Abstract
Mitochondrial functions are altered in many human diseases including cancer. Development of mitochondria-targeted therapies, either through restoring normal mitochondrial function or promoting mitochondrial-induced cell death, is one of the attractive strategies to improve the outcome of cancer treatment. Recent advances have revealed the important functional involvement of mitochondrial dynamics in cancer biology. Dynamin-related protein 1 (Drp1), a member of the dynamin family of GTPases required for mitochondrial fission, has been found upregulated in certain types of cancers, such as lung and breast cancers. In addition, the roles of Drp1 in cell cycle progression, genome instability, cell migration and apoptosis in cancer cells have also been recently uncovered. These findings raise the possibility of targeting Drp1-mediated mitochondrial fission as an effective therapy for treating cancer. This article explores the function of Drp1 in cancer cells and discusses the theoretical basis for the development of potential targeted therapy.
1. Introduction
Mitochondria are crucial organelles for a diverse range of important cellular functions, including cell metabolism, growth, differentiation, survival and programmed cell-death Citation[1]. Mitochondria are highly dynamic, constantly changing their morphology to suit the variable needs of the cell. Along with the recent advances in understanding the complex molecular machinery regulating mitochondrial dynamics, there are also expanding insights of how these processes are integrated into cellular physiology. This article describes recent important findings focusing on the role of Drp1 in regulating several important cancer-related processes. The rationale and potential of targeting mitochondrial fission protein Drp1 in cancer therapy will be discussed.
2. Mitochondrial dynamics and cancer progression
The mitochondrial network exists as mixed structures of long interconnected tubules with short isolated dot-like spheres. The status of mitochondrial network is regulated by highly dynamic processes involving fusion, fission and movement of the organelle along cytoskeleton tracks. Highly conserved dynamin-related GTPases (DRPs) have been identified as the primary regulators of mitochondrial dynamics Citation[2]. Notably, dynamin-related protein 1 (Drp1) is essential for mitochondrial fission. Mitofusins (Mfn1 and Mfn2) and Opa1 are required for mitochondrial fusion. By modifying the balance of mitochondrial fusion and fission events, the mitochondrial network remodels in response to a variety of signals derived from both endogenous and exogenous stimuli. Defects in the regulation of such balance have been correlated with human pathologies Citation[2].
Mitochondrial fission is required for certain normal cellular functions such as facilitating organelle transport in neurons Citation[3], removal of damaged mitochondria through mitophagy Citation[4] and proper mitochondrial distribution during cell division Citation[5]. This process is achieved through the coordination of Drp1 with other adaptor proteins including Mff, Fis1 and MiDs. Drp1 polymerizes into spirals around mitochondria, which constricts the organelle through GTP hydrolysis leading to mitochondrial division Citation[6]. The activity of Drp1 is under complex regulation through posttranslational modifications, including phosphorylation, S-nitrosylation, ubiquitination and SUMOylation Citation[7]. Excessive mitochondrial fission mediated by dysregulated Drp1 has been observed in diseases including cancer and neurodegenerative diseases.
2.1 The cycle of mitochondrial remodeling and the cell cycle
Cancer is characterized as a disease of inappropriate cell proliferation, with dysregulated cell cycle control as one of its fundamental aspects Citation[8]. Recent findings have revealed that mitochondrial fusion and fission cycles are integrated into the regulation of cell cycle progression Citation[9,10]. Mitochondria cannot be created de novo. They are formed from the division of existing organelles, and they must be inherited from mother cells through cell division. Mitochondria grow continuously throughout the cell cycle, and the organization of the mitochondrial network is sophisticatedly controlled across the different phases of the cell cycle. At the G1/S border, mitochondria form a single, giant tubular network Citation[9]. Mitochondria at this cell cycle stage are hyperpolarized and highly coupled, which is associated with increased energy production in order to prepare for the initiation of the highly energy consuming process – DNA synthesis Citation[9]. In addition, such mitochondrial hyperfusion also facilitates the mixing of mitochondrial contents such as mtDNA between adjacent mitochondria in order to maintain a homogenous mitochondrial network within the cell, thus ensuring proper inheritance following cellular division.
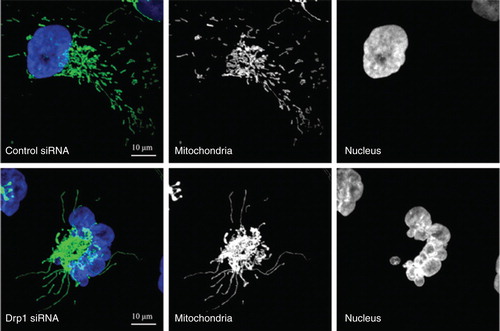
The formation of a highly connected mitochondrial network at G1/S border is transient. During the following S, G2 and M phases, the hyperfused mitochondrial network is then disassembled and becomes increasingly fragmented. Evidence has shown that Drp1-mediated mitochondrial fission is required for the proper progression of those cell cycle phases following G1/S transition Citation[10]. Extensive fragmentation of the mitochondrial network is achieved at mitosis and is required to facilitate the equal segregation of mitochondrial contents between daughter cells. Mitotic mitochondrial fission is directly regulated by the kinases involved in mitosis. Phosphorylation by mitotic kinase Aurora A promotes RalA relocalization to mitochondrial membranes, where it recruits the effector RalBP1. RalBP1 functions as a scaffold for cyclin B/cyclin-dependent kinase 1 (cdk1), which promotes phosphorylation of Drp1 at Ser616 resulting in mitochondrial fission Citation[5,11]. Sustained mitochondrial hyperfusion beyond the G1/S border induces replication stress causing ATM-dependent G2/M delay and chromosomal instability during mitosis Citation[10]. The replication stress was shown due to constant replication initiation signaling generated from hyperfused mitochondria Citation[9,10]. Prolonged mitochondrial fusion also causes mitochondrial bridges between daughter cells resulting in a defective cytokinesis, unequal distribution of mitochondria and even missegregation of chromosomes causing aneuploidy. These organelle segregation abnormalities in turn lead to mitochondrial dysfunction and delay in cell growth Citation[5]. The genome instability is well illustrated by the disorganized nuclear structure and micronuclei in cells that experience persistent mitochondrial hyperfusion (). Thus, the alterations in mitochondrial morphology may initiate a mitochondria-to-nucleus retrograde signaling in order to activate cell cycle checkpoint at G2/M, and thus ensure proper mitochondrial inheritance during every cell division cycle. These novel observations established functional links between mitochondrial morphology and cell cycle progression and have lead to the investigation as to whether cancer cells have dysregulated mitochondrial dynamics that contribute to cancer progression.
Figure 1. Mitochondrial hyperfusion induced by depletion of Drp1 causes genome instability. MDA-MB-231 breast cancer cells stably expressing mitochondria-targeted GFP were transfected with control or Drp1 siRNA for 4 days. DNA was visualized by DAPI staining. Control cells contained the relatively short and dispersed mitochondria. In contrast, Drp1 knockdown cells showed hyperfused mitochondria concomitant with the disorganized nuclear structure and micronuclei, a marker for genome instability.
2.2 Drp1-mediated mitochondrial fission in cancers
Cumulative evidence is beginning to reveal the links between cancer and dysregulated mitochondrial dynamics. It has been found that non-small-cell lung carcinoma A549 cells have markedly higher levels of Drp1 and lower levels of fusion protein Mfn-2 than normal airway cells Citation[12]. Such changes in the mitochondrial dynamics proteins were related with enhanced mitochondrial fission and reduced fusion in these tumor cells. In human tissues, it was found that lung adenocarcinoma sections had an increase in both the total expression and the phosphorylated form of Drp1, and reduced expression of Mfn-2 when compared to adjacent non-neoplastic lung tissue. Importantly, reversing mitochondrial fission in these tumor cells was able to suppress cell growth both in vitro and in vivo Citation[12]. Such dysregulated mitochondrial fission has also been observed in human breast carcinomas Citation[13]. Interestingly, Drp1-mediated mitochondrial fission is also required for cancer cell migration and invasion Citation[13]. Mitochondrial fission is a critical process for distribution of mitochondria into lamellipodial regions at the leading edge of breast cancer cells in response to chemoattractant-induced migration. The localization of mitochondria into the leading edge has been posited to provide necessary energy at the defined subcellular region. Further, it has also been reported that Drp1-mediated mitochondrial fission is enhanced by the overexpression of an oncogenic protein survivin, contributing to the development of glycolytic phenotype of neuroblastoma Citation[14]. Enhanced glycolysis is one of the hallmarks of many different cancer types, which promotes tumor cell progression and confers drug resistance.
Mitochondrial hyperfusion triggers S phase initiation, and this alone is sufficient to drive G0 quiescent cells into S phase of the cell cycle, suggesting mitochondrial morphology may be under direct regulation of growth factor and convey the growth signals to the cell cycle machinery. Both Aurora A and RalA, which regulates mitotic phosphorylation of Drp1, act downstream of oncogenic Ras, contributing to tumorigenesis through the induction of genome instability Citation[15,16]. Therefore enhanced mitochondrial fission may function as an important point of convergence in mediating oncogenic signaling and promoting cancer cell growth and metastasis. Since most of the Drp1 is cytosolic and overexpression of wild-type Drp1 alone does not seem to enhance mitochondrial fission Citation[17], enhanced Drp1 activation and recruitment may play more important roles for the increased mitochondrial fission observed in those cancer cells. The non-mitochondrial roles for Drp1 and the consequences of Drp1 upregulation due to non-mitochondrial function remain to be revealed.
2.3 Inhibiting Drp1 for cancer therapy
Despite the protective role of mitochondrial fusion against cytotoxic stimuli Citation[18], Drp1 knockout immortalized mouse embryonic fibroblasts (MEFs) maintain the ability to undergo efficient apoptosis albeit with a slight delay in the release of apoptotic factors such as cytochrome c from mitochondria Citation[19]. Intriguingly, persistent mitochondrial hyperfusion due to Drp1 depletion also induces cytochrome c release from mitochondria, leading to apoptosis Citation[20]. Reduced cancer cell growth and/or enhanced spontaneous apoptosis induced by inhibiting Drp1 have been observed both in vitro and in vivo in several cancer types, including colon, breast, lung and cervical cancers Citation[10,12,20,21]. The mechanism underlying the cellular dysfunction and cell death induced by mitochondrial hyperfusion seems to be the increased replication stress and mitotic defects that severely affect the genome integrity of cancer cells. Persistent mitochondrial fusion also causes mitochondrial dysfunction including accumulation of mtDNA mutation and generation of reactive oxygen species (ROS) Citation[21,22]. This combined effect of genome instability and mitochondrial dysfunction may eventually result in the activation of mitochondrial apoptotic pathway Citation[20]. This unique mechanism could potentially circumvent the resistance of cancer cells to conventional chemotherapy.
The first selective inhibitor for Drp1, mitochondrial division inhibitor-1 (mdivi-1), was identified from a chemical library screen using yeast-based assays Citation[23]. Mdivi-1 is a quinazolinone derivative, attenuating Drp1 self-assembly, thereby causing the inhibition of mitochondrial division. Due to its potential in preventing mitochondrial fragmentation-related apoptosis, mdivi-1 has shown protective efficacy in a number of disease models, including acute kidney injury, heart ischemia/reperfusion injury and Parkinson's disease Citation[18]. Thus mdivi-1 represents a potential class of therapeutics for stroke, myocardial infarction and neurodegenerative diseases Citation[23]. Importantly, in terms of cancer mdivi-1 also induces gross genome instability Citation[10] and was shown to have in vivo efficacy against lung tumor Citation[12]. In addition, there was no significant toxicity in rodent disease models observed after mdivi-1 therapy, although in-depth toxicology, pharmacodynamics and pharmacokinetics of mdivi-1 have not been reported. Recently, the possible off-target effects of mdivi-1 have been reported, such as Drp1-independent role of mdivi-1 in preventing mitochondrial outer membrane permeabilization (MOMP) Citation[24], and the effect of mdivi-1 on ion currents and cell membrane potential Citation[25]. While mdivi-1 may still serve as an important research tool, due to its undesired effects and relatively high IC50 in preventing mitochondrial fission in mammalian cells (IC50 ≈ 50 μM) Citation[23], the development of more potent and specific inhibitors is guaranteed. Mdivi-1 could possibly be employed as a lead compound for further optimization. Moreover, structural insights into the action of Drp1 will also undoubtedly facilitate the rational design and development of new generations of inhibitors. Excitingly, a novel Drp1 peptide inhibitor P110, which was designed to inhibit the interaction between Drp1 and Fis1, has been reported recently. P110 prevents mitochondrial fragmentation by selectively inhibiting the GTPase activity of Drp1 and blocks Drp1/Fis1 interaction, showing neuroprotectivity in a cell culture model of Parkinson's disease Citation[26]. It would also be interesting to test whether P110 has antitumor potential.
3. Expert opinion
As enhanced mitochondrial fission and impaired fusion appear to contribute fundamentally to the causes of certain aspects of cancer, Drp1-mediated mitochondrial fission thus may well represent a promising novel therapeutic target for cancers with excessive mitochondrial fission. Drp1 inhibition, either through genetic ablation or by pharmacological inhibitor mdivi-1 has shown initial proof of principle in cancer treatment. Nevertheless, many questions remain to be addressed in order to provide more comprehensive molecular basis for the development of effective Drp1-related targeted therapy. What is the frequency of Drp1 dysregulation across different types of human cancers, what is the molecular mechanism that leads to Drp1 dysregulation in cancers, do alterations in mitochondrial morphology directly contribute to tumorigenesis, and what are the molecular events that transduce signals derived from altered mitochondrial morphology? A better elucidation of the functional significance of mitochondrial dynamics in cancer physiology together with a clearer understanding of the molecular machineries that regulate mitochondrial dynamics will undoubtedly present a promising avenue for future exploration of mitochondrial dynamics-based therapeutic options.
Declaration of interest
This work was funded by Pennsylvania Department of Health, CURE, UPCI foundation and NIH P30 CA047904. The authors state no conflict of interest.
Bibliography
- Green DR, Van Houten B. SnapShot: mitochondrial quality control. Cell 2011;147(4):950; 950 e1
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11(12):872-84
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 1993;104(Pt 3):917-27
- Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27(2):433-46
- Kashatus DF, Lim KH, Brady DC, et al. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 2011;13(9):1108-15
- Mears JA, Lackner LL, Fang S, et al. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 2011;18(1):20-6
- Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 2013;1833(1):150-61
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646-74
- Mitra K, Wunder C, Roysam B, et al. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 2009;106(29):11960-5
- Qian W, Choi S, Gibson GA, et al. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci 2012;125(Pt 23):5745-57
- Taguchi N, Ishihara N, Jofuku A, et al. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 2007;282(15):11521-9
- Rehman J, Zhang HJ, Toth PT, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 2012;26(5):2175-86
- Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2012; doi: 10.1038/onc.2012.494
- Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2012; doi: 10.1038/onc.2012.500
- Yang G, Mercado-Uribe I, Multani AS, et al. RAS promotes tumorigenesis through genomic instability induced by imbalanced expression of Aurora-A and BRCA2 in midbody during cytokinesis. Int J Cancer 2013;133(2):275-85
- Lim KH, Mercado-Uribe I, Multani AS, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 2005;7(6):533-45
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 2001;12(8):2245-56
- Lackner LL, Nunnari J. Small molecule inhibitors of mitochondrial division: tools that translate basic biological research into medicine. Chem Biol 2010;17(6):78-83
- Ishihara N, Nomura M, Jofuku A, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 2009;11(8):958-66
- Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Biophys Res Commun 2012;421(1):81-5
- Parone PA, Da Cruz S, Tondera D, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 2008;3(9):e3257
- Malena A, Loro E, Di Re M, et al. Inhibitiony of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum Mol Genet 2009;18(18):3407-16
- Cassidy-Stone A, Chipuk JE, Ingerman E, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008;14(2):193-204
- Kushnareva Y, Andreyev AY, Kuwana T, Newmeyer DD. Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol 2012;10(9):e1001394
- So EC, Hsing CH, Liang CH, Wu SN. The actions of mdivi-1, an inhibitor of mitochondrial fission, on rapidly activating delayed-rectifier K(+) current and membrane potential in HL-1 murine atrial cardiomyocytes. Eur J Pharmacol 2012;683(1-3):1-9
- Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 2013;126(Pt 3):789-802