Abstract
Introduction: Mucopolysaccharidosis II (MPS II) is an X-linked lysosomal storage disorder caused by a deficiency in the lysosomal enzyme iduronate-2-sulfatase (I2S), leading to an accumulation of glycosaminoglycans within lysosomes. Patients experience progressive, multisystemic disease, significant morbidity, and early mortality.
Areas covered: Idursulfase, an I2S enzyme produced by recombinant DNA technology in a human cell line, was approved in 2006 in the United States and 2007 in the European Union for use in MPS II patients. The authors examine the published pharmacokinetic, safety, and efficacy data from the Phase I/II, Phase II/III, Phase II/III extension, and post-marketing surveillance studies of idursulfase.
Expert opinion: Idursulfase is generally well tolerated and produces measurable clinical improvements in walking ability and statistically significant reductions in mean liver and spleen volumes and urinary glycosaminoglycan levels. The impact of anti-drug antibodies upon efficacy is unclear but is an active area of research. Treatment should be offered to MPS II patients with or without cognitive involvement, including females, as soon as possible after diagnosis. Patients with the severe (neuropathic) phenotype may receive certain somatic benefits from treatment, supporting a test of idursulfase treatment with clear expectations and discontinuation criteria discussed with the family before treatment initiation.
1. Introduction
Mucopolysaccharidosis II (MPS II, Hunter syndrome; OMIM 309900) is an X-linked lysosomal storage disease (LSD) caused by a deficiency in the enzyme iduronate-2-sulfatase (I2S), leading to accumulation of the glycosaminoglycans (GAGs) heparan sulfate and dermatan sulfate within lysosomes Citation[1,2]. The incidence is from 0.2 to 1.07 per 100,000 live births Citation[3-9]. MPS II is a progressive, multisystemic disease with significant morbidity and early mortality. Patients generally appear normal at birth, with clinical signs and symptoms appearing at 2 – 4 years of age Citation[10,11]. The clinical features include hearing loss, recurrent ear and respiratory infections, coarse facial features, airway obstruction and restriction, communicating hydrocephalus, spinal cord compression, carpal tunnel syndrome, cardiac valve disease, hepatosplenomegaly, skeletal abnormalities, growth restriction, and joint stiffness Citation[1,12].
Patients with MPS II are often described as having either a severe or an attenuated phenotype, although patients may present at any point upon a spectrum of severity Citation[12,13]. About two-thirds of patients exhibit a severe (neuronopathic) phenotype, characterized by a developmental plateau in early childhood, followed by a relentless cognitive decline Citation[14]. Death typically occurs in the second decade Citation[15]. Patients with the attenuated phenotype remain cognitively intact but can display all of the somatic signs and symptoms of the disease, including neurological complications such as communicating hydrocephalus, spinal cord compression, and hearing loss Citation[16]. Such patients may survive into adulthood, although premature mortality occurs Citation[15].
In July 2006 in the United States and in January 2007 in Europe, enzyme replacement therapy (ERT) with idursulfase(), a recombinant human I2S (Elaprase®, Shire Human Genetic Therapies, Inc., Lexington, Massachusetts, USA), was approved. It is now available in 50 countries. Current treatment guidelines suggest the initiation of weekly idursulfase treatment as soon after diagnosis as possible for most patients Citation[17-21], although clinical trial data for the use in patients under the age of 5 years are not yet available. Safety and effectiveness data about the use of idursulfase in children who initiated therapy before 6 years of age have been described in data from a patient registry that collects observational data (see section 5.5, Postmarketing Surveillance).
Box 1. Drug summary.
2. Overview of the market
Historical treatment for MPS II was mainly palliative. Based on successes in MPS I-Hurler (OMIM 607014), hematopoietic stem cell transplantation (HSCT) has been performed in small numbers of patients with MPS II. Improvements in some somatic features have been reported Citation[22-25], but the neurocognitive decline is not ameliorated Citation[22,23,26,27]. Thus, HSCT is not recommended as a treatment option for MPS II Citation[28]. The most direct competitor to idursulfase is GC1111 (Hunterase®, Green Cross Corporation, Yongin, Korea), which has been approved by the Korea Food and Drug Administration based on unpublished Phase I/II clinical trial data (NCT01301898) Citation[29]. A second competitor is the soy isoflavone genistein, a natural compound that can inhibit the synthesis of GAGs Citation[30]. Genistein use has been studied in patients with the related disorders MPS IIIA, B, C, and D (OMIM 252900, OMIM 252920, OMIM 252930, and OMIM 252940). In an open-label study enrolling 19 patients, no clinical benefit was found after 1 year of treatment Citation[31]. A larger, randomized, placebo-controlled trial enrolling 30 patients with MPS III found that genistein marginally reduced urinary GAG (uGAG) and plasma heparan sulfate levels, but no clinical efficacy was detected Citation[32]. For MPS II, an open-label, 26-week study in seven previously untreated attenuated patients found that genistein treatment produced statistically significant improvements in mean active and passive shoulder flexion and abduction Citation[33]. Elbow, wrist, hip, and knee joint range-of-motion (JROM) were not improved by treatment.
3. Introduction to idursulfase
Idursulfase, a 525-amino acid glycoprotein with a molecular weight of approximately 76 kD, is produced by recombinant DNA technology in a human cell line. Idursulfase acts by cleaving the terminal 2-O-sulfate moieties from dermatan sulfate and heparan sulfate. The protein contains two disulfide bonds and eight N-linked glycosylation sites, each of which contains two bis-mannose-6-phosphate terminated glycans. These allow specific binding of the enzyme to the mannose-6-phosphate receptors on the cell surface, leading to cellular internalization and targeting to intracellular lysosomes. The enzyme activity of idursulfase is dependent on the post-translational modification of cysteine-59 to formylglycine; thus, idursulfase has a specific activity ranging from 46 to 74 U/mg of protein Citation[34,35].
Idursulfase is administered by continuous weekly intravenous infusion, using a 0.2 μm filter, at a recommended dose of 0.5 mg/kg of body weight weekly diluted in 100 mL of 0.9% sodium chloride. The total volume may be administered over 3 h, although a longer infusion time can be used if infusion-related reactions (IRRs) occur, as long as the infusion time does not exceed 8 h. A ramping protocol is suggested with an infusion rate of 8 mL/h for the first 15 min. If the infusion is well tolerated, the rate may be increased by 8 mL/h increments at 15-minute intervals. The infusion rate should never exceed 100 mL/h Citation[35].
4. Pharmacokinetics
The prescribing information states that the pharmacokinetic characteristics of idursulfase have been evaluated in several studies with MPS II patients Citation[35]. The area under the concentration-time curve increased in greater than a dose-proportional manner as the dose increased from 0.15 mg/kg to 1.5 mg/kg following a single 1-hour infusion. In a separate study, 10 patients aged 7.7 to 27 years received the recommended dose of 0.5 mg/kg as a 3-hour infusion weekly; pharmacokinetic parameters were determined at Week 1 and Week 27 (). The half-lives at Week 1 and Week 27 were 44 and 48 min, with an apparent volume of distribution of 21% and 25% of body weight, respectively, which were not significantly different.
Table 1. Pharmacokinetic parameters in 10 human patients receiving idursulfase (0.5 mg/kg weekly as a 3-hour infusion).
5. Clinical efficacy of idursulfase
5.1 Phase I/II study
This 24-week, double-blind, placebo-controlled trial enrolled 12 attenuated patients aged 6 to 20 years who were randomized to three dosing groups (0.15, 0.5, and 1.5 mg/kg every other week) of four patients each; one patient within each group received placebo Citation[36]. All patients continued in an open-label extension trial for at least 6 months. The primary endpoint was change in uGAG level from baseline. Secondary endpoints included changes in liver and spleen size, 6MWT distance, pulmonary function, JROM, heart size and function, and sleep study.
After 24 weeks in the double-blind phase plus 24 weeks in the open-label phase, reductions in uGAG levels of 47%, 43%, and 58% were seen in the 0.15, 0.5, and 1.5 mg/kg groups, respectively. Liver and spleen volumes were significantly decreased after 24 and 48 weeks in patients with organomegaly at baseline. No statistically significant changes in the mean 6MWT distance were seen in any group during the double-blind phase, but after 48 weeks of treatment, the mean distance among all treated patients was significantly improved from 398±117 to 445±124 m (p = 0.013, t-test). No statistically significant changes in pulmonary function testing, joint mobility, sleep study parameters, or cardiac parameters were demonstrated.
5.2 Phase II/III study
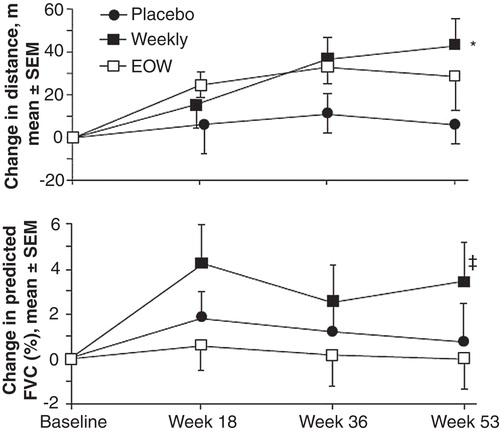
The Phase II/III study (NCT00069641) was a randomized, double-blind, placebo-controlled, multicenter, international trial enrolling 96 attenuated patients aged 5.0 – 30.9 years. Patients received either infusions of idursulfase (0.5 mg/kg every week or every other week [EOW]), or placebo for 53 weeks. Patients were stratified by age, 6MWT distance, and percent predicted forced vital capacity (%FVC) at baseline. The primary efficacy endpoint was a two-component score generated by summing the rank scores for change from baseline in the 6MWT distance and %FVC. Secondary endpoints included changes in 6MWT distance, %FVC, absolute FVC, liver and spleen volumes, uGAG excretion, and passive JROM.
After 53 weeks, the primary efficacy endpoint score was significantly higher in both idursulfase groups than in the placebo group (p = 0.0049 for weekly and p = 0.0416 for EOW ). Liver volume decreased from baseline in both treatment groups by about 25% (p < 0.0001 for both vs. placebo). Spleen volumes were decreased by about 25% in the weekly dosing group and 20% in the EOW group (p < 0.0001 for both vs. placebo). The uGAG level decreased by 44.7 ± 4.0% in the EOW group and 52.5 ± 5.3% in the weekly group (p < 0.0001 for both vs. placebo; p = 0.0394 for weekly vs. EOW). Elbow joint mobility was significantly improved in the weekly group as compared with placebo (p = 0.0476), but no other JROM differences between groups were seen.
Figure 1. Treatment efficacy in the Phase II/III clinical trial for change in 6MWT distance and change in %FVC from baseline (components of the primary composite endpoint score).
5.3 Phase II/III extension study
All 94 patients who completed the Phase II/III study enrolled in the extension study (NCT00630747) and were treated with intravenous idursulfase 0.5 mg/kg weekly for an additional 24 months Citation[37]. The primary efficacy endpoints of the extension study were changes from baseline in 6MWT distance and %FVC. Secondary endpoints included changes in liver and spleen volume, uGAG level, cardiac mass, JROM, linear growth velocity, and functional status (Child Health Assessment Questionnaire Disability Index Score [CHAQ DIS]).
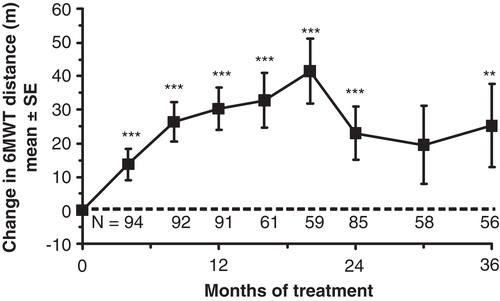
Statistically significant improvement in %FVC was observed at a single time point (Month 16) only, although absolute FVC showed sustained improvement throughout the study (). Improvements in 6MWT distance were statistically significant at all but one time point (). Reductions in liver and spleen volumes seen in the double-blind study were maintained throughout the extension. The mean uGAG level decreased 77% from baseline (p < 0.001) by Month 36. Statistically significant and clinically important improvements in JROM were seen only for the shoulder, but no other joints. The mean linear growth velocity in 15 patients who had not yet reached Tanner Stage 2 at baseline was 4.33 cm/year. The standard parent-assessed CHAQ DIS score showed statistically significant improvements from baseline at months 8, 16, 20, 24, and 30.
Figure 2. A. The effect of idursulfase on %FVC. Baseline %FVC = 56.2% ± 1.5% (mean ± SE). **p < 0.01. B. The effect of idursulfase on absolute FVC (p < 0.001 compared with baseline for each value of the total population). No statistical testing was performed on the subgroups. Baseline FVC was 1.18 ± 0.055 L (mean ± SE) for the total population.
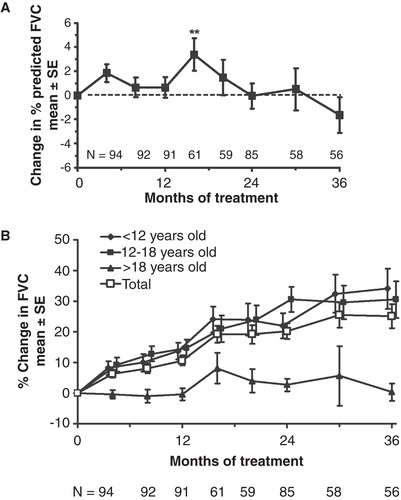
Figure 3. Effect of idursulfase on 6MWT distance in the Phase II/III extension study. Baseline 6MWT distance = 400 ± 10 m (mean ± SE). The dotted horizontal line represents the baseline.
5.4 Phase II/III and extension secondary analysis: effects of idursulfase on growth
A secondary analysis of height measurements from 18 attenuated patients who began treatment before the end of puberty and who were treated for at least 3 years in the Phase II/III and extension studies has been reported Citation[38]. Height measurements were taken the year before starting ERT, at baseline, and at years 1, 2, and 3. The patients were divided into two groups: those who had begun ERT at less than 10 years of age (n = 9) and those who had begun ERT at 10 years of age or older (n = 9). The mean increase in height at Year 3 in the younger group was 14.6 ± 5.5 cm, maintaining 8/9 patients' height within normal range (). There was an increase in height of about 3 cm (Z score = −0.5) the year before ERT, and 5.3 cm (Z score = 0.02), 4.5 cm (Z score = −0.07), and 5.7 cm (Z score = 0.08) for the first, second, and third years on ERT, respectively. In the older group, the mean increase in height over 3 years was 8.1 ± 1.6 cm; their growth curves remained below the 3rd percentile. There was an increase in height of 1.5 cm (Z score = −0.8) in the year before ERT, and 3.9 cm (Z score = −0.17), 3.6 cm (Z score = 0.23), and 1.3 cm (Z score = −0.06) for the first, second, and third years of ERT, respectively. The conclusion was that idursulfase improves growth velocity, but the most benefit is obtained when initiated before the age of 10 years.
Figure 4. Growth charts for MPS II patients A. under 10 years of age at the start of enzyme replacement therapy (ERT) with idursulfase and B. 10 years of age or older at the start of ERT. The dotted lines illustrate the growth before ERT and the continuous lines the growth on ERT. The shaded area represents the 3rd to the 97th percentiles of height in boys based on United States Centers for Disease Control and Prevention growth charts. With kind permission from Springer Science+Business Media: Citation[38].
![Figure 4. Growth charts for MPS II patients A. under 10 years of age at the start of enzyme replacement therapy (ERT) with idursulfase and B. 10 years of age or older at the start of ERT. The dotted lines illustrate the growth before ERT and the continuous lines the growth on ERT. The shaded area represents the 3rd to the 97th percentiles of height in boys based on United States Centers for Disease Control and Prevention growth charts. With kind permission from Springer Science+Business Media: Citation[38].](/cms/asset/e3c53e8a-58d2-4e3f-9944-9e5166a2688f/ieod_a_738182_f0004_b.jpg)
5.5 Post-marketing surveillance
As part of a commitment to post-marketing surveillance, Shire HGT supports the Hunter Outcome Survey (HOS), a global disease registry that is overseen by national, regional, and global scientific advisory boards Citation[10]. HOS was established in 2005 with the objectives to collect real-world data on the natural history of MPS II and on the long-term safety and effectiveness of the use of ERT in patients with MPS II. All data are anonymous.
A recent large analysis described data from 124 patients enrolled in HOS who had begun ERT with idursulfase under the age of 6 years (mean, 3.6 ± 1.6 years) Citation[39]. In the subgroup of 34 patients with elevated baseline uGAG levels, mean levels decreased by 37% from baseline after at least 6 months of idursulfase (p < 0.0001). Mean liver size as estimated by palpation was also significantly decreased after at least 6 months of idursulfase. These were comparable results to those seen in a similar cohort of patients in HOS who had begun ERT over the age of 6 years.
6. Safety and tolerability of idursulfase
6.1 Adverse events
The most common AEs in the clinical trials and post-marketing surveillance studies were IRRs, which were managed by slowing the infusion and/or premedication with antihistamines or steroids Citation[34,36-39]. The incidence of IRRs in the published studies varied between 20% and 75% (). Serious or potentially life-threatening IRRs were uncommon. Other AEs included limb pain, visual disturbance, chest wall pain, anxiety, and dyspepsia Citation[35].
Table 2. Summary of safety data in the Phase I/II, Phase II/III, and Phase II/III extension studies, and the HOS under 6 year analysis.
The idursulfase prescribing information carries a black-box warning that life-threatening anaphylactic reactions have been observed in some patients during infusions; therefore, appropriate medical support should be readily available during administration. Biphasic anaphylactic reactions have also been observed, which may require prolonged observation. Patients with compromised respiratory function or acute respiratory disease may be at risk of serious acute exacerbation of their respiratory compromise due to IRRs, and these patients require additional monitoring Citation[35].
Compliance with idursulfase treatment was reported to be good in the clinical trials and the extension studies. All patients completed the Phase I/II trial and extension. In the Phase II/III trial, all patients received at least 80% of infusions and no patient missed more than four consecutive infusions. Ninety-four of 96 patients completed the double-blind phase (two patients died of non-drug-related causes), and 85/94 patients completed the 2-year extension (one patient died of non-drug-related causes, and eight patients relocated). In the extension phase, only two patients received less than 80% of doses, and only five patients missed four or more consecutive doses.
6.2 Antibodies to idursulfase
IgG antibodies to idursulfase were detected at one or more time points in about half of treated patients across the clinical trials and post-marketing surveillance study (). Antibody positivity decreased over time. In the Phase II/III study, the incidence was 31.7% at Week 53, but only 27.1% of patients were antibody positive at Month 36 in the extension Citation[34,37]. In the Phase I/II study, antibody positivity was reported to have no significant effect on response as assessed by changes in uGAG levels, liver and spleen volumes, 6MWT distance, or %FVC volume Citation[36]. In the Phase II/III study, the reduction in uGAG levels in antibody-positive patients was reported to be about two-thirds of that seen in antibody-negative patients, but no correlation between antibody status and clinical assessments was seen Citation[34]. No anti-idursulfase IgE antibodies were detected at any time in these studies.
The presence of neutralizing antibodies was reported only in the Phase II/III extension study Citation[37]. Of the 94 patients, 22 (23.4%) were positive for neutralizing antibodies at any point. At the end of the study, 19/85 (22.3%) of patients were positive, indicating that tolerization had not occurred. As opposed to non-neutralizing antibodies, neutralizing antibodies were reported to be associated with a more muted absolute FVC response to idursulfase, although 6MWT, liver and spleen volume, and uGAG levels were not affected by neutralizing antibody status.
7. Access to treatment
Policies about eligibility for idursulfase treatment vary from country to country; these differences typically center around treating patients with cognitive involvement Citation[19,40-42]. One reason for these differences is that there is no available cost-effectiveness study of idursulfase for MPS II, which costs approximately $300,000 – $500,000 per patient per year. Such studies are very difficult to conduct for rare diseases and usually are not able to demonstrate cost-effectiveness Citation[43]. Thus, reimbursement agencies have diverse views on medical equity, health-care rights, and the “rule of rescue” Citation[43,44]. It has been argued that MPS II is a very rare disease, making the overall cost to a government or reimbursement agency for idursulfase treatment small compared with their total health care expenditures Citation[19]. The manufacturer of idursulfase has a patient assistance program in the United States meant to provide access for certain eligible patients Citation[45].
8. Conclusion
Clinical trial data from the pivotal Phase II/III trial and extension indicate that treatment with idursulfase produces measurable clinical improvements in a two-component score generated by summing rank scores for change from baseline in the 6MWT distance and %FVC; in liver and spleen volumes; and in standard parent-assessed CHAQ DIS score at some time points. In the Phase II/III trial, statistically significant improvements in elbow joint mobility were seen, while in the extension trial, only significant improvements in shoulder joint mobility were seen. A secondary analysis of the Phase II/III height data for 18 patients concluded that idursulfase improves growth velocity in a subset of patients, with the most benefit obtained when initiated before the age of 10 years. A post-marketing surveillance analysis based on disease registry data concluded that liver volume as measured by palpation decreased in a cohort of patients who initiated idursulfase treatment before 6 years of age. Consistent with these results, biochemical assays across these studies show statistically significant reductions in the uGAG levels of treated patients. IRRs were the most commonly reported AE across these studies, although their frequency decreased over time. These were managed by slowing or stopping the infusion and administering steroids and/or anti-histamines as necessary. Severe IRRs can be life-threatening; therefore, appropriate medical support needs to be available during infusions. Late-emergent anaphylactic reactions after an initial severe infusion reaction have been rarely reported and require prolonged observation.
9. Expert opinion
The approval of ERT with idursulfase for MPS II represented an advance in patient care for a disease that could previously only be managed palliatively. Based on data from clinical trials and an open-label extension study, we see that idursulfase treatment can improve or stabilize several somatic disease features and consider that stabilization of signs and symptoms is a therapeutic benefit in a progressive disease. These data, coupled with the secondary analysis of the growth data from the Phase II/III trial, support the conclusion that idursulfase treatment should be offered to MPS II patients with an attenuated or an indefinite phenotype as soon as possible after diagnosis. This has been recommended elsewhere as well Citation[17,20,21]. Sibling case studies support this conclusion Citation[46]. Should this recommendation extend to children under the age of 5 years? Although the safety and efficacy data from a 53-week, open-label trial of idursulfase in patients 5 years of age or younger (NCT00607386) have not yet been published, a recent abstract presented at the 12th International Symposium on MPS and Related Diseases reported similar effects upon uGAG levels and liver and spleen size in 27 patients as previously reported for older patients, with no new safety concerns Citation[47].
There has been some debate about the role of idursulfase treatment in the management of severe patients. Idursulfase does not cross the blood–brain barrier and does not alter cognitive decline Citation[19]. Nonetheless, patients with the severe phenotype may receive somatic benefits of treatment Citation[19,48]. A recent abstract detailing a case series of 22 patients concluded that severe patients could experience improved JROM, reduced liver and/or spleen size, fewer respiratory infections, resolution of diarrhea, and fewer hospitalizations Citation[48]. While such observations have not yet been confirmed by clinical trials, clinical experience would support a trial of idursulfase treatment in severe patients, with clear expectations and discontinuation criteria discussed with the family before treatment initiation.
MPS II in females, while uncommon, does occur, usually due to skewed X inactivation or complex genetic rearrangements Citation[49,50]. Treatment of females with idursulfase has not been studied in clinical trials. We found during magnetic resonance imaging (MRI) studies of the brain and cervical spine of an 11-year-old female with severe MPS II that the abnormalities did not differ from those detected in male patients Citation[51]. After 2.5 years of idursulfase treatment, brain atrophy on MRI progressed. Jurecka et al. found that the somatic signs and symptoms of two female Polish patients with severe disease responded to idursulfase treatment similarly to those of male patients Citation[52]. They received 24 and 20 months of idursulfase treatment, during which both patients experienced reduced uGAG levels. In addition, the first patient showed stabilization in cardiac disease, liver size, and JROM, while the other patient experienced decreased liver and spleen size and improved mobility.
Unanswered questions about idursulfase treatment remain. Approximately 50% of patients develop IgG antibodies to idursulfase, but limited data about the impact of antibody positivity upon efficacy are available. Indeed, measuring efficacy loss in clinical trials using established endpoints like the 6MWT and FVC is not straightforward given the progressive nature of the disease and the inability to use these tests in severe patients or young children. Assuming that efficacy loss can be demonstrated, would a tolerization regimen be feasible and clinically useful? Would an increase in dose overcome reduced efficacy? The impact of a drug holiday or ERT discontinuation upon patient outcomes is also unclear as yet. A very recent report of five Polish patients with MPS II who discontinued ERT for a median of 3 months (range 2 – 8 months) noted that worsening of the patients' clinical status was observed. Symptoms after ERT discontinuation included recurrent respiratory infections (severe pneumonia) with respiratory insufficiency (80%), difficulty with walking/standing (60%), increased joint stiffness (40%), decreased hematological parameters (40%), renal insufficiency (40%), and death (20%) Citation[53].
Looking forward, an investigational formulation of idursulfase has been designed for intrathecal delivery (idursulfase-IT, Shire Human Genetic Therapies, Inc., Lexington, Massachusetts, USA) in an attempt to alter the cognitive decline in severe patients. A safety study in monkeys found no clinical signs or gross central nervous system lesions in treated animals Citation[54]. A follow-up study demonstrated widespread cellular deposition of idursulfase-IT throughout the central nervous system Citation[55]. Idursulfase-IT is currently being investigated in a Phase I/II trial in severe patients (NCT00920647), and results are expected in the coming months. It is unclear whether idursulfase-IT will produce clinically significant plasma concentration of the drug, allowing uptake by somatic cells. Should idursulfase-IT prove safe and effective for altering cognitive decline and addressing somatic signs and symptoms, the role of intravenous idursulfase may shift to exclusive use in patients without central nervous system involvement.
When thinking about future goals for MPS II treatment, it is important to remember that the signs and symptoms are almost certainly not all directly caused by GAG storage. Many secondary pathogenic cascades appear to be triggered by accumulated GAGs Citation[56-60]. Future treatment will very likely involve not only ERT, but will take a synergistic approach that can address the pathological effects of secondary cascades. To this end, we encourage researchers in the search for biomarkers for the MPSs, not only to better study the efficacy and safety of ERT via surrogate endpoints in clinical trials, but also to better inform efforts toward a personalized medicine approach to treatment in these devastating disorders.
Declaration of interest
Editorial assistance to the author was provided by Jillian Lokere, MS, of The Curry Rockefeller Group, LLC, Tarrytown, New York, and was funded by Shire Human Genetic Therapies, Inc. The opinions expressed are solely those of the author, and the author confirms independence from the funding source. The author received no payment for his work. In the past Dr Scarpa has received honoraria, travel grants, and research grants from Shire Human Genetic Therapies, Inc.
Notes
Bibliography
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, editor. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. p. 3421-52
- Bach G, Eisenberg F Jr, Cantz M, Neufeld EF. The defect in the Hunter syndrome: deficiency of sulfoiduronate sulfatase. Proc Natl Acad Sci USA 1973;70:2134-8
- Poorthuis BJ, Wevers RA, Kleijer WJ, The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 1999;105:151-6
- Ben Turkia H, Tebib N, Azzouz H, Incidence of mucopolysaccharidoses in Tunisia. Tunis Med 2009;87:782-5
- Baehner F, Schmiedeskamp C, Krummenauer F, Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 2005;28:1011-7
- Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics 2000;105:e10
- Poupetova H, Ledvinova J, Berna L, The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010;33:387-96
- Pinto R, Caseiro C, Lemos M, Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet 2004;12:87-92
- Lin HY, Lin SP, Chuang CK, Incidence of the mucopolysaccharidoses in Taiwan. 1984-2004. Am J Med Genet A 2009;149A:960-4
- Wraith JE, Beck M, Giugliani R, Initial report from the Hunter Outcome Survey. Genet Med 2008;10:508-16
- Schwartz IV, Ribeiro MG, Mota JG, A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl 2007;96:63-70
- Martin R, Beck M, Eng C, Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008;121:e377-86
- Young ID, Harper PS, Archer IM, Newcombe RG. A clinical and genetic study of Hunter's syndrome. 1 Heterogeneity. J Med Genet 1982;19:401-7
- Young ID, Harper PS. The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Dev Med Child Neurol 1983;25:481-9
- Jones SA, Almassy Z, Beck M, Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis 2009;32:534-43
- Young ID, Harper PS. Mild form of Hunter's syndrome: clinical delineation based on 31 cases. Arch Dis Child 1982;57:828-36
- Wraith JE, Scarpa M, Beck M, Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 2008;167:267-77
- Muenzer J, Beck M, Eng CM, Multidisciplinary management of Hunter syndrome. Pediatrics 2009;124:e1228-39
- Muenzer J, Bodamer O, Burton B, The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr 2011;171(1):181-8
- Giugliani R, Federhen A, Rojas MV, Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol 2010;33:589-604
- Scarpa M, Almassy Z, Beck M, Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis 2011;6:72
- Coppa GV, Gabrielli O, Zampini L, Bone marrow transplantation in Hunter syndrome (mucopolysaccharidosis type II): two-year follow-up of the first Italian patient and review of the literature. Pediatr Med Chir 1995;17:227-35
- McKinnis EJ, Sulzbacher S, Rutledge JC, Bone marrow transplantation in Hunter syndrome. J Pediatr 1996;129:145-8
- Mullen CA, Thompson JN, Richard LA, Chan KW. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia. Bone Marrow Transplant 2000;25:1093-7
- Guffon N, Bertrand Y, Forest I, Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr 2009;154:733-7
- Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis 1995;18:413-29
- Vellodi A, Young E, Cooper A, Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis 1999;22:638-48
- Prasad VK, Kurtzberg J. Transplant outcomes in mucopolysaccharidoses. Semin Hematol 2010;47:59-69
- Green Cross Corporation. Green Cross gains market approval with the world's 2nd orphan drug for Hunter syndrome in South Korea. 12 January 2012. Available from: http://eng.greencross.com/ [Accessed 6 July] 2012
- Friso A, Tomanin R, Salvalaio M, Scarpa M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br J Pharmacol 2010;159:1082-91
- Delgadillo V, O'Callaghan M del M, Artuch R, Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis 2011;34:1039-44
- de Ruijter J, Valstar MJ, Narajczyk M, Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann Neurol 2012;71:110-20
- Marucha J, Tylki-Szymanska A, Jakobkiewicz-Banecka J, Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am J Med Genet A 2011;155A:2257-62
- Muenzer J, Wraith JE, Beck M, A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006;8:465-73
- Elaprase® (idursulfase) solution for intravenous infusion [prescribing information]. Shire Human Genetic Therapies, Inc; Cambridge, MA: 2011
- Muenzer J, Gucsavas-Calikoglu M, McCandless SE, A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab 2007;90:329-37
- Muenzer J, Beck M, Eng CM, Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011;13:95-101
- Schulze-Frenking G, Jones SA, Roberts J, Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis 2011;34:203-8
- Muenzer J, Beck M, Giugliani R, Idursulfase treatment of Hunter syndrome in children younger than 6 years: results from the Hunter Outcome Survey. Genet Med 2011;13:102-9
- Vellodi A. Mucopolysaccharidosis type II: guidelines for assessment, monitoring and enzyme replacement therapy (ERT). Available from: http://www.specialisedservices.nhs.uk/library/23/Guidelines_for_Mucopolysaccharidosis_Type_II.pdf [Accessed 2 July] 2012
- McGill JJ, Goldblatt J, Peters H, Guidelines for the treatment of mucopolysaccharidosis type II (MPS II) through the Life Saving Drugs Program. Available from: http://www.health.gov.au/internet/main/publishing.nsf/Content/lsdp-info/#Idursulfase [Accessed 2 July] 2012
- Ontario Public Drug Programs Exceptional Access Program: Elaprase (idursulfase) – Reimbursement Guidelines. Revision 3 (June 2011). Available from: http://www.health.gov.on.ca/english/providers/program/drugs/pdf/elaprase_reimburse.pdf [Accessed July 19] 2012
- Hughes DA, Tunnage B, Yeo ST. Drugs for exceptionally rare diseases: do they deserve special status for funding? QJM 2005;98:829-36
- McKie J, Richardson J. The rule of rescue. Soc Sci Med 2003;56:2407-19
- OnePath® Services. Available from: http://www.elaprase.com/healthcare/about_elaprase/onepath/ [Accessed 2 July 2012]
- Tylki-Szymanska A, Jurecka A, Zuber Z, Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3-year follow-up. Acta Paediatr 2012;101:e42-7
- Giugliani R, Hwu PWL, Tylki-Szymanska A, Whiteman DAH. A multi-center, open-label study evaluating safety and clinical outcomes in young children (1.4-7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. 12th International symposium on MPS and related diseases: 28 June - 1 July, 2012. Noordwijkerhout, the Netherlands; Abstract P100
- Lampe C, Bosserhoff A, Burton BK, Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe (neuronopathic) phenotype: an international case series. Final programme and abstracts from the 12th International symposium on MPS and related diseases; 28 June - 1 July, 2012. Noordwijkerhout, the Netherlands; Abstract S08.6
- Tuschl K, Gal A, Paschke E, Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol 2005;32:270-2
- Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis 2010;5:14
- Manara R, Rampazzo A, Cananzi M, Hunter syndrome in an 11-year old girl on enzyme replacement therapy with idursulfase: brain magnetic resonance imaging features and evolution. J Inherit Metab Dis 2010; DOI: 10.1007/s10545-009-9023-8
- Jurecka A, Krumina Z, Zuber Z, Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. Am J Med Genet A 2012;158A:450-4
- Jurecka A, Żuber A, Opoka-Winiarska V, Effect of rapid cessation of enzyme replacement therapy: a report of 5 cases and a review of the literature. Mol Genet Metab 2012; DOI: 10.1016/j.ymgme.2012.08.013
- Felice BR, Wright TL, Boyd RB, Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol Pathol 2011;39:879-92
- Calias P, Papisov M, Pan J, CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS One 2012;7:e30341
- Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J 2006;20:9-22
- Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med 2008;10:e1
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007;446:1030-7
- Kobialka S, Beuret N, Ben-Tekaya H, Spiess M. Glycosaminoglycan chains affect exocytic and endocytic protein traffic. Traffic 2009;10:1845-55
- Settembre C, Arteaga-Solis E, Ballabio A, Karsenty G. Self-eating in skeletal development: implications for lysosomal storage disorders. Autophagy 2009;5:228-9
- Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, CDC growth charts: United States. Adv Data 2000;1-27