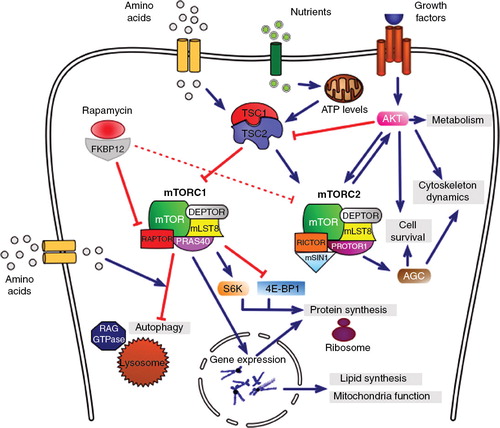
In contrast, mTORC2 seems to be insensitive to nutrients and energetic stress; it is activated mainly by growth factors via the TSC1-TSC2 complex which stimulates mTORC2 activity. The principal mTORC2 downstream substrate is the AKT-signaling pathway which promotes cell survival, proliferation and migration, and also regulates various metabolic processes following mTORC2 activation Citation[1,2]. Additionally, mTORC2 is able to activate other AGC kinases involved in cell survival and cytoskeletal organization ().
Thus, inhibition of mTOR signaling by rapamycin results in the promotion of catabolic processes, in the regulation of the protein synthesis necessary for neuronal plasticity responses and in the induction of autophagy that degrades long-lived organelles but also aberrant or misfolded proteins. All this evidence argues in favor of the potential use of rapamycin in neurodegenerative diseases, in which cellular energetic failure, synaptic dysfunction and accumulation of aberrant proteins are widely accepted to be crucial events leading to the progressive neuronal degenerative process Citation[3,4]. Due to the complexity of mTOR signaling pathways, distinct intermediates must be considered as potential drug targets of therapeutic intervention affecting specific mTOR activities.
2. Rapamycin in neurodegenerative diseases
Most of the interest in the potential utility of rapamycin in neurodegenerative diseases with accumulation of abnormal protein aggregates, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) and dominant spinocerebellar atrophy (SCA), has been focused on its ability to promote autophagy by inhibiting mTORC1. Several studies have demonstrated that the inhibition of mTORC1 with rapamycin promotes the degradation of aggregate-prone proteins in vitro and results in a reduction of the severity of neurodegeneration in several models in vivo Citation[3,4]. For instance, rapamycin attenuates dopaminergic nigrostriatal degeneration in toxin-induced PD murine models by promoting autophagy Citation[5] and it promotes degradation of ubiquitylated proteins resulting from the altered function of the ubiquitin-proteasome system Citation[6]. Similarly, rapamycin confers neuroprotection in Drosophila and mouse models of HD by reducing protein aggregates via autophagy Citation[7]. Rapamycin also improves cognition and reduces amyloid levels in the PDAPP transgenic mouse model of AD Citation[8]. In addition, rapamycin reduces TDP-43 protein aggregates in a cellular model of TDP-43 proteinopathy, thus having potential effects in frontotemporal lobar degeneration with TDP-43 protein aggregates and in sporadic amyotrophic lateral sclerosis Citation[9]. Finally, temsirolimus, a rapamycin analogue, improves motor function and reduces ataxin-3 inclusions in a transgenic mouse model of SCA3 by enhancing the autophagic clearance of toxic aggregate-prone proteins Citation[10]. However, the effects of rapamycin on autophagy are less prominent than those on peripheral tissues and non-neuronal cells. Everolimus, a rapamycin analogue, stimulates autophagy in muscle but not in brain and does not reduce mutant huntingtin levels in the R6/2 mouse model of HD Citation[11]. Another paper reported that mTOR-dependent but everolimus-independent mechanisms regulate autophagy and huntingtin-aggregate degradation in cells of neuronal origin Citation[12].
However, other rapamycin properties beyond its pro-autophagy activity deserve attention when considering its potential beneficial effect in neurodegenerative diseases. Since mTOR signaling controls cellular responses to nutrient availability by modulating energy production, it seems reasonable that rapamycin might also contribute to balancing the energetic deficiency in neurodegenerative processes. Moreover, several lines of evidence demonstrate the role of the mTOR pathway during inflammatory responses which are known to be involved in the progression of most neurodegenerative diseases. Another feature of rapamycin with potential interest vis-à-vis neurodegenerative processes is its capacity to modulate protein synthesis, which is relevant in synaptic plasticity and cell survival control Citation[5]. In line with these hypotheses, recent studies have shown that rapamycin blocks mTORC1-dependent translation of the pro-cell death protein RTP801, thereby conferring neuroprotection in a PD murine model Citation[13]. Moreover, rapamycin activation of the translation inhibitor 4E-BP suppresses pathology in PINK1/parkin Drosophila mutants used as models of inherited PD Citation[14]. Recent studies have shown a more sophisticated role for mTORC1 in cell signaling. UCHL1 protein, which is involved in the UPS, is decreased in sporadic PD, thus jeopardizing removal of abnormal proteins. Antisense Uchl1 function is under the control of stress signaling pathways, as mTORC1 inhibition by rapamycin causes an increase in UCHL1 protein that is associated with the shuttling of antisense Uchl1 RNA from the nucleus to the cytoplasm. Antisense Uchl1 RNA is required for the association of the overlapping sense protein-coding mRNA to active polysomes for translation. These data reveal another mechanism of gene expression control at the post-transcriptional level which can be modulated by rapamycin Citation[15].
In spite of these advantages, the effects of rapamycin are not always beneficial. Under certain conditions, rapamycin increases amyloid toxicity, reduces long-term potentiation and synaptic plasticity, and promotes brain atrophy, with probable deleterious effects in AD Citation[5,16]. Inhibition of synaptic plasticity by rapamycin is a major counter-argument to the use of rapamycin not only in AD but in most of, if not all neurodegenerative diseases. Finally, rapamycin exacerbates neuronal damage in a murine model of amyotrophic lateral sclerosis due to mutations in superoxide dismutase 1 Citation[17].
3. Conclusions and therapeutic implications
Experimental evidence regarding the beneficial effects of the inhibition of mTORC1 in cellular and animal models seems to point to rapamycin as a potential therapeutic agent against certain human neurodegenerative diseases. This is further supported by the demonstration that inhibition of mTOR pathway or ablation of the mTOR target p70S6K extends lifespan in mice; these effects are even observed when the drug is administered at relatively advanced ages Citation[2]. However, most experimental studies related to neurodegeneration have been carried out in cellular models, flies and rodents, and they have, with a few exceptions, been focused mainly on mechanistic aspects rather than long-lasting effects.
In addition to the limited information about rapamycin in neurodegenerative models, it is worth recalling that mTOR is implicated in a number of cellular processes including metabolic balance by regulating glucose, lipid and energy metabolism in liver, muscle, fatty tissue and pancreas; long-lasting treatment with rapamycin may negatively impact systemic homeostasis. Moreover, long-lasting rapamycin treatment may affect mTORC2 and the pro-survival pathways associated with its activity; therefore, long-term treatment with rapamycin may result in impaired neuron survival. Additional detrimental effects of chronic treatment with rapamycin include vascular dysfunction and vasculopathy and altered responses to insulin Citation[5].
Taken together, these observations illustrate the tremendous information that rapamycin has yielded toward increased understanding of one of the principal cellular signaling pathways which regulates variegated functions, from flies to humans. Because of its immunosupressor properties, rapamycin has been used to prevent rejection in organ transplantation; and because of its depressing effects on cell proliferation and growth, rapamycin is a useful therapeutic tool in certain types of cancer and in the local prevention of cellular proliferation in coronary stents. Yet the wide-ranging potent effects of rapamycin also emerge as the main objection to its use as a long-term therapeutic agent in the treatment of neurodegenerative diseases. At first glance, control of the dosage and standard administration of rapamycin should be established in very precise settings. Increased understanding of the physiological role of mTOR pathways in normal brain function is required in order to prevent potential side-effects of chronic mTOR inhibition. A more realistic approach might stress that the lack of specificity of rapamycin highlights the need to develop new mTOR-specific inhibitors (i.e., those directed against PI3K). Several drugs are currently available to researchers that fit this description including Torin1, Torin2, WAY-600 and WYE-354. Finally it is worth remembering that human neurodegenerative diseases are characterized by long preclinical stages lasting for years during which the major components of the disease develop together with plastic changes to correct altered metabolic pathways and neuronal connections until reaching thresholds leading to neurological deficits. The appearance of neurological symptoms in these particular chronic settings represents an advanced stage in the progression of the degenerative process that is difficult to reconcile with observations in experimental models. This is the reason, in part, for the repeated failure of treatments in humans that are successful in animal models. As an example, rapamycin activation of autophagy in clinical stages of AD seems unrealistic, as autophagy is already in motion in millions of aberrant neurites of beta-amyloid plaques. However, mTOR inhibition by more specific rapamycin analogues may have therapeutic benefits at early stages of neurodegenerative processes by striking specific targets (e.g., alteration of autophagy in early plaques in AD) years before the appearance of clinical symptoms.
Declaration of interest
All the authors declare no conflict of interest.
Acknowledgment
Authors thank T Yohannan for editorial assistance.
Bibliography
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011;12:21-35
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274-93
- Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011;12:437-52
- Chong ZZ, Shang YC, Wang S, Maiese K. Shedding new light on neurodegenerative diseases through the mammalian target rapamycin. Prog Neurobiol 2012;99:128-48
- Dehay B, Bové J, Rodríguez-Muela N, et al. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci 2010;30:12535-344
- Pan T, Kondo S, Zhu W, et al. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis 2008;32:16-25
- Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36:585-95
- Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One 2010;5:e9979
- Caccamo A, Majumder S, Deng JJ, et al. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem 2009;284:27416-24
- Menzies FM, Huebener J, Renna M, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010;133:93-104
- Fox JH, Connor T, Chopra V, et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Mol Neurodegener 2010;5:26
- Roscic A, Baldo B, Crochemore C, et al. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem 2011;119:398-407
- Malagelada C, Jin ZH, Jackson-Lewis V, et al. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci 2010;30:1166-75
- Tain LS, Mortiboys H, Tao RN, et al. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci 2009;12:1129-35
- Carrieri C, Cimatti L, Biagioli M, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012;491:454-7
- Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci 2012;4:941-52
- Zhang X. Rapamycin treatment augments motor neuron degeneration in SOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011;7:412-25