Abstract
Attrition is a major issue in anticancer drug development with up to 95% of drugs tested in Phase I trials not reaching a marketing authorisation making the drug development process enormously costly and inefficient. It is essential that this problem is addressed throughout the whole drug development process to improve efficiency which will ultimately result in increased patient benefit with more profitable drugs. The approach to reduce cancer drug attrition rates must be based on three pillars. The first of these is that there is a need for new pre-clinical models which can act as better predictors of success in clinical trials. Furthermore, clinical trials driven by tumour biology with the incorporation of predictive and pharmacodynamic biomarkers would be beneficial in drug development. Finally, there is a need for increased collaboration to combine the unique strengths between industry, academia and regulators to ensure that the needs of all stakeholders are met.
1. Introduction
Over the past decade, a large number of novel anticancer drugs have been developed and many are now implemented into routine clinical practice Citation[1]. Some of these drugs have made improvements in overall survival for all patients with a given condition, whereas others have only shown benefit in smaller groups of patients with known molecular aberrations.
However, the development of new anticancer drugs remains an expensive and inefficient process. In anticancer drug development, attrition rate is the major factor that reflects the level of loss of new candidate drugs during the process from pre-clinical to clinical and through their clinical development. Less than 5% of drugs that reach Phase I gain a marketing authorisation (MA) Citation[2]. Even more, it has been reported that only 1 in 10,000 pre-clinical compounds ever reach the market Citation[3].
It is envisioned that a more scientific and biology-driven drug development practice would lead to more efficiency, but to date this has only been successful for relatively small populations with known molecular aberrations by using predictive biomarkers in the development of such potent inhibitors. Giving drugs that match key genomic aberrations in the patients promise to produce a much greater benefit in smaller patient populations Citation[4]. This has been defined as the ‘inverted pyramid' paradigm, where for drugs developed classically a large population is required to achieve a small benefit (the standard pyramid). Using agents targeted against specific molecular aberrations, a small population is targeted but the benefit obtained is large (the inverted pyramid).
Numerous solutions have been proposed to tackle the issue of attrition in anticancer drug development by many authors Citation[5-11], which has even been defined by some as ‘The Valley of Death in anticancer drug development' Citation[12]. Reported factors are innumerable and include scientific and financial or non-scientific reasons. The latter might include lack of resources, wrong incentives, aggressive pricing strategies or adverse regulatory environment, while the scientific reasons include considering tumour microenvironment, cross-talk and negative feedback loops, development of resistance, exposure time, drug delivery or the choice of pre-clinical models. While many of these issues have been reviewed elsewhere extensively, this editorial assesses and comments on the most relevant and promising scientific strategies to improve attrition rates in the development of anticancer drugs in the authors' opinion.
2. Better pre-clinical drug development
Ideally, robust pre-clinical studies should identify the best drugs with the highest likelihood of efficacy and the least possible toxicity before starting clinical trials. There are major areas where improvements in pre-clinical testing would lead to more efficient drug development: first, a better identification and qualification of the targets that are of relevance to each tumour type is essential. Despite the difficulties related to the biological heterogeneity of cancers, some efforts have been made to achieve a consensus on the required data to pursue a set target both in adults and paediatric patients Citation[13].
Second, better pre-clinical models more representative of human tumour biology need to be pursued. While pre-clinical studies in cell lines and xenografts are a useful tool to screen compounds and might provide useful early signs of interest, they have not shown good correlation with efficacy in Phase II trials or survival advantage in Phase III trials Citation[14].
Although their final ability to predict success is still unproven, new models will better recapitulate tumour biology and microenvironment than multiple passaged cell lines or cell line-derived xenografts Citation[15].
Robust examples of genetically engineered murine models (GEMM) are: KRAS-driven models of pancreatic cancer; MYC/MYCN-driven models of lymphomas/neuroblastoma or sonic hedgehog-driven models of medulloblastoma Citation[16-18]. But recently also patient-derived cell lines and patient-derived xenografts are paving the way for a true personalised medicine approach Citation[19].
While pre-clinical testing packages have significantly improved, evaluation of potential mechanisms of resistance is generally lacking. The development of feedback loops, new mutations, drug resistance or blockade of drug uptake should be incorporated in the evaluation of new therapies Citation[20-24]. For example, a resistant smoothened mutation developed on a patient with medulloblastoma shortly after very successful treatment with a sonic hedgehog inhibitor Citation[21,22]. It has been shown how the addition of MEK inhibitors overcomes the resistance to single-agent BRAF inhibition for BRAF-mutated melanomas Citation[23-25].
In the clinic, anticancer agents are mostly given in combination schedules. The issue of combinations should be addressed in pre-clinical studies upfront in order to rationally design and guide clinical trials Citation[26,27]. Finally, better pre-clinical models to assess toxicity or test different formulations are also needed. Too many drugs that go into Phase I/II clinical trials still have excessive toxicity or formulation problems that preclude further clinical development of potent inhibitors.
It is envisioned that a better selection of the drugs that reach Phase I trials with more stringent criteria to qualify targets, show antitumour activity or tolerability and develop formulations would reduce the number of failures during early clinical trials. Careful studies need to be established to relate efficacy in pre-clinical models with efficacy in Phase II trials.
3. Incorporation of biomarkers
Predictive biomarkers that select patients who are most likely to benefit from a targeted therapy based on the patient's molecular characteristics have already been shown to reduce attrition. Only 5% of drugs without patient selection reach registration as opposed to 47% of selected kinase inhibitors targeting specific patient genomic aberrations Citation[4,28]. For these agents, response rates in Phase I/II clinical trials were above 50%, while 10% has been reported as the average response rate for Phase I trials without patient selection Citation[4]. Nevertheless, it is important to note that premature decisions on biomarkers that have not been adequately validated and qualified might mislead the development of molecularly targeted agents Citation[29].
More importantly, not all drugs and targets may have a simple ideal predictive biomarker. There will still be many drugs that make modest contributions to improving outcome that are valid when agents are combined into multimodal regimens for which there will not be a validated predictive biomarker Citation[30]. For some conditions or targets mRNA signatures might provide better prediction than other genomic aberrations Citation[31]. Where no predictive biomarker is still identified, collection of biological material and pilot analyses of tertiary biomarker end points is highly recommendable in order to identify novel biomarkers of response.
The issue of tissue heterogeneity remains unsolved. Differences between primary tumour and metastases Citation[32] or circulating tumour cells Citation[33,34] have been described and recently deep sequencing analyses have shown significant differences between regions of the same tumour Citation[35] including the detection of good and poor prognosis signatures within the same tumour mass.
But as a result of the interest in predictive biomarkers, indications with known molecular aberrations might become overcrowded and competitive whereas fewer drugs are developed for most common heterogeneous cancers.
Similar to the pre-clinical setting, it would appear essential to avoid taking forward drugs that do not achieve the necessary target inhibition or downstream pharmacodynamic (PD) effects. The use of PD biomarkers provides the proof of principle of target modulation and should be a requirement prior to embarking on larger Phase II trials, following the ‘Pharmacologic Audit Trail', a concept proposed by Workman and collaborators Citation[36]. But, it still remains crucial to qualify biomarkers appropriately Citation[29].
Phase 0 trials have been proposed by some authors as a bridge between pre-clinical and clinical drug development to accelerate and improve the efficiency of this process. The primary objective of these trials is to obtain pilot proof-of-mechanism/pharmacokinetic data at non-therapeutic drug exposures (e.g., by giving a single dose of a new investigational agent) in a small subset of patients, therefore identifying those drugs that do not achieve expected biological effects that would then be de-prioritised without further trials. A significant number of such clinical trials have been conducted to date, with some successful examples, such as the PARP (poly-ADP ribose polymerase) inhibitor ABT-888 Citation[37]. However, ethical concerns about the complete lack of benefit and the repeated tumour biopsies involved are not resolved. Proving target inhibition with a single dose of a single drug might not be a good model to mimic the clinical setting, when sustained inhibition is required to maintain benefit from the agents or they have to be given in combination schedules to prevent the development of resistance Citation[12,38,39].
Most PD biomarker studies require repeated tumour biopsies thus increasing the burden to the patients. It is therefore crucial to make the most rationale and efficient use of tumour material. A recent publication has shown how less than half of early clinical trials collecting PD analyses include that data in the final manuscript Citation[40,41]. In many cases, surrogate biomarkers can be used to avoid sampling primary tumours Citation[42].
It is essential that the selection of agents to take forward into early clinical trials is based on the availability of predictive and PD biomarker data which are developed pre-clinically and implemented into early clinical trials.
4. Efficient transition from early to late phase clinical trials
The decision to progress a drug from early Phase I/II/proof-of-concept clinical trials into randomised Phase III trials for the purpose of registration is extremely difficult, complex and costly. Companies have to consider not only scientific rationale but also the financial investment required, the available market and/or the anticipated returns more commonly referred to as the benefit to risk ratio Citation[3]. This commits the company to continue the development of the agent(s) over the next quinquennium with the clinical trials enrolling hundreds if not thousands of patients. Hypothesis-driven biomarker-rich studies will importantly smoothen the progress of these decisions.
A number of innovative designs now facilitate the maximising of the information obtained from early clinical trials. Increasingly, Phase I trials include expansion cohorts at the recommended Phase II dose (RP2D) that target the population of interest in the search for early signals of activity. Well-designed Phase I trials incorporating tumour biology, predictive and PD biomarkers surely promise to detect ineffective or toxic drugs that should not progress further.
Randomised Phase II trials can provide more robust activity and efficacy data before proceeding to larger Phase III trials compared with single arm Phase II trials with historic controls Citation[43]. Novel adaptive designs or Bayesian statistics allow randomised comparisons with smaller numbers of patients Citation[44,45]. Some designs will allow testing several drugs, doses or combinations more efficiently: first, in a pick-a-winner design drugs are tested in several stages: in the first of which, patients are randomised between a number of different novel treatments and a control arm. Only those drugs that show a pre-specified degree of benefit at the end of the first stage will proceed to the next stage(s). This allows testing a number of novel strategies in a randomised fashion without the limitation of conducting several larger-scale Phase III trials Citation[46]. Second, drop-the-loser designs were developed to monitor multiple doses of an experimental treatment compared with a control arm before proceeding to a large randomised Phase III trial Citation[47,48]. depicts the differences between classic Phase II trial designs and novel adaptive designs.
Figure 1. Differences between classic and adaptive randomised clinical trial designs.
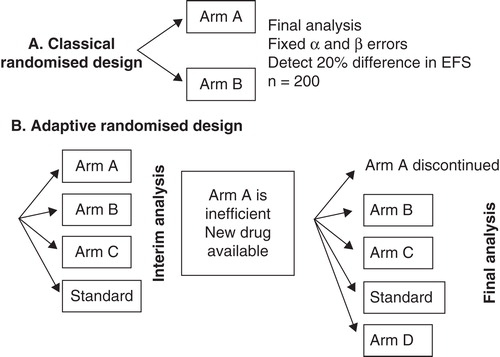
In summary, novel designs will provide an efficient way of identifying ‘winners' or dropping ‘losers' in trials with a small number of patients. Where possible, all decisions should be evidence-based using information gathered from randomised trials, even in the Phase II setting with reduced numbers and more flexible power calculations.
5. Collaboration: industry, academia, regulators
Fundamentally all stakeholders involved in the development of anticancer drugs: industry, academia, regulators, patient advocates and policymakers must work together. The authors strongly believe that close collaboration will improve the efficiency of the drug development process and reduce attrition.
Academic partnerships, designation of orphan drugs, elaboration of Paediatric Investigational Plans (PIPs) are strategies that increase the revenues or decrease the costs of developing agents, therefore ensuring that drugs are profitable Citation[49].
Regulatory bodies are now increasingly offering collaboration at multiple levels, and they provide scientific and regulatory advice that ensures that good and safe drugs are ultimately delivered to patients Citation[50].
Additionally, academic partnerships can help provide access to larger cohorts of patients and thus bring new drugs forward into frontline treatment that would benefit the wider population of patients with cancer.
6. Expert opinion
Attrition is a significant and costly problem for anticancer drug development and must be addressed at all levels and stages of the drug development process.
Pre-clinically, better models are needed that will be more predictive of success in clinical trials and these need to be scientifically evaluated. GEMMs and patient-derived xenografts better recapitulate the patient's tumour biology. More efforts in drug discovery units will lead to less toxic, better formulated drugs and predictive biomarkers selecting patients with known molecular aberrations for specific kinase inhibitors have already been shown to reduce attrition, but are not applicable to all cancers.
PD biomarkers help in the go–no go decisions at the end of Phase I and ensure that new drugs modulate the target(s) as expected.
The decisions to take forward drugs from Phase I/II (proof-of-concept) to large Phase III randomised trials have to be taken carefully. Innovative trial designs such as RP2D expansion cohorts in biomarker-driven Phase I trials and randomised Phase II trials provide better information. These decisions must be based on robust scientific data and advice should be sought from academia and regulators alike.
Declaration of interest
All authors acknowledge support from the NHS for providing funding to the NIHR Biomedical Research Centre and the Experimental Cancer Medicine Centre Network (ECMC). L Moreno is funded by the Oak Foundation and ADJ Pearson is funded by Cancer Research UK (Grant C1178/A10294). The authors want to thank R Barfoot for the assistance in the preparation of the manuscript. L Moreno has participated in advisory boards for Roche Genentech and AstraZeneca. ADJ Pearson has participated in advisory boards for Roche Genentech, Celgene and AstraZeneca.
Bibliography
- Trotta F, Leufkens HG, Schellens JH. Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: when differences have an impact on clinical practice. J Clin Oncol 2011;29(16):2266-72
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004;3(8):711-15
- Tonkens R. An overview of the drug development process. Physician Exec 2005;31(3):48-52
- Garrido-Laguna I, Hidalgo M, Kurzrock R. The inverted pyramid of biomarker-driven trials. Nat Rev Clin Oncol 2011;8(9):562-6
- Scannell JW, Blanckley A, Boldon H, Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov 2012;11(3):191-200
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov 2011;10(7):507-19
- Reed JC. NCATS could mitigate pharma valley of death. Genet Eng Biotechnol News 2011;31(10):6
- Williams M. Productivity shortfalls in drug discovery: contributions from the preclinical sciences? J Pharmacol Exp Ther 2011;336(1):3-8
- Pammolli F, Magazzini L, Riccaboni M. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov 2011;10(6):428-38
- Paul SM, Mytelka DS, Dunwiddie CT. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 2010;9(3):203-14
- Hait WN. Anticancer drug development: the grand challenges. Nat Rev Drug Discov 2010;9(4):253-4
- Adams DJ. The Valley of death in anticancer drug development: a reassessment. Trends Pharmacol Sci 2012;33(4):173-80
- Goodwin R, Giaccone G, Calvert H. Targeted agents: how to select the winners in preclinical and early clinical studies? Eur J Cancer 2012;48(2):170-8
- Johnson JI, Decker S, Zaharevitz D. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer 2001;84(10):1424-31
- Moreno L, Chesler L, Hargrave D. Preclinical drug development for childhood cancer. Expert Opin Drug Discov 2011;6(1):49-64
- Lee Y, Kawagoe R, Sasai K. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007;26(44):6442-7
- Chesler L, Weiss WA. Genetically engineered murine models–contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Semin Cancer Biol 2011;21(4):245-55
- Hingorani SR, Petricoin EF, Maitra A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4(6):437-50
- Hidalgo M, Bruckheimer E, Rajeshkumar NV. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther 2011;10(8):1311-16
- Moore AS, Faisal A, de Castro DG. Selective FLT3 inhibition of FLT3-ITD(+) acute myeloid leukaemia resulting in secondary D835Y mutation: a model for emerging clinical resistance patterns. Leukemia; 2012;26(7):1462-70
- Yauch RL, Dijkgraaf GL, Alicke B. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009;326(5952):572-4
- Rudin CM, Hann CL, Laterra J. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361(12):1173-8
- Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer 2013. DOI: http://dx.doi.org/10.1016/j.ejca.2012.11.019
- Das Thakur M, Salangsang F, Landman AS. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013. DOI: 10.1038/nature11814
- Britschgi A, Andraos R, Brinkhaus H. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012;22(6):796-811
- Humphrey RW, Brockway-Lunardi LM, Bonk DT. Opportunities and challenges in the development of experimental drug combinations for cancer. J Natl Cancer Inst 2011;103(16):1222-6
- LoRusso PM, Canetta R, Wagner JA. Accelerating cancer therapy development: the importance of combination strategies and collaboration. Summary of an institute of medicine workshop. Clin Cancer Res 2012;18(22):6101-9
- Walker I, Newell H. Do molecularly targeted agents in oncology have reduced attrition rates? Nat Rev Drug Discov 2009;8(1):15-16
- Cummings J, Raynaud F, Jones L. Fit-for-purpose biomarker method validation for application in clinical trials of anticancer drugs. Br J Cancer 2010;103(9):1313-17
- Basu B, Olmos D, de Bono JS. Targeting IGF-1R: throwing out the baby with the bathwater? Br J Cancer 2011;104(1):1-3
- Valentijn LJ, Koster J, Haneveld F. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci USA 2012;109(47):19190-5
- Dupont Jensen J, Laenkholm AV, Knoop A. PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res 2011;17(4):667-77
- Gasch C, Bauernhofer T, Pichler M. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem 2013;59(1):252-60
- Strati A, Markou A, Parisi C. Gene expression profile of circulating tumor cells in breast cancer by RT-qPCR. BMC Cancer 2011;11:422
- Gerlinger M, Rowan AJ, Horswell S. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366(10):883-92
- Yap TA, Sandhu SK, Workman P, Envisioning the future of early anticancer drug development. Nat Rev Cancer 2010;10(7):514-23
- Kummar S, Kinders R, Gutierrez ME . Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol 2009;27(16):2705-11
- Kummar S, Kinders R, Rubinstein L. Compressing drug development timelines in oncology using phase ‘0’ trials. Nat Rev Cancer 2007;7(2):131-9
- Rodriguez-Pascual J, Sha P, Garcia-Garcia E. A preclinical and clinical study of mycophenolate mofetil in pancreatic cancer. Invest New Drugs 2012;31(1):14-9
- Freeman GA, Kimmelman J. Publication and reporting conduct for pharmacodynamic analyses of tumor tissue in early-phase oncology trials. Clin Cancer Res 2012;18(23):6478-84
- Iannone R. Improving publication rates of biomarker results from cancer trials. Clin Cancer Res 2012;18(23):6398-9
- Yap TA, Olmos D, Brunetto AT. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol 2011;29(10):1271-9
- Rubinstein L, Crowley J, Ivy P. Randomized phase II designs. Clin Cancer Res 2009;15(6):1883-90
- Seymour L, Ivy SP, Sargent D. The design of phase II clinical trials testing cancer therapeutics: consensus recommendations from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin Cancer Res 2010;16(6):1764-9
- EMA. E.M.A., EMA/EFPIA 2nd Workshop: adaptive Design in Confirmatory Trials (EMA/779520/2009). 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Minutes/2010/04/WC500089206.pdf [Last accessed 12 November 2012]
- Hills RK, Burnett AK. Applicability of a “Pick a Winner” trial design to acute myeloid leukemia. Blood 2011;118(9):2389-94
- Joshua Chen YH, Demets DL, Gordon Lan KK. Some drop-the-loser designs for monitoring multiple doses. Stat Med 2010;29(17):1793-807
- Mahajan R, Gupta K. Adaptive design clinical trials: methodology, challenges and prospect. Indian J Pharmacol 2010;42(4):201-7
- Saint-Raymond A, Herold R. Medicines for pediatric oncology: can we overcome the failure to deliver? Expert Rev Clin Pharmacol 2012;5(5):493-5
- EMA. E.M.A. European Medicines Agency guidance for companies requesting scientific advice and protocol assistance. EMA Scientific Advice 21 May 2010. EMEA-H-4260-01-Rev. 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004089.pdf [Accessed 26 November 2012]