Abstract
Introduction: Amyotrophic lateral sclerosis (ALS) is a progressive and devastating neurodegenerative disease resulting from injury and death of upper and lower motor neurons. Symptoms initially include muscle weakness and twitching and subsequently progress to muscle atrophy, complete loss of limb use, respiratory difficulties and ultimately death. Multiple biological mechanisms have been implicated in ALS and a complex etiology has been described. As a result, drug discovery researchers have few validated targets to pursue and patients have few therapeutic options.
Areas covered: Identification of new drug targets in ALS can be facilitated by a detailed understanding of the processes and genes that contribute to pathogenesis. Accordingly, this review summarizes current hypotheses regarding underlying mechanisms for motor neuron susceptibility in ALS. An overview of emerging and tractable drug targets that could result in therapeutic breakthroughs is provided.
Expert opinion: Despite the immense progress that has been made in understanding ALS over the last decade, riluzole remains the only approved drug to treat ALS. Combining structure-guided drug design applied to validated and pharmaceutically tractable targets with disease-relevant phenotypic screens will allow for the identification of novel drug targets and potentially breakthrough therapeutics for ALS.
1. Introduction
1.1 Motor neuron susceptibility in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a progressive and devastating neurodegenerative disease resulting from injury and death of upper and lower motor neurons. This degeneration of motor neurons causes weakness and atrophy of limb musculature progressing to a complete loss of arm and leg function, speech and swallowing difficulties and ultimately death, usually from respiratory failure. The prognosis is bleak for ALS patients whose average survival after first symptom onset is 3 years Citation[1].
Multiple underlying factors responsible for the selective motor neuron degeneration in ALS have been described. Motor neurons are large cells and have extensive dendritic and axonal processes. While the cell body of an alpha motor neuron is 50 – 70 μm in diameter, the axon length can oftentimes be greater than 1 m. Motor neurons conduct action potentials to muscle fibers where larger axon diameters increase the rate of neuronal conduction velocity. Motor neuron neuronal filaments (NFs) function to increase axonal diameter in myelinated axons in a process known as radial axonal growth Citation[2]. A hallmark of both sporadic and familial ALS (FALS) is the presence of cytoplasmic inclusions, many of which contain phosphorylated NFs Citation[3,4].
The metabolic demands in maintaining an axon of 1 m length or more requires a high level of mitochondrial activity. This high metabolic demand is consistent with the observation that motor neurons are particularly susceptible to hypoxia. For example, deletion of the hypoxia responsive element in the vascular endothelial growth factor (VEGF) promoter caused a selective degeneration of motor neurons and concomitant ALS-like phenotype in mice Citation[5]. Motor neurons are involved in neurotransmission to skeletal muscle involving both glutamatergic and acetylcholine neurotransmitter systems. Thus, motor neurons are susceptible to oxidative stress. Elevated protein carbonyl levels, increased 3-nitrotyrosine levels and high 4-hydroxynonenal levels (an indicator of lipid peroxidation) are observed in ALS patients Citation[6-8]. Despite high metabolic activity and exposure to oxidative stress, motor neurons do not have high levels of glutathione compared to hepatocytes or astrocytes Citation[9] and thus appear particularly vulnerable to oxidative stress Citation[10]. High levels of oxidative stress can predispose proteins to unfolding and aggregation, yet in motor neurons there is a high threshold for induction of the stress response and expression of heat shock proteins (Hsps), which was associated with a failure to activate HSF1 Citation[11]. Similarly, challenge of spinal motor neurons with ZnCl2, paraquat or toxic glutamate concentrations did not lead to induction of metallothioneins (zinc-binding proteins with antioxidant properties) where, in contrast, other neuronal populations did induce metallothioneins Citation[12]. The attenuated Hsp response of motor neurons could represent an opportunity for therapeutic intervention as it has been shown that treatment with arimoclomol, a coinducer of Hsps, delays disease progression in ALS mice Citation[13]. Arimoclomol is now in the clinic for ALS patients Citation[14]. It is not clear why a neuronal type that is exposed to high levels of stress has difficulty mounting a heat shock or other protective responses to injury. Nonetheless, the high threshold necessary to express Hsps may contribute, to some extent, to the vulnerability of motor neurons in ALS and may represent an opportunity for therapeutic intervention.
Lower motor neurons receive extensive glutamatergic input from upper motor neurons and excitatory interneurons. Motor neurons express alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), kainate and N-methyl-D-aspartate (NMDA) glutamate receptors (GluRs) where glutamate is the neurotransmitter used for fast synaptic transmission in the CNS. AMPA receptors are highly expressed on motor neurons and consist of four homo- or hetero-oligomeric receptor subunits (GluR1, GluR2, GluR3, and GluR4). It has been shown pharmacologically that there is a significant pool of AMPA receptors expressed in motor neurons that are permeable to calcium Citation[15-17]. Glutamate released into the synaptic cleft stimulates AMPA receptors where signaling is terminated through uptake of glutamate by the excitatory amino acid transporter (EAAT2). It has been shown that there is a decrease in EAAT2 levels in ALS patients Citation[18,19], which may lead to overstimulation of AMPA receptors and excessive calcium influx, initiating a number of detrimental processes and excitotoxicity in motor neurons Citation[20].
Natural cellular processes exist to counteract excessive calcium influx from overstimulation of AMPA receptors. Cytoplasmic calcium can be transported into the mitochondria by the mitochondrial uniporter. Calcium is then slowly released through the mitochondrial Na/Ca exchanger and then ultimately leaves the cytoplasm through a number of mechanisms including the plasma membrane Na/Ca exchanger Citation[21,22] and sarcoendoplasmic reticulum calcium transport ATPase (SERCA) Citation[23]. It has been shown that mitochondria from spinal cord have decreased calcium-buffering capacity compared to other brain mitochondria Citation[24] which could be a result of a lower density of mitochondria in motor neurons compared to other neurons.
The endoplasmic reticulum (ER) is also an important and high capacity storage site for calcium. Calcium plays an important role in protein folding and processing within the ER Citation[25,26]. Elevated intracellular calcium will cause calcium-induced calcium release through the ER ryanodine and triphosphoinositol (IP3) receptors and potentially deplete ER calcium stores Citation[21,27]. There is an important crosstalk between ER and mitochondrial calcium stores as ER calcium released into the cytosol can be uptaken by the mitochondria and then ultimately reuptaken into the ER by SERCA pumps Citation[23,27]. This calcium signaling between compartments could become disturbed in ALS where it is tempting to speculate, that the depletion of ER calcium stores that could happen with glutamate-receptor overstimulation will lead to activation of the unfolded protein response (UPR) Citation[27]. Although this seems to be a valid hypothesis, it has yet to be unequivocally proven Citation[27].
Intracellular calcium can also be buffered by calcium-binding proteins (CaBPs). Of note, motor neuron populations, such as the oculomotor and abducens nuclei, which express CaBPs such as parvalbumin, calbindin, calretinin, and neurocalcin are spared in ALS patients, while other motor neurons that do not express these proteins are not Citation[28-31]. Expression of CaBPs in cultured motor neurons are also protective against mutant superoxide dismutase (SOD), excessive glutamate and oxidative stress Citation[32], which is consistent with the clinical observation that neurons expressing CaBPs are spared in ALS.
The ER orchestrates a complex symphony of protein distribution for axonal transport, exocytosis, and for transport to Golgi, mitochondria and nuclear compartments of the cell. In the context of a motor neuron, with the demands of maintaining large cells with long axons and extensive dendritic morphology, it is not surprising that these cells could be susceptible to ER stress. It has been noted that ER stress, as indicated by activation of the UPR, occurs in ALS Citation[21,33]. The UPR inhibits protein synthesis and results in the expression of ER resident chaperones to assist in protein folding. Quantitative Western blotting of human spinal cord tissue from sporadic ALS (SALS) patients showed that three UPR sensors, inositol-requiring enzyme1 (IRE1), activating transcription factor 6 (ATF6) and protein kinase RNA-like ER kinase (PERK) were upregulated from fourfold to sixfold compared to control patients Citation[34]. Also in this study, the UPR chaperones protein disulfide isomerase (PDI) and PDI-related protein Erp57 were upregulated in ALS patients. These results were consistent with an earlier study showing increased expression of PDI and eukaryotic initiation factor 2-alpha (eIF2α) in the spinal cord of ALS patients compared to control patients Citation[35]. Similarly, in 60-day-old mutant superoxide dismutase 1 (SOD1) mice, before ALS symptoms emerged, UPR sensor proteins IRE1, ATF6 and PERK were elevated and a high level of expression of PDI was observed compared to WT mice Citation[21,34].
In summary, motor neuron susceptibility in ALS may have many contributing factors. Motor neurons are large in size and have long axons. The metabolic requirements to sustain these cells are inherently large. Motor neurons express high levels of calcium permeable AMPA receptors and by nature of their function, are often exposed to elevated levels of calcium, particularly during intense stimulation. Motor neurons may have a limited ability to induce Hsps, reduced glutathione levels, and many motor neurons do not express CaBPs which reduce calcium-buffering capacity. In light of the enhanced susceptibility of motor neurons to stress, it is perhaps no surprise that there are more known mutated genes causing FALS () then there are for more common neurodegenerative diseases such as Alzheimer's disease (AD) or Parkinson's disease (PD).
Table 1. Genes associated with FALS.
1.2 Current and future therapy for ALS patients
Riluzole, a potent anticonvulsant drug Citation[36], has been approved to treat ALS since 1995, yet despite its approval for this use, the efficacy of riluzole is modest. Riluzole will extend the lifespan of ALS patients by a few months on average Citation[37]. Riluzole may have several different mechanisms of action; however, the biological relevance of these for its effects in ALS is not completely understood. Riluzole noncompetitively blocks excitatory amino acid receptors Citation[38], inhibits glutamic acid release and inactivates voltage-dependent sodium channels Citation[39]. Some of the electrophysiological effects of riluzole can be blocked by pertussis toxin suggesting that it may interact with a G-protein driven mechanism and pathway Citation[40].
There are a number of therapeutics that are currently in clinical trials for ALS and these have been reviewed elsewhere Citation[37,41-43] and therefore will not be discussed in this present report. Additional development candidates not reviewed elsewhere include GSK1223249, an anti-Nogo-A antibody Citation[44]. Nogo-A is a high molecular weight membrane protein of spinal cord myelin. Nogo-A functions to inhibit neurite outgrowth and can trigger growth cone collapse Citation[45,46] which suggests that Nogo-A is an important factor involved in limiting neurite regeneration and repair of injured axons. In preclinical studies, Anti-Nogo-A antibody treatment promotes recovery of manual dexterity after cervical lesion in primates Citation[47]. Genetic ablation of Nogo-A in SOD mice resulted in a moderate but significant increase in lifespan, increased the number of motor neurons, and eliminated ubiquitin inclusions (a marker of cell stress) Citation[48]. Nogo-A expression has also been shown to be a prognostic marker for ALS early in the course of the disease Citation[49]. A Phase I trial of GSK1223249 in ALS patients has been completed where we await safety and pharmacokinetic (PK) results of this clinical study.
Tirasemtiv, formerly known as CK-2017357, is an imidazo-pyrazine with a molecular mass of 230 Da. Tirasemtiv is a fast skeletal muscle troponin activator which increases fast skeletal muscle sensitivity to calcium, resulting in enhanced skeletal muscle force and slowing of time to muscle fatigue Citation[50]. Tirasemtiv demonstrated potentially clinically relevant pharmacodynamic effects in a completed Phase IIa evidence of effect clinical trial in ALS patients Citation[51]. At 6 h after dosing, patients demonstrated a positive change in overall status and decreased muscle fatigability. Data also demonstrated a statistically significant increase in the maximum volume of air patients could inhale and exhale Citation[51]. A Phase IIb clinical trial for Tirasemtiv is expected in the near future as this looks to be a promising, albeit palliative approach to treating ALS.
Histone deacetylase (HDAC) inhibitors have been discussed as potential therapeutics for neurodegenerative diseases for some time where their limitation lies in their lack of selectivity and concomitant toxicity Citation[52]. Inhibition of histone deacetylation can modulate gene expression and protein synthesis Citation[53] and thus presents a biological basis for potential treatment of ALS. HDAC inhibitors have also been shown to be efficacious in ALS mouse models in a number of studies Citation[54-58]. A Phase II clinical trial of sodium phenylbutyrate in patients with ALS demonstrated safety and tolerability at high doses, as well as a trend toward an increase in histone acetylation in patients' blood samples Citation[59]. As this clinical trial was not powered to observe a potential clinical benefit, it remains to be seen whether phenylbutyrate will show efficacy in ALS patients.
2. Genes associated with FALS
Approximately 5 – 10% of ALS patients have a family history and/or the presence of a known genetic mutation and thus has familial ALS (FALS), while the remainder of patients are characterized with SALS Citation[60]. Symptoms first appear for patients with FALS in the fourth to fifth decade of life, while features of the condition for SALS usually appear later. FALS and SALS are indistinguishable based on clinical symptoms. Genes associated with FALS can be grouped into five categories: i) those that effect RNA processing, ii) those that effect vesicle trafficking, iii) those that affect oxidative stress, iv) those that effect the ubiquitin proteasome system (UPS) and autophagy, and v) those that currently have an unknown function ().
2.1 Genes associated with RNA processing
TAR DNA-binding protein of 43 kDa (TDP-43) is a major component of ubiquitinated protein aggregates found in the CNS of ALS patients and those suffering from frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) Citation[61,62]. To date, 44 different mutations of this gene have been identified that account for close to 5% of all FALS patients Citation[53]. TDP-43 binds to an estimated one-third of all mouse and human brain RNAs Citation[63,64]. TDP-43 has a profound effect on mRNA splicing and mature mRNA levels as determined from TDP-43 knockdown studies Citation[63,64]. TDP-43 participates in the processing of many mRNAs that are involved in neuronal function, such as neurexins, the NMDA receptor and NF-L Citation[63]. Fused in sarcoma or translocated in liposarcoma (FUS/TLS) is another gene involved in RNA processing, which has structural and functional similarities to TDP-43, where a total of 46 mutations have been identified accounting for ∼5% of all FALS patients Citation[65]. In ALS, wild type and mutant TDP-43, as well as FUS accumulate in the cytoplasm and lose their nuclear localization Citation[65]. Interestingly, TDP-43 and FUS mislocalization and aggregation is observed in other diseases such as FTLD-U, AD and Huntington's disease (HD) Citation[65]. One unanswered question is why are TDP-43 and FUS so susceptible to aggregation? They are certainly large, multi-domain proteins that have poly-functional roles in mRNA processing. It is possible that posttranslational modifications such as phosphorylation and proteolysis may enhance the susceptibility of these proteins to aggregation Citation[65]. Interestingly, one ALS-associated mutation in TDP-43 enhances its stability and promotes a complex with FUS, suggesting that this mutation in TDP-43 may perturb FUS function Citation[66]. This result leaves the door open for a potential toxic gain of function activity for some TDP-43 mutations.
Other proteins that are genetically linked to ALS that are involved in RNA processing include Senataxin (SETX) which is a DNA/RNA helicase protein that affects gene transcription Citation[67]. It has been demonstrated that depletion of SETX in HeLa cells causes an increase in readthrough RNA and Pol II density downstream of the polyA site, suggesting an involvement in transcriptional termination Citation[68]. Mutations in SETX account for <1% of FALS cases. Angiogenin (ANG) is a secreted RNase that accounts for <1% of FALS cases. ANG has been shown to induce RNA cleavage in astrocytes where FALS associated mutations in ANG abrogate its RNase activity Citation[69].
2.2 SOD1
Cu/Zn SOD was the first gene to be linked to ALS. SOD is responsible for the catabolism of superoxide radicals to hydrogen peroxide and oxygen. SOD1 is a 153 amino acid, functional homodimer that binds to Cu and Zn. SOD is ubiquitously expressed and comprises ∼ 1% of all cytoplasmic proteins Citation[70]. Mutations that occur in the SOD gene account for ∼ 20% of all FALS cases Citation[71]. The concept that mutant SOD1 causes aberrant oxyradical reactions, explaining its toxic gain of function, is controversial and has been reviewed elsewhere Citation[71,72]. However, of note, it has also been suggested that the oxidative damage caused by mutant SOD1 may be attributed to Cu binding outside of the active site Citation[73].
2.3 Genes involved in vesicle trafficking and other genes of interest
The size of a motor neuron and length of its axon can cause increased metabolic burden. Thus, it is not surprising that mutations in genes involved in vesicle trafficking may be linked to ALS. One such gene is Alsin (ALS2), which functions as a Rab5 guanine nucleotide exchange factor Citation[74]. Mice deficient in ALS2 exhibit age-dependent neurological and motor neuron deficits that are associated with altered endosome trafficking Citation[75]. For example, in wild type cortical neurons that were stimulated with brain-derived neurotrophic factor (BDNF), we observed a typical accumulation of Trk-B in the perinuclear area, which is indicative of retrograde transport. In contrast, in neurons derived from Als2−/− mice, this was not observed Citation[75]. Vesicle-associated membrane protein-associated protein B (VAPB), factor-induced gene 4 (FIG4), and Optineurin are other FALS genes that are also involved in vesicle trafficking Citation[76-78] and have been similarly linked to FALS ().
Mutations in the C9Orf72 gene have been shown to cause up to 40% of all FALS cases and are also associated with FTLD-U Citation[79]. Although the function of the C9Orf72 gene is not known, the presence of RNA nuclear foci in C9Orf72 FALS patient brain suggests that alternative mRNA splicing is dysregulated Citation[80]. Most recently, mutations within the profilin 1 (PFN1) gene have been shown to cause FALS Citation[81]. PFN1 is an actin-binding protein that plays an important role in regulating actin dynamics. PFN1 enhances the rate of actin polymerization by binding to actin-ADP to catalyze nucleotide exchange facilitating formation of actin-ATP. PFN1 bound actin-ATP is a substrate of Formin which adds actin-ATP to the barbed ends of nascent actin fibers, with a subsequent release of PFN1 Citation[82]. From a functional perspective, a mutation in PFN1 that blocked actin-binding activity was shown to inhibit neurite outgrowth in a neuroblastoma cell line Citation[83]. In a follow-up study to determine the prevalence of PFN1 gene mutations in FALS, this gene was sequenced in a cohort of 94 FALS patients of European ancestry. No PFN1 mutations were identified in this cohort suggesting that PFN1 mutations are a rare cause of FALS Citation[84]. Despite that mutations in PFN1 may account for a low frequency of FALS patients; PFN1 gene mutations and their linkage to FALS have demonstrated a novel mechanism that can cause the disease.
3. New targets for ALS
3.1 EphA4
Ephrin type-A receptor 4 (EphA4) is a receptor tyrosine kinase that consists of an extracellular ligand-binding domain, a transmembrane region, and an intracellular kinase domain that can accommodate multiple docking sites for downstream signaling Citation[85]. Upon ligand binding, the EphA4 receptor becomes activated and initiates signaling in the forward direction. However, ephrins, the ligands of the Eph receptor, are attached to the cell membrane through a glycosylphosphatidylinositol link which enables signal transduction in the reverse direction Citation[86]. This bidirectional signaling is a key feature of Ephrin biology.
EphA4 is enriched at excitatory synapses in motor neurons and in the hippocampus Citation[87,88]. Genetic knockdown of EphA4 in the mouse suggests that ephrin-A:EphA forward signaling is required for selection of lateral lower motor column neurons toward the dorsal limb, thus contributing to the topographic organization of motor neuron projections Citation[87,89]. It has also been shown that the EphA4 receptor tyrosine kinase regulates spine morphology where its activation reduces spine length and density in hippocampal slices Citation[90]. Subsequently EphA4 forward signaling was shown to decrease phosphorylation of focal adhesion kinase (FAK), and proline-rich tyrposine kinase 2 (Pyk2), thus regulating dendritic spine remodeling by affecting β1 integrin signaling pathways Citation[91]. In the hippocampus, EphA4 receptor reverse signaling may overactivate its ligand glial ephrinA3 and inhibit glutamate uptake by the glutamate transporter Citation[88]. Recently, in a genetic screen using a SOD1 zebrafish model, the most protective knockdown identified was of the gene Rtk2, which has 67% identity to human EphA4 Citation[92]. Further validation of the importance of this target were the results in SOD1G93A mice where a 50% knockdown of EphA4 expression significantly enhanced motor function (rotarod performance) and increased survival time Citation[92]. In patients with ALS, EphA4 expression inversely correlated with disease onset and survival where loss of function and EphA4 mutations are associated with longer survival Citation[92]. These findings suggest that EphA4 represents a compelling biological target for ALS.
From a drug discovery perspective, EphA4 is a tractable drug target. Compounds of the pyrrolyl benzoic acid scaffold have been identified that block the interaction of ephrins with the EphA4 receptor Citation[93]. In addition, small molecules have been identified that bind to the ATP pocket and block EphA4 kinase activity with high affinity Citation[94]. A crystal structure of the EphA4 tyrosine kinase domain in the apo- and dasatinib-bound state has also been determined which will facilitate structure-guided drug design Citation[95]. Key insights from the crystal structure suggest a path forward for kinase receptor selectivity between EphA4 and other Ephrin receptors Citation[95]. There are a number of commercially available EphA4 biochemical kinase assays that are amenable to high throughput screening (HTS) which should facilitate the identification of leads for drug discovery efforts. Further, a number of antibody reagents are readily available suggesting that the development of cell-based characterization assays will not represent an obstacle for drug discovery. In addition to the potential role for EphA4 as a drug target for ALS, it has been implicated to play a role in gastric cancer Citation[96] and blocking EphA4 is effective in preclinical models of spinal cord injury Citation[97]. At present, however, it does not appear that EphA4 has been intensely investigated in other neurodegenerative indications.
3.2 Nuclear erythroid 2-related factor 2
Nuclear erythroid 2-related factor 2 (Nrf2) is a transcription factor, which under non-stressed resting conditions, is normally located in the cytoplasm bound to Kelch-like ECH-associated protein 1 (Keap1). Under conditions of oxidative stress, oxidative modification of Keap1 leads to the dissociation of Nrf2 from Keap1. Nrf2 can then translocate to the nucleus, where Nrf2 binds the antioxidant response element (ARE) and facilitate the expression of over 250 cytoprotective genes including heme oxygenase-1 (HO-1), glutathione, thioredoxin (TRX) and Hsps. The Nrf2/Keap1/ARE system is a major cellular defense system for cells undergoing oxidative stress Citation[98]. As described above, oxidative stress has been implicated to play a role in ALS Citation[99] and the Nrf2/Keap1 system was shown to be impaired in motor neurons in a SOD1 mouse model Citation[100]. Interestingly, selective overexpression of Nrf2, in astrocytes, using double transgenic SOD1G93A/GFAP-Nrf2 mice, extended survival by over 20 days compared to SOD1G93A mice Citation[101]. This protective effect was mediated in part by an increase of astrocyte-secreted glutathione Citation[101]. Triterpenoid small molecule activators of the Nrf-2/Keap1 system have extended survival in SOD1G93A mice Citation[102]. Bardoxolone Methyl (formerly RTA402), a triterpenoid has completed a Phase II clinical trial for stage 4 chronic kidney disease (CKD) Citation[103] and is currently in a Phase III trial for this indication. As there is good rationale that activation of Nrf2/Keap1 system would be helpful to ALS patients, clinical testing using a CNS permeable Nrf2 activator could be considered.
3.3 DJ-1
DJ-1 is a 20 kDa protein that is ubiquitously expressed and widely conserved throughout species Citation[104]. DJ-1 was identified as an oncogene and subsequently linked to familial PD Citation[105]. DJ-1 is a multifunctional oxidative stress response protein that confers protection to cells undergoing oxidative and or mitochondrial stress Citation[106,107]. Multiple functions for DJ-1 have been described. For example, DJ-1 has been shown to negatively regulate the apoptotic signaling kinase1 (ASK1) Citation[108,109]. DJ-1 has also shown to suppress the proapoptotic PTEN phosphatase and thus is an enhancer of the Akt survival pathway Citation[109,110]. DJ-1 is a redox sensitive protein where it has been shown that oxidation of cysteine 106 on the DJ-1 protein is required for its protective function Citation[106,107]. Interestingly, excessive oxidation of this cysteine to the sulfonic acid renders the protein inactive Citation[106]. DJ-1 is also linked to ALS where DJ-1 mutations in one Italian family caused Parkinsonism-Dementia ALS (PDALS) complex Citation[111]. PDALS complex has been particularly well characterized in patients in the Kii peninsula of Japan and in Guam Citation[112] where environmental as well as genetic factors may play a role in the development of disease. Thus, ALS and PD pathogenesis, may in some cases share a common etiology. At the molecular level, DJ-1 forms complexes with mutant SOD1 and ameliorates its toxicity Citation[113]. As oxidative stress may play a role in many neurodegenerative diseases, it is not surprising that a protein like DJ-1 could be implicated in both ALS and PD. Although DJ-1 is protective in cellular and animal models of oxidative stress, it has not proved simple to assay for small molecule binders in a high-throughput biochemical format. One virtual screen identified potential binders of DJ-1 whose binding properties were confirmed using a quart crystal microbalance Citation[114]. These compounds exhibited specificity for DJ-1 and prevented oxidative stress-induced cell death in SH-SY5Y and in primary ventral mesencepahlon cells Citation[114]. In addition, these DJ-1-binding compounds were active in blocking dopaminergic cell death and blocked movement abnormalities in a 6-hydroxydopamine injected PD rat model Citation[114]. It was reported that the protective effects of these compounds were mediated through binding to an oxidized form of DJ-1. It remains of interest to test these compounds in cellular and animal models of ALS.
DJ-1 levels are reported to be lower in DJ-1 familial PD patients and, as such, increasing levels of DJ-1 could be a therapeutic strategy for familial and sporadic PD and ALS Citation[115,116]. Thus, compounds that bind and stabilize DJ-1 may be useful therapeutics. Binding assays such as those that use surface plasmon resonance (SPR) could also be used to identify small molecules that interact with DJ-1 in a medium throughput manner. A structure for DJ-1 has been solved by X-ray crystallography which may further facilitate a drug discovery program for this target Citation[117]. Compounds of interest could be run through cellular oxidative stress protection assays in cells expressing mutant or wild type DJ-1. Although there are many inherent risks with this approach, DJ-1 is a validated target for PD and may be of relevance for ALS.
3.4 Apoptotic signaling kinase1
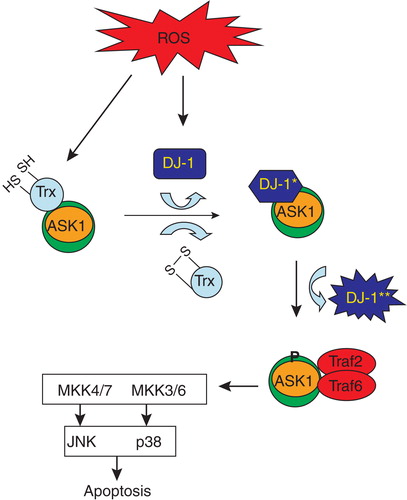
Apoptotic signaling kinase1 (ASK1) is a 1374 amino acid polypeptide consisting of a central serine/threonine kinase domain and coiled-coil domains at the N- and C-terminus Citation[118,119]. ASK1 is a MAP3K that plays an essential role in cellular stress. In its resting state, ASK1 is a homodimer stabilized by its C-terminal coiled-coil domain. The N-terminal domain is bound by the redox regulatory protein, thioredoxin, which prevents its activation Citation[120,121]. ASK1 is activated by ROS, ER stress, calcium ion influx and other stressful stimuli Citation[122]. ASK1 is a regulator of the JNK and p38 MAP kinase cascades in stress signaling Citation[118,122]. Oxidative stress leads to disulfide bride formation of thioredoxin and dissociation from ASK1, triggering a conformational change in the ASK1 dimer leading to phosphorylation and activation of its kinase activity Citation[120,121]. DJ-1 can bind to ASK1 at its N-terminal domain and negatively regulate ASK1 phosphorylation activity (). Further levels of oxidative stress lead to diminished DJ-1 levels, unregulated ASK1 activity, and subsequent cell death (). ASK1 knockout is protective in ALS and PD mouse models Citation[121,123]. Specifically, in SOD1G93A/ASK1−/− mice, mean survival times were significantly longer and neuronal cell death was ameliorated in the spinal cord compared to SOD1G93A mice Citation[123]. Furthermore, ASK1 has been reported to negatively regulate the proteasome, possibly by phosphorylating and inhibiting the ATPase activity of Rpt5 Citation[124]. Thus, there is good biological rationale suggesting that well-tolerated ASK1 inhibitors may be useful for the treatment of ALS Citation[125].
Figure 1. Hypothetical scheme for activation of ASK1 by oxidative stress. In resting conditions, ASK1 is bound to and negatively regulated by TRX. Under conditions of oxidative stress, TRX becomes oxidized and dissociates from the ASK1 signaling complex (signalosome). DJ-1 becomes oxidized under conditions of oxidative stress (*) and subsequently binds to ASK1, negatively regulating its activity. Continued oxidation of DJ-1 can lead to a conformational change (**) where it dissociates from the ASK1 signalosome, relieving the negative inhibition. ASK1 becomes phosphorylated and is now activated resulting in the activation of JNK and p38 pathways. This schematic connects ASK1, DJ-1 and p38 targets in a common pathway initiated by oxidative stress.
From a drug discovery perspective, ASK1 appears to be a tractable target. A crystal structure of ASK1 has been determined Citation[119] which should facilitate structure-based drug discovery. ASK has three isoforms (ASK1-3) and preliminary analysis suggests that selectivity between ASK1 and ASK2 is possible, yet selectivity between ASK1 and ASK3 is unlikely Citation[126]. It is not known whether selectivity between the three ASK isoforms is necessary for an appropriate tolerability/efficacy profile. Selectivity between ASK1 and the broader kinome also appears possible from a preliminary analysis Citation[126]. ASK1−/− mice are healthy and viable, yet studies have shown that ASK1 does play a role in innate immunity Citation[127]. Thus, the benefit to cost ratio of an ASK1 inhibitor may override any underlying safety concerns, particularly in ALS patients, where there is a dire unmet medical need. Although there have been reports of ASK1 inhibitors in development for insulin resistance and cardiac indications, their use in neurodegeneration seems relatively unexplored Citation[128].
3.5 HDAC6 stabilizers/inhibitors
Currently the nonselective HDAC inhibitor, phenylbutyrate, is undergoing clinical evaluation for the treatment of ALS Citation[59]. HDAC6, a class II HDAC, represents a particularly intriguing target for ALS as there appears to be rationale for both a HDAC6 stabilizer/activator and a HDAC6 inhibitor for therapeutic application to ALS. HDAC6 has been shown to facilitate the formation of aggresomes, activate autophagy, and may be involved in the expression of Hsps Citation[129,130]. Some of these neuroprotective functions may be independent of the deacetylating activity of HDAC6 Citation[129-131]. TDP-43 and FUS may function in a complex to regulate HDAC6 mRNA expression where a loss of TDP-43 function may decrease levels of HDAC6 expression in motor neurons Citation[132]. In this manner, a HDAC6 stabilizer, which could increase HDAC6 levels, would be helpful in ALS. Small molecule chaperones or stabilizers have been successfully pursued for some targets Citation[133]. However, this approach is not universally applicable to all proteins.
There is also rationale for potential therapeutic role for a HDAC6 inhibitor in ALS. HDAC6 is the major α-tubulin deacetylating enzyme Citation[134-136]. HDAC6 inhibition protects against oxidative stress-induced neurodegeneration and promotes neurite outgrowth in cortical neurons co-cultured with Chinese hamster ovary (CHO) cells expressing myelin-associated glycoprotein (MAG) Citation[137]. In addition, HDAC6 inhibition promotes neurite outgrowth in dorsal root ganglion neurons exposed to MAG Citation[137]. Furthermore, in a mouse model of mutant Hsp27-induced Charcot-Marie-Tooth (CMT) disease (a peripheral neuropathy), HDAC6 inhibitors restored axonal transport, and partially restored the CMT phenotype both at the behavioral and electrophysiological levels Citation[138]. From a pharmacological perspective, HDAC6 represents a tractable target. The HDAC6 knockout mice are fully functional and viable, suggesting that the reduction of HDAC6 activity may be less likely to induce mechanism-based toxicity Citation[139]. In addition, selective HDAC6 inhibitors have been synthesized Citation[140,141], thereby obviating the potential for nonspecific toxicity that may be observed following treatment with CNS penetrant pan-HDAC inhibitors.
3.6 Nuclear factor-kappaB
Nuclear factor-kappaB (NF-κB) consists of a dimer of the p65 and p50 proteins. In the inactivated state, NF-κB is kept in the cytoplasm complexed with the inhibitory protein inhibitor of kappaB (IκBα). Extracellular stimuli such as TNF, reactive oxygen species and cytokines can activate many transmembrane receptors which can then activate IκB kinase which then phosphorylates the inhibitory protein IκBα. After phosphorylation, IκBα becomes ubiquitinated and then degraded. The NF-κB transcription factor then translocates to the nucleus to turn on protein expression Citation[142]. Aberrant regulation of NF-κB has been linked to cancer, inflammatory disease, septic shock and autoimmune disease Citation[142,143]. Recently, TDP-43 was shown to interact with the p65 subunit of NF-κB in mouse microglia cells after LPS simulation Citation[144]. From a functional perspective, a gene reporter assay was used to show that TDP-43 acts as a coactivator of p65. Furthermore, this interaction was shown to occur in the context of human ALS patients where TDP-43 and p65 were coimmunoprecipitated from spinal cord extracts of ALS patients but not from spinal cord extracts of control patients. Consistent with an inflammatory component of ALS, it was also demonstrated that TDP-43 overexpression in glia or macrophages causes hyperactive inflammatory responses following a LPS challenge. Treatment of TDP-43 transgenic mice with Withaferin-A (WA), an NF-κB inhibitor, significantly improved motor function as demonstrated by enhanced rotorod performance and WA-treated mice had a 40% reduction in the number of partially denervated neuromuscular junctions Citation[144]. Corroborating the results from this study, high levels of circulating TNFα have also been observed in other cohorts of ALS patients and in other animal models of ALS Citation[145-148]. Using spinal cord organotypic cultures, it was shown that TNFα potentiates glutamate-induced motor neuron cell death which was blocked by inhibition of the NF-κB pathway Citation[149]. Thus, chronic NF-κB activation may be a contributing factor to ALS disease and or disease progression and as such is a relevant therapeutic target. There are many approaches to successfully inhibit the NF-κB pathway with small molecules and these have been thoroughly reviewed elsewhere Citation[150,151]. The advancement of these compounds toward clinical testing for ALS may offer hope to patients in the future.
3.7 P38α
The P38α is a member of the mitogen-activated protein kinase (MAPK) family of serine/threonine kinases. The p38α is widely expressed in endothelial, inflammatory cells, microglia and in the spinal cord Citation[152]. The p38α is activated in response to stressful stimuli such as osmotic stress, oxidative stress, LPS, growth factors and inflammatory cytokines (TNFα, IL-1β, IL6) Citation[152,153]. The p38 is activated in motor neurons and microglia of SOD1G93A mice Citation[154-157] and in ALS patients Citation[157,158]. The p38α directly phosphorylates NFs and aberrant accumulation of phosphorylated NFs are hallmark pathological features of ALS Citation[157]. Deficits in axonal transport are an early event observed in the disease course of SOD1 mutant transgenic mice Citation[159]. Additional substrates for p38α include the heavy chain of the molecular motor kinesin Citation[160]. In a series of studies, it was shown that one functional consequence of p38-mediated phosphorylation of kinesin was to inhibit fast axonal transport, an effect that was blocked by pharmacological p38 inhibition Citation[160,161]. Of interest, p38 inhibition had a marked effect on motor neuron survival in the SOD1G93A mouse model, yet only elicited a modest effect on survival Citation[162]. It has been shown that riluzole protects against glutamate-induced deficits in NF axonal transport Citation[163,] and it is therefore tempting to speculate that a combination of riluzole with a p38 inhibitor may work synergistically to reverse axonal transport deficits. There are a number of p38 inhibitors that have now entered into clinical testing for inflammatory indications and these have been reviewed extensively elsewhere Citation[164,165]. It would be of interest to understand if a p38 inhibitor in combination with riluzole could provide significant benefit to ALS patients.
3.8 Activation of autophagy and the UPS for protein clearance
The UPS and autophagy/lysosome pathways are the two major cellular systems that degrade and clear proteins. In ALS, cytoplasmic inclusions consisting of hyperphosphorylated neurofilaments, TDP-43 and other proteins are a hallmark of the disease Citation[61,62]. Clearance of these protein inclusions could be helpful in ameliorating symptoms of the disease as we do not know for certain whether inclusions are directly toxic themselves or represent an attempt to package aberrantly folded proteins that may not be cleared otherwise, into an innocuous compartmentalized form. Proteasomes predominantly degrade shorter half life nuclear and cytoplasmic proteins that need to be unfolded. In contrast, lysosomes degrade larger membrane proteins, oligomers and aggregates, and organelles in a process called autophagy. When proteins oligomerize, aggregate and form large inclusions, they become inaccessible to the proteasome. HD is a neurodegenerative disorder caused by a long CAG repeat in the Huntington (Htt) protein. Thus this mutant protein contains an abnormally long polyglutamine tract where after cleavage of the mutant protein, fragments containing the polyglutamine have a tendency to aggregate, forming inclusions. These inclusions, which are a hallmark of HD, can interfere with neuronal function, ultimately leading to cell death Citation[166]. It was shown that inhibition of mTOR by rapamycin induces autophagy and reduces toxicity of mutant Htt in fly and mouse models of HD Citation[167]. In addition, a screen for autophagy enhancers was performed which identified a number of small molecule modulators of autophagy Citation[168]. It remains to be determined whether a similar approach will yield benefits in ALS. However, evidence of the relevance of enhancing autophagy as a potential therapeutic approach for ALS comes from a report by Hetz et al. Citation[33]. In this study, they investigated the contribution of X-box binding protein-1 (XBP-1), a transcription factor involved in the UPR, for its contribution to FALS. They observed that knockdown of XBP1 in NSC34 cells transiently transfected with mutant SOD enhanced clearance of mutant SOD aggregates by macroautophagy Citation[33]. This finding in a cellular system was validated in vivo where genetic knockdown of XBP1 in female mutant SOD mice prolonged survival by 10 days and increased autophagy and SOD1 degradation Citation[33]. Thus, we look forward to the results of testing small molecule enhancers of autophagy in ALS animal models.
It has similarly been postulated that enhancing proteasomal clearance of proteins would have broad benefit across neurodegenerative diseases. Proteasomal degradation is a complex and highly regulated process. Proteins are brought to the proteasome after ubiquitination for degradation. Once at the proteasome, proteins must be deubiquitinated and unfolded before entry into the proteasome. The deubiquitinating function is performed by a family of proteins termed deubiquitinating enzymes (DUBs). There are three resident DUBs that are associated with the proteasome: Usp14, Uch37 and Rpn11. Usp14 has an ubiquitin chain trimming activity which in biochemical experiments slowed down proteasomal degradation of ubiquitinated protein substrates Citation[169]. In addition, it was shown that small molecule inhibitors of Usp14 enhanced the clearance of TDP-43, Ataxin-3 and other proteins of relevance for neurodegenerative diseases in cellular assays Citation[169]. It remains to be seen whether small molecule inhibitors of Usp14 will be efficacious in in vivo models of neurodegeneration.
3.9 Phenotypic screens aimed at identifying novel ALS targets
Despite some potentially promising clinical candidates currently under evaluation and some of the new promising drug targets listed above, a lack of a detailed understanding of the underlying mechanisms that cause ALS has hampered drug discovery. Cellular phenotypic screens can help facilitate the identification of new drug targets for ALS. Phenotypic screening is a type of screening used in drug discovery to identify small molecules that alter a cellular phenotype such as neurite outgrowth, expression of a particular protein or cell death. In a blinded phenotypic screen of 1040 FDA-approved drugs, it was discovered that many β-lactam antibiotics are potent stimulators of the glutamate transporter, EAAT2 (also called GLT1) expression Citation[170]. Glutamate uptake by the transporter is one of the major modes of inactivation of glutamate signaling and thus increased expression of this transporter may be of benefit for ALS. Ceftriaxone, one of the β-lactam antibiotics identified in that study, elevated GLT1 expression threefold in cellular models and also raised GLT1 expression in rodents. In SOD1G93A mice, administration of Ceftriaxone significantly spared motor neurons and had a modest effect on prolonging survival Citation[170]. In a Phase II study for ALS, Ceftriaxone demonstrated good tolerability and is now in an ongoing Phase III study Citation[171].
Similarly, cellular phenotypic screens can be used to identify compounds that enhance vesicle trafficking, RNA processing and clearance of aberrantly folded proteins. Not only can previously approved drugs be used in these screens, but screening could also be performed on larger and more diverse collections of compounds. One limitation of phenotypic screens is the lack of identification of the molecular drug target. This lack of understanding may make a traditional medicinal chemistry-based structure activity relationship (SAR) effort more difficult. However, novel chemical proteomics technologies have emerged that could help in target deconvolution following a phenotypic screen Citation[172]. Stable isotope labeling for cell culture (SILAC) is one such technique based on mass spectrometry that allows for identification and quantification of proteins. Of relevance for identifying targets from phenotypic screens, using SILAC, cells can be metabolically labeled with different isotopes of arginine and lysine. The compound of interest can then be chemically tethered to sepharose beads and added to the cellular lysate. Specifically bound proteins can be identified, and their K d values determined using relative quantitative mass spectrometry. Although careful controls must be considered with this technique, it has been successful in identifying novel binding targets of small molecule compounds.
One potential limitation in the development of new therapeutics targeting ALS is the observed discordance between interventions that demonstrate activity in ALS animal models and efficacy in the clinical setting. The reasons for this discrepancy are manyfold and have been highlighted elsewhere and are therefore not in the scope for this review Citation[173]. As a potential mechanism for mitigating this issue, a drug repositioning approach has been suggested Citation[173]. This approach would test as many FDA approved drugs as possible, in a small number of patients to observe if a marked clinical benefit was noted in individual patients. Endpoints in such a trial would include improvement of strength or cessation of disease progression. Using cellular phenotypic screens or directly testing a number of previously approved drugs in a smaller number of ALS patients may decrease the overall time to realize success in the search for new therapeutics to treat this disease.
4. Conclusions
Numerous mechanisms have been described that may contribute to the pathogenesis of ALS. The primary target of degeneration in ALS, the motor neuron, is large and has a high metabolic requirement. Motor neurons are exposed to a high level of oxidative stress as a result of primary signaling using excitatory neurotransmitters; yet these neurons appear particularly vulnerable in that additional protective mechanisms to counterbalance this stress appear to be lacking. Many motor neurons do not express high levels of calcium-buffering proteins which likely also contribute to their susceptibility in ALS.
Identification of mutations in TDP-43, FUS and most recently C9Orf72 that cause FALS have changed the way the field thinks about this disease from a mechanistic and clinical perspectives. There are currently 15 described ALS gene subtypes () and many more unnamed genes that contribute to FALS. As this field of study progresses, it appears probable that many more genes will be categorized in the near future. Many of the ALS-associated genes can be broadly categorized into those that effect RNA processing, vesicular trafficking and oxidative stress. The recently discovered FALS gene PFN1 has illustrated a new mechanism of motor neuron degeneration. As additional genes are identified, these will further help to advance our understanding of ALS in the future.
As described above, there are a number of promising novel ALS drug targets on the horizon that may lead to breakthrough therapeutics. Genetic knockdown studies evaluating EphA4 and ASK1 kinases have significant biological effects in prolonging survival in the SOD animal models. Inhibition of HDAC6 has also shown efficacy in CMT animal models. New technology platforms such as relative quantitative mass spectrometry using SILAC will further help to deconvolute hits from phenotypic screens which should lead to additional ALS drug targets in the future, as well as provide further mechanistic understanding around this complex, multifactorial disease.
5. Expert opinion
There are many challenges facing researchers attempting to identify and develop new therapeutics for ALS. First, as demonstrated from ALS genetic studies, it is clearly a multifactorial disease with multiple potential underlying pathological mechanisms that can converge at the common endpoint of motor neuron degeneration. Despite the potential multifactorial nature of the pathogenesis of the disease, clinical studies typically utilize a heterogeneous group of ALS patients for clinical trials. Thus, it remains formally possible that compounds that have previously failed in clinical trials may exhibit efficacy in a small subset of ALS patients that are appropriately stratified. Further complicating the ability to translate nonclinical findings to effective treatments is the lack of universal concordance between results using current animal models and clinical outcomes. Given the large number of molecules that are reported to provide efficacy in nonclinical models, it may be argued that these models and/or the endpoints used for assessment of efficacy are set at a lower hurdle relative to clinical studies in patients. Another possibility that must be considered is whether the current clinical study paradigms are sufficiently sensitive and/or whether it is too late to therapeutically intervene with current treatment approaches once patients are symptomatic. Similar arguments have been forwarded in the treatment of other neurodegenerative disorders such as AD, where it has been suggested that earlier intervention may be needed to effectively impact the course of the disease. Treatment studies that allow for earlier intervention in the course of ALS, before unequivocal diagnosis, could result in an enhanced efficacy profile for the current standard of care, riluzole, as well as potentially allow compounds that have previously failed in clinical trials to demonstrate efficacy. In order to enable this approach, new biomarkers that can accurately predict disease progression need to be identified.
Despite the fact that genetics play a prominent role in ALS research, particularly with the discovery of mutations in TDP-43 and FUS that cause FALS and more recently with C9Orf72 and PFN1, these discoveries have not yet translated into new therapeutic approaches. New animal models such as the TDP-43 transgenic mouse should allow us to test new hypotheses and gain additional insight into mechanisms underlying ALS pathology. Such models may also prove to be more predictive for clinical efficacy. The zebrafish model of ALS allows for both small molecule and genetic screens directly in a medium throughput mode in vivo. Such a model system provides the opportunity to screen low-to-medium-sized compound or siRNA libraries in a reasonable time frame. Drug screening using ALS patient-specific induced pluripotent stem cells represents an alternate, albeit exciting approach that has tremendous potential from both a research and clinical perspective. Use of stem cells in this manner could allow for the high-throughput screening of large chemical libraries to identify compounds of relevance. Once a target is identified, the combination of traditional drug discovery approaches such as structure-guided drug design on compelling new targets like EphA4, ASK1, Nrf2 and HDAC6 with phenotypic screening approaches holds the promise to enhance the rate at which new drug targets for ALS are discovered and translated into clinical testing paradigms. Phenotypic screening historically has been less well utilized due to the fact that it is often difficult to deconvolute the molecular target for compounds of interest that emerge after a screening campaign. However, with new technologies such as relative quantitative mass spectrometry using SILAC, the ability to identify a compound's cellular-binding partners in a reasonable time frame is significantly enhanced. Phenotypic screens with endpoints focused on protein clearance, RNA processing, vesicle trafficking and motor neuron survival have the potential to result in multiple new targets for ALS.
Finally, the growing emphasis on drug repositioning approaches for the treatment of ALS represents a significant opportunity to expedite possible treatments to patients. Testing previously FDA-approved compounds in smaller numbers of ALS patients has the potential to lead to exciting new findings. Also, using combination therapy, taking advantage of pharmacological synergy, as is increasingly accepted as standard of care for cancer and hypertension, is an additional approach that should be considered. By combining the recent advances in genetics, chemical proteomics and informatics, we expect a number of new and exciting drug targets for this indication, and hope for ALS patients in the future.
Article highlights.
Overview of potential mechanisms underlying motor neuron susceptibility in ALS is provided.
Current genes linked to FALS including recently identified Profilin 1 and C9Orf72 are covered.
In-depth discussion of the use of phenotypic screens to identify new small molecules that could be of benefit in ALS and new technology useful in deconvoluting hits from phenotypic screens.
Discussion of the tractability, from a drug discovery perspective, of emerging targets for ALS such as EphA4, ASK1, DJ-1 and HDAC6.
Insight into the challenges facing those developing therapeutics for ALS and how some of these challenges may be mitigated from a clinical and research perspective is provided.
Acknowledgments
The authors thank E Johnson and D Walker for helpful discussions and Z Ren and R Artis for the critical reading of this manuscript. This manuscript is dedicated to the memory of L Gehrig and H Katz, may we all follow our dreams.
Declaration of interest
M Bova and G Kinney are both employees of ELAN Pharmaceutical.
Notes
This box summarizes key points contained in the article.
Bibliography
- del Aguila MA, Longstretch WT Jr, McGuire V, Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology 2003;60(5):813-19
- Lee MK, Cleveland DW. Neurofilament function and dysfunction:involvement in axonal growth and neuronal disease. Curr Opin Cell Biol 1994;6(1):34-40
- Manetto V, Sternberger NH, Perry G, Phosphorylation of neurofilaments is altered in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1988;47:642-53
- Itoh T, Sobue G, Ken E, Phosphorylated high molecular weight neurofilament protein in the peripheral motor, sensory and sympathetic neuronal perikarya: system-dependent normal variations and changes in amyotrophic lateral sclerosis and multiple system atrophy. Acta Neuropathol 1992;83(3):240-5
- Oosthuyse B, Moons L, Storkebaum Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 2001;28:131-8
- Shaw PJ, Ince PG, Falkous G, Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol 1995;38:691-5
- Abe K, Pan LH, Watanabe M, Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci Lett 1995;199:152-4
- Smith RG, Henry YK, Mattson MP, Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 1998;44:696-9
- Durham HD. In: Shaw PJ, Strong MJ, editors. Motor neuron disorders: bluebooks of practical aneurology. Butterworth-Heinemann; Philadelphia: 2003. p. 379-400
- Wang X, Michaelis EK. Selective vulnerability to oxidative stress in the brain. Front Aging Neurosci 2010;2(12):1-13
- Batulan Z, Shinder GA, Minotti S, High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 2003;23(13):5789-98
- Taylor DM, Minotti S, Agar JN, Overexpression of metallothionein protects cultured motor neurons against oxidative stress, but not mutant Cu/Zn-superoxide dismutase toxicity. Neurotoxicology 2004;25:779-92
- Kiernan D, Kalmar B, Dick JR, Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 2004;10(4):402-5
- Arimoclomol clinical trials information. Available from: www.Clinical.Trials.gov [Last accessed 24 October 2012]
- Williams TS, Day NC, Ince PG, Calcium permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann Neurol 1997;42:200-7
- Greig A, Donevan SD, Mujtaba TJ, Characterization of the AMPA-activated receptors present on motoneurons. J Neurochem 2000;74:179-91
- Van Damme P, Van Den BL, Van Houtte E, GluR2-dependent properties of AMPA receptors determines the selective vulnerability of motor neurons to excitotoxicity. J Neurophysiol 2002;88:1279-87
- Rothstein JD, Van Kammen M, Levey AI, Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 1995;38:73-84
- Ferraiuolo L, Kirby J, Grierson AJ, Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:616-30
- Carriedo SG, Sensi SL, Yin JH, AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in-vitro. J Neurosci 2000;20:240-50
- Lautenschlaeger J, Prell T, Grosskreutz J. Endoplasmic reticulum stress and the ER mitochondrdia calcium cycle in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2012;13:166-77
- Arnaudeau S, Kelley WL, Walsh JV, Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem 2001;276:29430-9
- Brini M, Carafoli E. Calcium pumps in Health and Disease. Physiol Rev 2009;89:1341-78
- Panov AV, Kubalik N, Zinchenko N, Metabolic and functional differences between brain and spinal cord mitochondria underlie different predisposition to pathology. Am J Physiol Regul Integr Comp Physiol 2011;300:R844-54
- Lodish HF, Kong N, Wikstrom L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J Biol Chem 1992;267:12753-60
- Kuznetsov G, Brostrom MA, Brostrom CO. Demonstration of a calcium requirement for secretory protein processing and export. Differential effects of calcium and dithiothreitol. J Biol Chem 1992;267:3932-9
- Grosskreutz J, Van Den Bosch L, Keller BU. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010;47:165-74
- Ince P, Stout N, Shaw P, Parvalbumin and calbindin D-28K in the human motor system and in motor neuron disease. Neuropathol Appl Neurobiol 1993;19:291-9
- Alexianu ME, Ho B-K, Mohamed AH, The role of calcium binding proteins in selective motor neuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol 1994;36:846-58
- Elliott JL, Snider WD. Parvalbumin is a marker of ALS-resistant motor neurons. Neuroreport 1995;6:449-52
- Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol 2000;247(Suppl):I17-27
- Roy J, Minotti S, Dong L, Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by post-synaptic calcium-dependent mechanisms. J Neurosci 1998;18:9673-84
- Hetz C, Thielen P, Matus S, XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009;23:2294-306
- Atkin JD, Farg MA, Walker AK, Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis 2008;30:400-7
- Ilieva EV, Ayala V, Jove M, Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007;30:3111-23
- Mizoule J, Meldrum B, Mazadier M, 2-amino-6-trifluoromethoxybenzothiazole, a possible antagonist of excitatory amino acid neurotransmission. I. Anticovulsant properties. Neuropharmacology 1985;24:767-83
- Habib AA, Mitsumoto H. Emerging drugs for amyotrophic lateral sclerosis. Expert Opin Emerg Drugs 2011;16(3):537-58
- Debono MW, Le Guern J, Canton T, Inhibition by riluzole of electrophysiological responses mediated by rat kainite and NMDA receptors expressed in Xenopus oocytes. J Pharmacol 1993;235:283-9
- Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996;47(Suppl4):S233-41
- Doble A, Hubert JP, Blanchard JC. Pertussis toxin pretreatment abolishes the inhibitory effect of riluzole and carbachol on D-[3H]-aspartate release from cerebellar granule cells. Neurosci Lett 1992;140:251-4
- Siciliano G, Carlesi C, Pasquali L, Clinical trials for neuroprotection in ALS. CNS Neurol Disord Drug Targets 2010;9(3):305-13
- Traynor BJ, Bruijn L, Conwit R, Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology 2006;67:20-7
- Bruijn LI, Cudkowicz M. Therapeutic targets for amyotrophic lateral sclerosis: current treatments and prospects for more effective therapies. Expert Rev Neurother 2006;6:417-28
- GSK1223249 clinical trials information. Available from: www.Clinical.Trials.gov [Last accessed 17 September 2012]
- Prinja R, Moore SE, Vinson M, Inhibitor of neurite outgrowth in humans. Nature 2000;403:383-4
- Oertle T, van der Haar ME, Bandtlow CE, Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci 2003;23(13):5393-406
- Freund P, Schmidlin E, Wannier T, Anti-Nogo-A antibody treatment promotes recovery of manual dexterity after unilateral cervical lesion in adult primates-re-examination and extension of behavioral data. Eur J Neurosci 2009;29:983-96
- Jokic N, de Aguilar J-LG, Dimou L, The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep 2006;7:1162-7
- Pradat P-F, Bruneteau G, de Aguilar J-LG, Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Ann Neurol 2007;62:15-20
- Russell AJ, Hartman JJ, Hinken AC, Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat Med 2012;3:452-6
- Shefner J, Cedarbaum JM, Cudkowicz ME, Safety, tolerability and pharmacodynamics of a skeletal muscle activator in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2012;13(5):430-8
- Schmalbach S, Petri S. Histone deacetylation and motor neuron degeneration. CNS Neurol Disord Drug Targets 2010;9(3):279-84
- Echaniz-Laguna A, Bousiges O, Loeffler JP, Histone deacetylase inhibitors: therapeutic agents and research tools for deciphering motor neuron diseases. Curr Med Chem 2008;15:1263-73
- Yoo Y-E, Ko CP. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 2011;231:147-59
- Ryu H, Smith K, Camelo SI, Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem 2005;93:1087-98
- Petri S, Kiaei M, Kipiani K, Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2006;22:40-9
- Del Signore SJ, Amante DJ, Kim J, Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler 2009;10:85-94
- Chuang D-M, Leng Y, Marinova Z, Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 2009;32:591-601
- Cudkowicz ME, Andres PL, Macdonald SA, Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler 2009;10:99-106
- Siddique T, Ajroud-Driss S. Familial ALS, a historical perspective. Acta Myol 2011;30:117-20
- Lagier-Tourenne C, Polymedidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010;19:R46-64
- Lagier-Tourenne C, Cleveland DW. Rethinking the fuss about TDP-43. Cell 2009;136:1001-4
- Tollervey JR, Curk T, Rogelj B, Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 2011;14(4):452-8
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Long pre-mRNA depletion and RNA missplicing contribute to the neuronal vulnerability from loss of TDP-43. Nat Neurosci 2011;14:459-68
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol 2011;21:904-19
- Ling S-C, Albuququerque CP, Han JS, ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. PNAS 2010;107:13318-23
- Kawauchi J, Mischo H, Braglia P, Budding yeast RNA polymerases I and II employ parallel mechanisms of transcriptional termination. Genes Dev 2008;22:1082-92
- Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human Senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011;42:794-805
- Skorupa A, King MA, Aparicio IM. Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J Neurosci 2012;32:5024-38
- Ticozzi N, Tiloca C, Morelli C, Genetics of familial amyotrophic lateral sclerosis. Arch Ital Biol 2011;149:65-82
- Boillee S, Velde CV, Cleveland DW. ASL: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52:39-59
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006;7:710-23
- Bush AI. Is ALS caused by an altered oxidative activity of mutant superoxide dismutase? Nat Neurosci 2002;5:919
- Hadano S, Benn SC, Kakuta S, Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet 2006;15:233-50
- Devon RS, Orban PC, Gerrow K, Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. PNAS 2000;103:9595-600
- Morotz GN, De Vos KJ, Vagnoni A, Amyotrophic lateral sclerosis-associated mutant VABP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet 2012;21(9):1979-88
- Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 2009;18(24):4868-78
- Nagabhushana A, Chalasani ML, Jain N, Regulation of endocytic trafficking of transferring receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol 2010;11(4):1-19
- Renton AE, Majounie E, Waite A, A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72(2):257-68
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res 2012;1462:3-15
- Wu C-H, Fallini C, Ticozzi N, Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012;488:499-505
- Horrevoets AJG. Profilin-1: an unexpected molecule linking vascular inflammation to the actin cytoskeleton. Circ Res 2007;101:328-30
- Suetsugu S, Miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J 1998;17:6516-26
- Daoud H, Dobrzeniecka S, Camu W, Mutation analysis of PFN1 in familial amyotrophic lateral sclerosis patients. Neurobiol Aging 2012;12: In press
- Hirai H, Maru Y, Hagiwara K, A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987;238:1717-20
- Qin H, Noberini R, Huan X, Structural characterization of the EphA4-Ephrin-B2 complex reveals new features enabling Eph-ephrin binding promiscuity. J Biol Chem 2010;285:644-54
- Kao T-J, Law C, Kania A. Eph and ephrin signaling: lessons learned from spinal motor neurons. Semin Cell Dev Biol 2012;23:83-91
- Chen Y, Fu AKY, Ip NY. Eph receptors at synapses: implications in neurodegenerative diseases. Cell Signal 2012;24:606-11
- Helmbacher F, Schneider-Maunoury S, Topilko P, Targeting of EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development 2000;127:3313-24
- Murai KK, Nguyen LN, Irie F, Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci 2003;6:153-60
- Bourgin C, Murai KK, Richter M, The EphA4 receptor regulates dendritic spine remodeling by affecting beta1-integrin signaling pathways. J Cell Biol 2007;178:1295-307
- Van Hoecke A, Schoonaert L, Lemmens R, EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 2012
- Noberini R, Koolpe M, Peddibhotla S, Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. 2008;283(43):29461-72
- Noberini R, Lamberto I, Pasquale EB. Targeting Eph receptors with peptides and small molecules: progress and challenges. Semin Cell Dev Biol 2012;23:51-7
- Farenc C, Celie PNH, Tensen CP, Crystal structure of the EphA4 protein tyrosine kinase domain in the apo- and dasatinib-bound state. FEBS Lett 2011;585:3593-9
- Oki M, Yamamoto H, Taniguchi H, Overexpression of the receptor-tyrosine kinase EphA4 in human gastric cancers. World J Gastroenterol 2008;14(37):5650-6
- Goldshmit Y, Spanevello MD, Tajouri S, EphA4 blockers promote axonal regeneration and functional recovery following spinal cord injury in mice. PLoS ONE 2011;6(9):e24636
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 2006;46:113-20
- Barber SC, Shaw PJ. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic Biol Med 2010;48:629-41
- Mimoto T, Miyazaki K, Morimoto N, Impaired antioxydative Keap1/Nrf2 system and the downstream stress protein responses in the motor neuron of ALS model mice. Brain Res 2012;1446:109-18
- Vargas MR, Johnson DA, Sirkis DW, Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 2008;28(50):13574-81
- Neymotin A, Calingasan NY, Willie E, Neuroprotective effect of Nrf2/ARE activators, CDDO-ethylamide and CDDO-trifluoroethylamide in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med 2011;51(1):88-96
- Pergola PE, Raskin P, Toto RD, Bardoxolone methyl and kidney function in CKD with Type 2 diabetes. NEJM 2011;365(4):327-36
- Bandyopadhyay S, Cookson MR. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol Biol 2004;4:6
- Bonifati V, Rizzu P, van Baren MJ, Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299:256-9
- Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal 2011;15:111-22
- Canet-Aviles RM, Wilson MA, Miller DW, The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. PNAS 2004;101:9103-8
- Im JY, Lee KW, Junn E, DJ-1 protects against oxidative damage by regulating the thioredoxin/ASK1 complex. Neurosci Res 2010;67:203-8
- Gorner K, Holtorf E, Waak J, Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J Biol Chem 2007;282:13680-91
- Kim YC, Kitaura H, Taira T, Oxidation of DJ-1 dependent cell transformation through direct binding of DJ-1 to PTEN. Int J Oncol 2009;35(6):1331-41
- Annesi G, Savettieri G, Pugliese P, DJ-1 mutations and Parkinsonism-Dementia-Amyotrophic Lateral Sclerosis complex. Ann Neurol 2005;58:803-7
- Kaji R, Izumi Y, Adachi Y, Kuzuhara S. ALS-Parkinsonism-Dementia complex of Kii and other related diseases in Japan. Parkinsonism Relat Disord 2012;18S1:S190-1
- Yamashita S, Mori A, Mita S, DJ-1 forms complexes with mutant SOD1 and ameliorates its toxicity. J Neurochem 2010;113:860-70
- Miyazaki S, Yanagida T, Nunome K, DJ-1 binding compounds prevent stress-induced cell death and movement defect in Parkinson's disease model rats. J Neurochem 2008;105:2418-34
- Macedo MG, Anar B, Bronner IF, The DJ-1L166P mutant protein associated with early onset Parkinson's disease is unstable and forms higher-order protein complexes. Hum Mol Genet 2003;12(21):2807-16
- Alvarez-Castelao B, Munoz C, Sanchez I, Reduced protein stability of human DJ-1/Park7 L166P, linked to autosomal recessive Parkinson disease, is due to direct endoproteolytic cleavage by the proteasome. Biochim Biophys Acta 2012;1823(2):524-33
- Wilson MA, Collins JL, Hod Y, The 1.1 A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. PNAS 2003;100(16):9256-61
- Ichijo H, Nishida E, Irie K, Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997;275:90-4
- Bunkoczi G, Salah E, Filippakopoulos P, Structural and functional characterization of the human protein kinase ASK1. Structure 2007;15:1215-26
- Saitoh M, Nishitoh H, Fujii M, Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998;17(9):2596-606
- Hu X, Weng Z, Chu CT, Peroxiredoxin-2 protects against 6-Hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci 2011;31(1):247-61
- Tobiume K, Matsuzawa A, Takahashi T, ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2(3):222-8
- Nishitoh H, Kadowaki H, Nagai A, ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 2008;22:1451-64
- Um JW, Im E, Park J, ASK1 negatively regulates the 26S proteasome. J Biol Chem 2010;285(47):36434-46
- Cuny GD. Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Des 2009;15:3919-39
- Personal communication from Marc Adler, based on homology modles of ASK2 and ASK3 built on the ASK1 structure 2CLQ
- Matsuzawa A, Saegusa K, Noguchi T, ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol 2005;6(6):587-92
- Norman P. Evaluation of WO2012003387, Gilead's ASK1 inhibitors. Expert Opin Ther Patents 2012;22(4):455-9
- Boyault C, Zhang Y, Fritah S, HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 2007;21:2172-81
- Matthias P, Yoshida M, Khochbin S. HDAC6 a new cellular stress surveillance factor. Cell Cycle 2008;7(1):7-10
- Li G, Jiang H, Chang M, HDAC6 alpha-tubulin deacetylase: A potential therapeutic target in neurodegenerative diseases. J Neurol Sci 2011;304:1-8
- Kim SH, Shanware NP, Bowler MJ, Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC-6 mRNA. 2010;285(44):34097-105
- Yu Z, Sawkar AR, Whalen LJ, Isofogamine- and 2,5-Anhydro-2,5-Imino-D-Glucitol-based Glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J Med Chem 2007;50(1):94-100
- Hubbert C, Guardiola A, Shao R, HDAC6 is a microtubule-associated deacetylase. Nature 2002;417:455-8
- Boyault C, Sadoul K, Pabion M, HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007;26:5468-76
- Zilberman Y, Ballestrem C, Carramusa L, Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci 2009;122:3531-41
- Rivieccio MA, Brochier C, Willis DE, HDAC6 is a target for protection and regeneration following injury in the nervous system. PNAS 2009;106(46):19599-604
- d'Ydewalle C, Krishnan J, Chiheb DM, HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011;17(8):968-75
- Zhang Y, Kwon S, Yamaguchi T, Mice lacking histone deacetylase 6 have hyperphosphorylated tubulin but are viable and develop normally. Mol Cell Biol 2008;28(5):1688-701
- Butler KV, Kalin J, Brochier C, Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J Am Chem Soc 2010;132(31):10842-6
- Inks ES, Josey BJ, Jesinkey SR, A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol 2012;7:331-9
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006;25:6680-4
- DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev 2012;246:379-400
- Swarup V, Phaneuf D, Dupre N, Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med 2011;208(12):2429-47
- Babu GN, Kumar A, Chandra R, Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem Res 2008;33:1145-9
- Cereda C, Baiocchi C, Bongioanni P, TNF and sTNFR1/2 plasma levels in ALS patients. J Neuroimmunol 2008;194:123-31
- Poloni M, Facchetti D, Mai R, Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett 2000;287:211-14
- Ghezzi P, Bernardini R, Giuffrida R, Tumor necrosis factor is increased in the spinal cord of an animal model of motor neuron degeneration. Eur Cytokine Netw 1998;9:139-44
- Tolosa L, Caraballo-Miralles V, Olmos G, Llado J. TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB. Mol Cell Neurosci 2011;46:176-86
- Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta 2010;1799:775-87
- Kwak J-H, Jung J-K, Lee H. Nuclear factor-kappa B inhibitors: a patent review (2006-2010). Expert Opin Ther Patents 2011;21(12):1897-910
- Schieven GL. The p38alpha kinase plays a central role in inflammation. Curr Top Med Chem 2009;9(11):1038-48
- Coulthard LR, White DE, Jones DL, p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol Med 2009;15(8):369-79
- Hu JH, Chernoff K, Pelech S, Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem 2003;85:422-31
- Tortarolo M, Veglianese P, Calvaresi A, Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci 2003;23:180-92
- Xu L, Guo Y-S, Liu Y-L, Oxidative stress in immune-mediated motoneuron destruction. Brain Res 2009;1302:225-32
- Ackerley S, Grierson AJ, Banner S, p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci 2004;26:354-64
- Hu JH, Zhang H, Wagey R, Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem 2003;85:432-42
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 1999;2:50-6
- Morfini GA, Burns M, Binder LI, Axonal transport defects in neurodegenerative diseases. J Neurosci 2009;29(41):12776-86
- Bosco DA, Morfini G, Karabacak NM, Wild type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 2010;13(11):1396-403
- Dewil M, dela Cruz VF, Van Den Bosch L, Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1G93A-induced motor neuron death. Neurobiol Dis 2007;26:332-41
- Stevenson A, Yates DM, Manser C, Riluzole protects against glutamate-induced slowing of neurofilament axonal transport. Neurosci Lett 2009;454:161-4
- Yasuda S, Tanaka H, Takigami S, Yamagata K. p38 MAP kinase inhibitors as potential therapeutic drugs for neural disease. Cent Nerv Syst Agents Med Chem 2011;11(1):45-59
- Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem 2010;53:2345-53
- Bano D, Zanetti F, Mende Y, Nicotera P. Neurodegenerative processes in Huntington's disease. Cell Death Disease 2011;2:e228
- Ravikumar B, Vacher C, Berger Z, Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington's disease. Nat Genet 2004;36(6):585-95
- Sarkar S, Peristein EO, Imarisio S, Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol 2007;3(6):331-8
- Lee B-H, Lee MJ, Park S, Enhancement of proteasome activity by a small-molecule inhibitor of Usp14. Nature 2010;467:179-87
- Rothstein JD, Patel S, Regan MR, beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005;433:73-7
- Ceftriaxone clinical trial information. Available from: www.ClinicalTrials.gov [Last accessed 25 October 2012]
- Sharma K, Weber C, Bairlein M, Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat Meth 2009;6(10):741-4
- Glass JD. New drugs for ALS: how do we get there? Exp Neurol 2012;233(1):112-17