Abstract
Introduction: Hairy cell leukemia (HCL) is a rare leukemia derived from mature B lymphocytes. Advances in the understanding of its pathogenesis have translated into the development of effective therapies. Interferon was the first agent that showed activity in HCL. However, most responses were partial and after stopping the drug, relapses occurred. The introduction of 2-deoxycoformycin (DCF) and 2-chlorodeoxyadenosine (CDA) represented a major progress in the management of HCL. Responses to these agents are seen in 95% of patients and > 75% are durable complete responses. However, 40% of patients relapse and although a second response may be achieved, this is of shorter duration. In such setting, strategies using Rituximab targeting CD20 combined with DCF or CDA and/or immunotoxins such as the anti-CD22 McAb linked to the truncated pseudomonas exotoxin have shown promising results in relapsed patients. The recent discovery of the presence of BRAF mutations in HCL has opened avenues for targeting this deregulated gene with BRAF inhibitors.
Areas covered: This manuscript summarises the disease features, pathogenesis, diagnosis and current and new targeted treatments for HCL. A literatura search on pub med has been undertaken and the most relevant references have been considered.
Expert opinion: HCL is a potentially curable disease with the available treatments. DCF or CDA should be the standard first line treatment. Patients with suboptimal responses to these drugs or multiple relapses should be treated with Rituximab plus DCF or CDA or immunotoxins.
1. Introduction
Hairy cell leukemia (HCL) is a distinct disease entity defined by its clinical, morphological, pathological, immunological and molecular features Citation[1]. In the 1970s the therapeutic options for HCL were very limited as these patients were refractory to the available drugs used in other chronic lymphoid leukemias and lymphomas such as chronic lymphocytic leukemia (CLL) or follicular lymphoma. Splenectomy was the sole therapeutic approach that was partially effective in a subset of HCL patients in whom bone marrow infiltration was mild and cytopenias were mainly due to hypersplenism. Over the last three decades, a significant progress in the treatment of HCL has been made as a result of the introduction of drugs such the purine analogs 2´deoxycoformycin (DCF) and 2´chlorodeoxyadenosine (CDA) that have shown a remarkable activity in this disease. The use of DCF or CDA has contributed to change the natural history of HCL. Further to this, new targeted agents directed against cell surface antigens conjugated or not to toxins have emerged Citation[2,3]. These treatments are commonly used in the small proportion of patients who are refractory to the purine analogs or who experience only partial responses (PR) or multiple relapses. An excellent review on the efficacy of the various treatments in HCL has been published by Grever and Lozanski Citation[4]. Such progress has not been taken place in the variant form of HCL (HCL-variant), a disease that will not be discussed here since, although it shares some pathological features with HCL, it is at present considered biologically unrelated to the classical form Citation[5-7].
The present therapies available for HCL, the potential use of other drugs and novel therapeutic avenues for this disease would be discussed here. In order to better understand the management of these patients, the use and efficacy of the drugs and development of targeted treatments, we will also briefly describe the clinical manifestations and pathogenic pathways underlying the development or progression of HCL.
2. Hairy cell leukemia
2.1 Clinical features
HCL is a clinicopathological entity with distinct clinical and pathological features. It was first reported by Ewald in 1923 but described as a pathological entity by Bertha Bouroncle in 1958 under the name of leukemic reticuloendotheliosis Citation[8]. HCL occurs preferentially in middle age males with a male:female ratio of 5:1 and manifests with splenomegaly, pancytopenia and bone marrow infiltration. Cytopenias, particularly monocytopenia, are the most common manifestations but other features such as vasculitis, autoimmune hemolytic anemia or bone lytic lesions may be present in a minority of patients. The main sites of disease are the spleen and bone marrow, while peripheral lymphadenopathy is rare. Abdominal lymphadenopathy may be present in ∼20% of patients, particularly during the evolution of the disease Citation[9]. These patients tend to have some resistance to treatment with purine analogs Citation[10] and therefore a whole body computerized tomography (CT scan) should be considered in the work-up to estimate the extent of the disease.
2.2 Laboratory features
The morphology, the immunophenotypic makeup of the neoplastic cells and the histology of the bone marrow and spleen are characteristic of this disease. Hairy cells are of medium size, with an oval or kidney-shaped rarely convoluted nucleus with a cotton–wool chromatin pattern and an abundant pale cytoplasm with irregular borders or hair-like projections (). Immunophenotyping by flow cytometry shows that hairy cells are mature clonal B-lymphocytes that express a single immunoglobulin (Ig) light chain and that are preferentially IgG positive or usually express multiple Ig heavy chains. They express at a high density the antigens recognized by the monoclonal antibodies (McAb): CD19, CD20, CD22 and FMC7, while CD5 and CD23 are often negative. Expression of the antigens recognized by the McAb CD11c, CD25 against the Interleukin (IL)-2 receptor, CD103 and CD123 (anti-IL3 receptor) is characteristic of HCL () Citation[11,12].
Figure 1. May Grunwald Giemsa stained peripheral blood film from a patient with HCL showing circulating hairy cells.
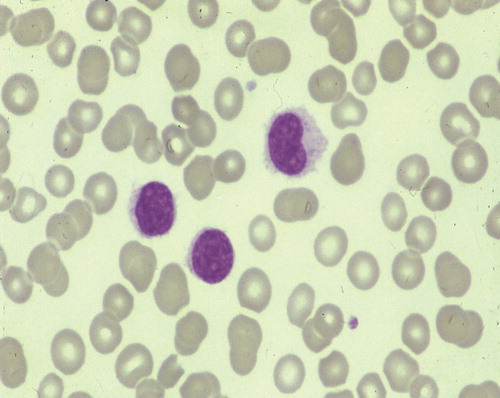
Table 1. Laboratory features of HCL and other splenomegalic B-cell disorders with villous lymphocytes.
Examination of the bone marrow trephine biopsy is essential for the diagnosis and assessment of response following therapy. The bone marrow often cannot be aspirated because of an increase in reticulin fibers (dry-tap bone marrow). If the bone marrow is aspirable, hairy cells may be seen admixed with hemopoietic precursors. In most patients, the bone marrow is hypercellular and the pattern of infiltration by hairy cells in the trephine biopsy is variable ranging from subtle to diffuse. The so-called fried-egg pattern, leaving clear spaces around the cells, is typical of this disease. In some cases, there is only a mild intrasinusoidal and/or interstitial bone marrow involvement that can be overlooked if immunohistochemical techniques are not carried out. Markers that are useful in tissue sections to detect hairy cells are Annexin A1, DBA44 and the McAb that detects tartrate-resistant acid phosphatase (TRAP). Of these, the most specific for lymphoid disorders is Annexin A1 Citation[13]. Hairy cells in most cases weakly express cyclin D1 without evidence of the translocation t(11;14)(q13;q32) Citation[14]. The spleen histology is characteristic of HCL with exclusive involvement of the red pulp and a naked white pulp. Pseudolake formation and extravasation of red cells are frequently seen.
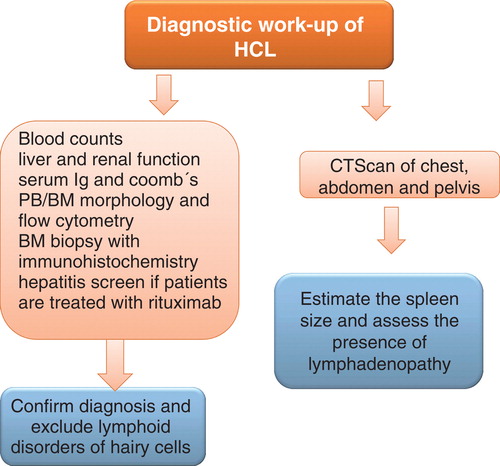
A diagnostic workup on a patient with suspected HCL is shown in .
Figure 2. Diagnostic work up of a patient with HCL.
2.3 Molecular genetics
HCL is not characterized by a recurrent chromosomal abnormality. Chromosomal abnormalities often involve the chromosome 5 with deletions or pericentric inversion, chromosome 7 and chromosome 14 at 14q32, the locus for the Ig heavy chain. High resolution genomic profiling in HCL, comparing it to HCL-variant, has demonstrated in both diseases a number of chromosome gains and losses, namely in chromosomes 5 and 7q Citation[15]. However and in contrast to HCL-variant, losses at 17p – a region that codes for the tumor suppressor gene TP53 or mutations of this gene – were not found in HCL and have rarely been reported in this disease Citation[16]. The high frequency of TP53 abnormalities in HCL-variant may well underline its refractoriness to agents that are very effective in HCL.
The putative cell of origin in HCL where the neoplastic transformation has probably taken place has been described in a number of studies and these suggested that transformation occurs in a post-germinal center memory cell. Most HCL cases have somatic mutations of the IGHV gene; unlike HCL-variant or splenic marginal zone lymphoma (SMZL), hairy cells lack specific IGHV/IGHD/IGHJ repertoires or stereotypes Citation[17]. Microarray gene profiling has shown that HCL has a unique gene signature. Cyclin D1, annexin A1, IL-3 receptor and FLT-3 are upregulated and a variety of other genes are deregulated Citation[18]. Although not yet explored, agents that target FLT-3 could be potentially used in the treatment of refractory cases. A recent exciting finding is the presence of mutations on the BRAFV600E gene in virtually all HCL cases investigated, while mutations of this gene are very rare in other lymphoid disorders with only occasional case of SMZL, CLL or B-cell prolymphocytic leukemia (B-PLL) being documented Citation[19-22]. Only one study documented that a significant minority of HCL cases are wild type for the BRAF gene Citation[23]. However, most of these cases had IGHV4-34 rearrangement – a feature more characteristic of HCL-variant and not of HCL. The BRAFV600E mutation has been shown to be involved in the pathogenesis of various cancers such as melanoma and thyroid cancer and it appears to be the case in HCL. The mutation of the gene activates the Raf/MEK-ERK pathway providing an enhanced proliferation and longer survival to the cells. New and more feasible methods to be carried out on a routine basis detecting the mutation have been described such as quantitative real-time polymerase chain reaction (PCR) Citation[22] and immunohistochemistry with a McAb that selectively detects the BRAFV600E gene when is mutated Citation[21.] These latter techniques not only have a diagnostic potential but also may be useful to assess the quality of response following therapy by allowing to detect minimal residual disease (MRD). There are ongoing trials using inhibitors of the BRAFV600E gene in patients with BRAF mutated metastatic melanoma. In HCL this could also be a potential therapeutic target in patients who have a suboptimal response or are refractory to purine analogs or immunotherapy.
2.4 Differential diagnosis ()
The differential diagnosis of HCL mainly arises with other splenomegalic lymphoid disorders and particularly with those with circulating villous lymphocytes such as SMZL and splenic B-cell lymphoma/leukemia unclassifiable (HCL-variant and splenic diffuse red pulp small B-cell lymphoma). Monocytopenia unlike in HCL is not a feature of these diseases that in addition have a morphology and immunophenotype different from HCL. The pattern of bone marrow and spleen involvement in HCL is also different from that seen in SMZL although it overlaps with that of splenic B-cell lymphomas unclassifiable. Rarely the differential diagnosis of HCL arises with aplastic anemia in patients with a hypocellular bone marrow, myelofibrosis in those with subtle bone marrow involvement or mast cell disease.
2.5 Prognostic factors and outcome
The outcome of HCL patients has significantly changed due to the availability of effective therapies and better knowledge of the biology of this disease. This has resulted in median survivals of > 20 years compared to < 5 years in the 1980s. A number of clinical parameters shown to be predictors for a poor outcome are age, anemia or thrombocytopenia, splenomegaly, shorter time from diagnosis to treatment, lower performance status and failure to achieve a complete response (CR) to purine analogs. Citation[24-31]. However, it needs to be considered that most of these predictive parameters were established in the past before effective treatments were available. Patients with bulky abdominal lymphadenopathy tend to have suboptimal responses to purine analog therapy Citation[10]. One study has suggested that the uncommon HCL cases with IgHV4-34 usage or with unmutated IgVH are those that more often do not respond to the purine analog CDA Citation[16,32]. The very minority of HCL that have TP53 abnormalities may also have an unfavorable outcome.
2.6 Treatment
2.6.1 Indications
Treatment is indicated in patients with symptomatic disease as manifested by cytopenias, bulky or progressive splenomegaly, recurrent infections and/or systemic symptoms. A minority of patients is asymptomatic at diagnosis and a policy of watch and wait can be adopted – such is the case of asymptomatic stage A CLL patients.
The progress and landmarks of treatment in HCL are summarized in .
Table 2. Progress of treatments in HCL.
2.6.2 Splenectomy
In the past, splenectomy was the only effective therapeutic modality in HCL, particularly in those patients with bulky spleens and minor bone marrow involvement in whom cytopenias were mainly the result of hypersplenism. These patients achieve hematological responses following splenectomy and although no complete remissions are observed, a small proportion may not require treatment for many years Citation[33,34]. Splenectomized patients should receive vaccinations and prophylaxis with penicillin to prevent infections by encapsulated bacteria. Although at present there is no definitive role for splenectomy in HCL, patients with profound cytopenias and very bulky spleens might benefit from it prior purine analog therapy as it will facilitate the delivery of this drug.
2.6.3 Interferon-alpha
Quesada et al. first documented in 1984 the efficacy of Interferon-alpha (IFN-α) in HCL Citation[35]. Out of the seven patients treated with IFN-α, three achieved a CR and the remaining four achieved a PR. Subsequent studies in large cohorts of patients confirmed the activity of IFN-α in HCL Citation[25,36-41]. The mechanism of action of IFN-α in HCL is not fully understood, may be multiple and in part by inducing the production of cytokines and growth factors. In most studies IFN-α has been administered at a dose of 2 or 3 × 106 IU subcutaneously (s.c.) three times a week for 12 to 18 months. Main side effects are flu-like symptoms, gastrointestinal symptoms and more rarely neurological symptoms including depression or memory loss. Myelosuppression during the first 3 months is common and there may be impairment of the liver function tests. Although the overall response rate (ORR) to IFN-α is high ranging from 75 to 90%, most responses are partial and only a minority of patients (5 – 30%) achieves a CR with still detectable hairy cells in the bone marrow. Furthermore, discontinuation of the therapy is almost always associated to recurrence of the disease within 6 to 25 months. Patients who undergo maintenance therapy may experience a longer remission but the disease ultimately will recur or will become resistant to IFN-α. A prospective randomized trial comparing DCF with IFN-α has shown the superiority of DCF over IFN-α Citation[25]. The CR rate was 76% in patients treated with DCF versus 11% for patients treated with IFN-α. Therefore, this agent is no longer used as a first-line treatment in HCL considering the greater efficacy of purine analogs (see below) neither seems to add a benefit when used as a consolidation or maintenance in patients who achieved a response to DCF. Marotta et al. randomized 145 patients that had a response to DCF (CR or PR) to receive IFN-α or not. The results showed that there were no differences between the two arms regarding improvement of response or progression free of disease Citation[42].
2.6.4 Purine analogs
The introduction of the two purine nucleoside analogs first DCF by Spiers et al. in 1984 Citation[43] and subsequently CDA by Piro et al. in 1990 Citation[44] represented a remarkable advance in the treatment of HCL and changed the natural history of HCL. Therefore at present, purine analog is considered the first-line treatment in HCL. A number of reports including large number of patients treated with one of these two purine analogs have substantiated the early findings as described below.
2.6.4.1 DCF
This is a purine analog that inhibits the enzyme adenosine deaminase (ADA) that is essential to the purine metabolism in lymphoid cells. Inhibition of ADA results on the accumulation of deoxyadenosine triphosphate metabolites that likely are responsible for the cytotoxicity in hairy cells.
A number of clinical studies have shown the efficacy of DCF in HCL. Different schedules for delivery of this drug have been used such as 4 mg/m2/intravenously (i.v.) as a bolus every two or four weeks, 5 mg/m2 on two consecutive days every two weeks, or 4 mg/m2/weekly for 3 weeks and subsequent therapy every 8 weeks. The schedule of 4 mg/m2/i.v. every two weeks is currently adopted in the routine practice since it is shown to be effective and has less side effects than other schedules, particularly opportunistic infections.
The ORR in patients with HCL either untreated or previously treated ranges from 79 to 100% with a CR rate of 33 – 93% () Citation[24,25,29,30,45-53]. The responses are long lasting and if relapse occurs, the patient may achieve a second response to DCF and/or responds to the other purine analog CDA. There is no evidence for a cross-resistance to these two drugs. Differences in the ORR and quality of responses (CR vs PR) in the different studies may well relate to the suboptimal drug delivery and/or disease burden.
Table 3. Responses to deoxycoformycin in HCL.
2.6.4.2 CDA
CDA is a purine analog that, unlike DCF, is not an inhibitor of ADA. This drug is cytotoxic to lymphocytes as its accumulation in the cells creates a deoxynucleotide that induces DNA strand breaks and depletion of NAD and ATP.
Like DCF, a number of studies have demonstrated the efficacy of CDA in HCL Citation[24,26,27,31,54-61]. Different routes and schedules of administration of CDA have been used. In the first study by Piro et al., CDA was administered i.v. at a dose of 0.1 mg/kg/day for 7 days by continuous infusion as a single cycle. Other schedules employed have been 0.15 mg/kg i.v. weekly/ 6weeks, 3.4 mg/m2/day s.c. per 7 days and 0.14 mg/Kg/day s.c. per 5 days. A prospective randomized study on 132 HCL patients who received CDA daily for 5 days versus once a week for 6 weeks have not shown differences in toxicity between the two arms Citation[62]. However, data is not available on the duration of response using these two different schedules to deliver the drug.
The ORR to CDA in the different clinical studies is shown in . The ORR ranged from 75 to 100% and the CR rate from 75 to 98%. It is worth to note that patients that achieve a PR following one course of CDA may get a CR following a second course.
Table 4. Responses to CDA in HCL.
There have been no prospective randomized trials comparing DCF and CDA. This is likely due to the rarity of the disease and the fact that the two purine analogs are highly effective in HCL. However, from all data published it appears that there are no significant differences in terms of response duration, quality of response and ORR. Long-term follow-up studies have shown that responses to either drug are long lasting; however, it has also become apparent that there is no plateau for relapse-free survival and, in a long term, a substantial proportion of patients will relapse. Data for DCF shows an estimated disease-free survival at 10 years of 68.8 and 67% Citation[28,29] and for CDA of 63 and 64% at a median follow-up of 9 and 9.7 years Citation[27,63]. The study with a longer follow-up comes from Else et al. Citation[24] and shows that at 15 years, 47 and 48% of patients treated with DCF and CDA, respectively, will relapse. Although a second or even a third response can be achieved with either purine analog, the proportion of patients achieving a second or third CR is less than when these drugs are used as first line; despite of this, there are no differences in the duration of response when used as a second or third time. Therefore in such patients with one or more relapses and, particularly in those in whom the response is not long-lived, there is a need for modern therapies aimed to cure the disease as described below (see Sections 2.6.5 and 2.6.6).
2.6.4.3 MRD and adverse events
Two issues that need to be considered are the following: i) the prognostic impact of residual hairy cells in the bone marrow following a response to the purine analog in terms of disease free survival and ii) the adverse events derived from this treatment.
Despite of the high CR rate with these two drugs, a proportion of HCL patients may have low level of residual disease detected either by immunohistochemistry, multicolor flow cytometry or molecular studies with consensus primers PCR Citation[64-66]. There has been no consensus in the definition of MRD in patients with HCL. Large studies comparing the different methods to detect MRD and/or correlating with clinical outcome are lacking. The scenario of CR (MRD+) is different to that of PR in which residual leukemic cells are easily detected by conventional methods. A PR achievement predicts for a less durable response compared to CR Citation[24] and therefore it is important to continue with further therapy until a maximal response is obtained. In contrast, there are uncertainties concerning the clinical significance of the presence of MRD in HCL and whether its detection would predict for an early relapse. This has not been evaluated in prospective randomized studies including a large number of patients with a long follow-up and therefore results need to be taken with caution.
The two purine analogs result in a profound immunosuppression with lymphopenia and particularly affecting the CD4+ T-lymphocytes. Opportunistic infections and particularly reactivation of the varicella zoster virus may be seen. Although there has not been a study showing that antiviral prophylaxis with acyclovir and pneumocystis carinii prophylaxis (PCP) with cotrimoxazole may prevent these infections, it will be advisable to administer cotrimoxazole and acyclovir to all HCL patients during the treatment with the purine analog and 6 to 12 months after stopping therapy. Other potential side effects, which are very uncommon with these drugs, are renal failure and hypoplasia of the bone marrow that usually recovers in a long term.
2.6.5 Purine analog plus antibody therapy
These schedules include targeted therapies directed against cell surface antigens. Rituximab targeting the CD20 antigen has been used as a single agent in a few studies Citation[67,68]. However, results with Rituximab as single agent are less impressive that those obtained with single purine analog agent. In contrast to the purine analogs, responses to Rituximab are achieved in 30 – 50% of patients and less than 20% are CR. By contrast the combination of chemoimmunotherapy (Rituximab plus DCF or CDA) in patients who experience early relapse, multiple relapses or do not achieve a response to single agent purine analog demonstrates that this combination is highly effective Citation[69,70]. Else et al. reported the efficacy of a combination of DCF or CDA with Rituximab in eight multiple relapsed or refractory HCL patients with impressive results Citation[69]. An update of this study in 18 patients that had previously received one to six therapies has shown an ORR of 100%, a CR rate of 89% and that 16 of 18 patients treated with this combination remain in CR with a median follow-up of 36 months; one PR patient relapsed at 10 months Citation[71]. Therefore the vast majority of these patients achieve a long-lasting CR to this combination. However there are uncertainties as to whether which is the best schedule to deliver these drugs (combined or sequential) and whether this combination is superior to single purine analog when used as first line.
Single agent Fludarabine, another purine analog that has shown to have a high activity in CLL, has been rarely used in HCL given the efficacy of the two other purine analogs. However, a recent study by Gerrie et al. Citation[72] in 15 HCL patients with refractory/relapsed disease using a combination of Fludarabine and Rituximab has shown an ORR of 100% and, at a median follow-up of 35 months, 14 of the 15 patients maintain the response. The 5-year progression-free survival and overall survival are 83 and 89%.
2.6.6 Immunochemotherapy and immunotoxins
New therapeutic strategies, essentially recombinant immunotoxins conjugates have been developed for HCL patients with truly refractory disease. These are fusion proteins that link variable fragment domains (Fv) of an antibody directed to a B-cell antigen strongly expressed by hairy cells with the toxic portion of a toxin. The Fv of the antibody binds to the antigen in the cell surface and delivers the toxin into the cytosol where it exerts cytolytic effect. Citation[73-76]. Since 1999 several immunotoxins have been used in Phase I and Phase II trials in HCL and most if not all studies come from the National Cancer Institute in Bethesda (USA). One of the first drugs developed was the compound LMB-2 that fuses the Fv of the McAb anti-CD25 (anti-IL-2 receptor) with a 38KD truncated pseudomonas exotoxin. Although the anti-CD25 McAb does not internalize when delivered by its own, it does when conjugated to an exotoxin. A Phase I trial in four patients with resistant HCL showed responses in all of them including a long-lasting CR. Citation[77]. Another agent designated BL22 that is an immunotoxin conjugate that delivers the pseudomonas toxin into cells expressing CD22 was later developed. A Phase I trial using this agent at a dose of 3 to 50 µg/kg/every other day × 3doses in 31 patients with refractory HCL showed a CR rate of 61% increasing to 86% when a higher dose was used and a PR rate of 19% Citation[78]. The median duration of response was 36 months. Most common side effects were hypoalbuminemia, fatigue, edema and, in four patients, a reversible hemolytic uremic syndrome that required dialysis. Neutralizing antibodies occurred in 24% of patients A subsequent Phase II trial with this drug in 36 patients who had a short response duration (less than 12 months) or lasting for 1 – 4 years to CDA and/or were refractory to this purine analog showed a CR rate of 47% and a PR rate of 25% after two cycles of treatment. The ORR (CR and PR) was higher in patients who had non-bulky splenomegaly (spleens < 14 cm) compared to those with bulky spleens (> 20 cm) or those who did not have splenomegaly Citation[79]. A modified drug mutant of BL22, with mutations in the heavy chain complementary determinant region 3 (CDR3) designated moxetumomab pasudotox (HA-22), has been produced and shown to have an increased cytotoxic activity Citation[80,81]. A Phase I dose–escalation trial to assess the safety and response to this drug in 28 HCL patients who had multiple relapses has been conducted. The patients had two or more previous lines of chemotherapy. The compound was delivered at doses of 5, 10, 30, 40 and 50 µg/kg q.o.d. × 3 for 1 to 16 cycles. The median of cycles received was 4. The ORR was 83% and seen at all doses and the CR rate was 46%. Citation[82,83]. The latter were long-lasting with only one patient having relapsed within one year. Further, the profile was safe at all doses. Grades 1 – 2 hypoalbuminemia, edema, nausea and fatigue were documented in 25 – 64% of patients. Two patients had also grade 2 uremic hemolytic syndrome. Therefore moxetumomab pasudotox administered at a dose of 50 µg/kg/q.o.d. × 3 has activity in refractory HCL with a safe profile Citation[83].
New therapeutic avenues need to be developed for HCL. Thus, despite of the exquisite sensitivity of HCL to the available purine analog drugs, most of the patients will experience a relapse, and repeated courses of purine analogs might carry the risk of developing myelodysplasia or solid tumors. Drugs targeting the bone marrow microenvironment which may be the source of residual and/or refractory disease or those directed against genes that are deregulated in HCL, such as FLT-3 and BRAF, need to be considered in the therapeutic scenario. The rationale for employing a BRAF inhibitor has been established by Tiacci et al. Citation[19] that showed that in vitro treatment of hairy cells with a BRAF inhibitor causes dephosphorilation of MEK and ERK, thereby turning off the oncogenic Raf-MEK-ERK pathway. Recently, a HCL patient refractory to purine analog therapy responded to vemurafenib, a BRAFV600E inhibitor reinforcing that HCL is likely a kinase-dependent disease that can be treated with a targeted drug Citation[84].
3. Expert opinion
HCL is a rare distinct and well-defined clinicopathological entity for which very effective treatments have become available for the last three decades. Hence, the outcome and survival of these patients has dramatically changed from a median survival of < 5 years in the 1970s to > 20 years at present. Indeed the life expectancy of a patient with HCL is not different to the general population. Two issues that need to be considered when first dealing with a patient with HCL are the following: i) Do all patients require treatment at diagnosis? ii) Which is the best therapeutic option and how we need to assess response to the treatment? With regards to the first question, treatment may well be postponed in a small proportion of patients without cytopenias and who do not have or have mild splenomegaly. In my experience, some of these patients may never require treatment in their life span. Treatments are not harmless and therefore a policy of watch and wait with close monitoring of the blood counts is reasonable. Considering the second question, it is clear that the purine analogs DCF or CDA are the gold standard first-line treatment for this disease. The fact that there have been not randomized clinical trials comparing these two drugs makes difficult the choice of one or another as initial therapy. The response rates and quality and duration of responses appear to be similar although the form of delivery is different. DCF is given sequentially i.v. every 2 weeks over a period of months and has the advantage of the possibility of a more closer monitoring in terms of delaying or reducing a dose and/or giving growth factors if the patient experiences profound cytopenias (neutropenia) due to drug toxicity rather than disease activity. CDA is usually given i.v. or s.c. as a block during 5 or 7 days. The advantages are the easier delivery via the s.c. form and that only needs one or perhaps two courses. Since the efficacy of these two drugs is similar, all these practical issues should be discussed individually with the patient. Prophylaxis with acyclovir and cotrimoxazole is recommended all through the treatment and up to 6 – 12 months after stopping the drugs. It is also important to assess the response to the purine analogs adequately. A bone marrow trephine biopsy with immunohistochemistry following treatment is mandatory regardless a complete resolution of the cytopenias is achieved. This will allow assessing the quality of response and, if a significant disease is left (i.e., PR), it is important to pursue with the treatment in order to achieve the best quality of response (CR). A practical aspect is that the bone marrow assessment should be carried out 3 or 4 months after therapy since the purine analogs work along several months. Another issue is how we manage patients that despite an optimal treatment with the purine analog only achieve a PR, those that relapse and/or have truly refractory disease. There are various alternatives to approach these patients, as the scenarios are different. Patients who achieve a PR following the optimal doses of a purine analog might well benefit from Rituximab as a consolidation without increasing toxicity and resulting in the conversion of the PR to a CR. Although there have not been studies in a large cohort of these patients, it seems that this approach is safe and feasible. Patients who had achieved a durable response and of good quality to a purine analog and relapse, may well be retreated with the same or the alternative purine analog. However, if the initial response has been of short duration, < 2 years, they should be treated with a combination of a purine analog and Rituximab. There is evidence albeit in small series of patients that this combination results in a very high and durable CR rate. The minority of patients who are purine analog refractory should be enrolled, when possible, on clinical trials that use immunotoxin conjugates. Hopefully in a near future we will be able to use new agents targeting the mutated BRAF gene in refractory patients and it will not be surprising that clinical trials are set up to see whether this agent or immunotoxin conjugates when used as first line are even superior to the current standard therapies in HCL.
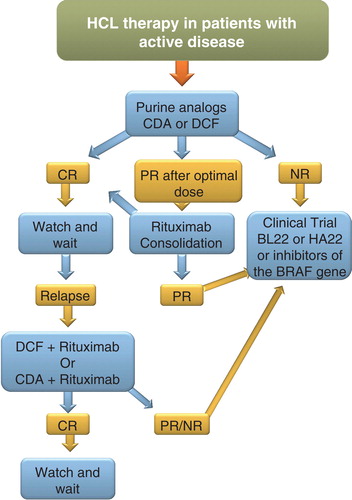
A flow chart on the management and therapeutic options in HCL is illustrated in .
Figure 3. Flow chart illustrating the treatment pathways in HCL.
Article highlights.
HCL has distinct clinical and laboratory features and a unique genetic signature.
Mutations of the BRAF gene play a role in the pathogenesis of HCL and can be exploited for targeted therapies.
The purine analogs DCF and CDA have a high efficacy in HCL and their use results in a high and durable CR.
Prophylaxis with acyclovir and cotrimoxazole is recommended in patients receiving treatment with purine analogs.
The clinical significance of MRD detection following treatment with purine analogs is uncertain.
HCL patients who are refractory to purine analogs, or who have multiple relapses, should be treated with chemoimmunotherapy (Rituximab plus DCF or CDA), immunotoxins (antibodies conjugated to toxins) and/or inhibitors of the BRAF gene.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Notes
This box summarizes key points contained in the article.
Bibliography
- Swerdlow SH, Campo E, Harris NL, editors. WHO classification of tumours of haematopoietic and lymphoid tissues IARC. IARC Press; Lyon: 2008
- Robak T. Novel drugs for chronic lymphoid leukemias: mechanism of action and therapeutic activity. Curr Med Chemistry 2009;16:2212-34
- Matutes E. Novel and emerging drugs for rarer chronic lymphoid leukaemias. Curr Cancer Drug Targets 2012;12:484-504
- Grever MR, Lozanski G. Modern strategies for hairy cell leukemia. J Clin Oncol 2011;29:583-90
- Matutes E, Wotherspoon A, Catovsky D. The variant form of hairy cell leukaemia. Best Pract Res Clin Haematol 2003;16:41-56
- Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico -pathological features of the variant form of hairy cell leukemia. Leukemia 2001;15:184-6
- Robak T. Hairy cell leukemia variant: recent view on diagnosis, biology and treatment. Cancer Treat Rev 2011;37:3-10
- Bouroncle BA, Wiseman BK, Doen CA. Leukemic reticuloendotheliosis. Blood 1958;13:609-30
- Mercieca J, Matutes E, Moskovic E, Massive abdominal lymphadenopathy in hairy cell leukaemia: a report of 12 cases. Br J Haematol 1992;82:547-54
- Mercieca J, Matutes E, Emmett E, 2-Chlorodeoxyadenosine in the treatment of hairy cell leukaemia: differences in response in patients with and without abdominal lymphadenopathy. Br J Haematol 1996;93:409-11
- Del Giudice I, Matutes E, Morilla R, The diagnostic value of CD123 in B-cell disorders with hairy or villous lymphocytes. Haematologica 2004;89:303-8
- Matutes E. Immunophenotyping and differential diagnosis of hairy cell leukemia. Hematol. Oncol. Clin. North Am 2006;20:1051-63
- Falini B, Tiacci E, Liso A, Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet 2004;363:1869-70
- Miranda RN, Briggs RC, Kinney MC, Immunohistochemical detection of cyclin D1 using optimized conditions is highly specific for mantle cell lymphoma and hairy cell leukemia. Mod Pathol 2000;13(12):1308-14
- Hockley S, Morgan GJ, Leone PE, High-resolution genomic profiling in hairy cell leukemia-variant compared with typical hairy cell leukemia. Leukemia 2011;25:1189-92
- Forconi F, Sozzi E, Cencini E, Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009;114:4696-702
- Hockley SL, Giannouli S, Morilla A, Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. Br J Haematol 2010;148:666-9
- Basso K, Liso A, Tiacci E, Gene expression profiling of hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J Exp Med 2004;199:59-68
- Tiacci E, Trifonov V, Schiavoni G, BRAF mutations in hairy-cell leukemia. N Engl J Med 2011;364:2305-15
- Blombery PA, Wong SQ, Hewitt CA, Detection of BRAF mutations in patients with hairy cell leukemia and related lymphoproliferative disorders. Haematologica 2012;97:780-3
- Andrulis M, Penzel R, Weichert W, Application of a BRAF V600E mutation-specific antibody for the diagnosis of hairy cell leukemia. Am J Surg Pathol 2012; Epub ahead of print
- Schnittger S, Bacher U, Haferlach T, Development and validation of a real-time quantification assay to detect and monitor BRAFV600E mutations in hairy cell leukemia. Blood 2012;119:3151-4
- Xi L, Arons E, Navarro W, Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012;119:3330-3
- Else M, Dearden CE, Matutes E, Long-term follow-up of 233 patients with hairy cell leukaemia treated initially with pentostatin or cladribine at a median of 16 years. Br J Haematol 2009;145:733-40
- Grever M, Kopecky K, Foucar MK, Randomized comparison of pentostatin versus interferon alpha in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol 1995;13:974-82
- Cheson BD, Sorensen JM, Montello MJ, Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine via the Group C protocol mechanism of the National Cancer Institute: a report of 979 patients. J Clin Oncol 1998;16:3007-15
- Goodman RG, Burian C, Koziol JA, Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol 2003;21:891-6
- Maloisel F, Benboubker L, Gardembas M, Long-term outcome with pentostatin treatment in hairy cell leukemia patients. A French retrospective study of 238 patients. Leukemia 2003;17:45-51
- Flinn IW, Kopecky KJ, Foucar MK, Long-term follow-up of remission duration, mortality, and second malignancies in hairy cell leukemia patients treated with pentostatin. Blood 2000;96:2981-6
- Rafel M, Cervantes F, Beltran JM, Deoxycoformycin in the treatment of patients with hairy cell leukemia: results of a Spanish collaborative study of 80 patients. Cancer 2000;88:352-7
- Saven A, Burian C, Koziol JA, Piro LD. Long term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood 1998;92:1918-26
- Arons E, Suntum T, Stetler-Stevenso M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood 2009;114:4687-95
- Coad J, Matutes E, Catovsky D. Splenectomy in lymphoproliferative disorders. A report on 70 cases and review of the literature. Leuk Lymphoma 1993;10:245-64
- Matutes E, Catovsky D. The role of splenectomy in hairy cell leukemia. Chapter 10 In: Tallman M, Polliack A, editors. Hairy cell leukemia. Harwood Academic Publishers; 2000. p. 127-39
- Quesada JR, Reuben J, Manning JT, Alpha interferon for induction of remission in hairy cell leukemia. N Engl J Med 1984;310:15-18
- Golomb H, Fefer D, Golde H, Sequential evaluation of alpha-2b interferon treatment in 128 patients with hairy cell leukemia. Semin Oncol 1987;14:13-17
- Golomb HM, Fefer D, Golde H, Report of a multiinstutional study of 193 patients with hairy cell leukemia treated with interferon alpha 2b. Semin Oncol 1988;15:7-9
- Castaigne S, Sigaux F, Degos G, Flandrin G. Hairy cell leukemia. Follow-up after interferon treatment. Nouv Rev Fr Hematol 1989;31:321-5
- Berman E, Heller G, Kempin S, Incidence of response and long term follow up in patients treated with recombinant interferon-alpha-2b. Blood 1990;75:839-45
- Rai K, Davey F, Peterson B, Recombinant alpha-2b interferon in therapy of previously untreated hairy cell leukemia. Long term results of a study by the Cancer and Leukemia Group B. Leukemia 1995;9:1116-20
- Golomb HM. Hairy cell leukaemia. Treatment successes in the past 25 years. J Clin Oncol 2008;26:2607-9
- Marotta G, Frassoldati A, Zinzani P, Italian Cooperative group for HCL (ICGHCL). Role of interferon-alpha administration after 2-deoxycoformycin in the treatment of hairy cell leukemia patients. Eur J Haematol 2006;77(2):109-13
- Spiers AS, Parekh SL, Bishop MB. Hairy cell leukaemia: induction of complete remission with pentostatin (2’-deoxycoformycin). J Clin Oncol 1984;2:1336-42
- Piro LD, Carrera CJ, Carson DA, Beutler E. Lasting remissions in hairy cell leucemia induced by a single infusion of 2-chlorodeoxyadenosine. N Engl J Med 1990;322:1117-21
- Seymour JF, Talpaz M, Kurzrock R. Response duration and recovery of CD4+ lymphocytes following deoxycoformycin in interferon-alpha resistant hairy cell leukemia: 7 year follow-up. Leukemia 1997;11:42-7
- Kraut EH, Bouroncle BA, Grever MR. Pentostatin in the treatment of advanced hairy cell leukemia. J Clin Oncol 1989;7:168-72
- Johnston JB, Eisenhauer E, Corbett WE, Efficacy of 2´deoxycoformycin in hairy cell leukemia. A study of the National Cancer Institute of Canada Clinical trials Group. J Natl Cancer Inst 1988;80:765-9
- Ho AD, Thaler J, Mandelli F, Response to pentostatin in hairy cell leukemia refractory to interferon-alpha. The European Organization for Research and Treatment of Cancer. J Clin Ocol 1989;7:1533-8
- Ribeiro P, Bouaffia F, Peaud PY, Lond term outcome of patients with hairy cell leukemia treated with pentostatin. Cancer 1999;88:65-71
- Cassileth PA, Cheuvart B, Spiers ASD, Pentostatin induces durable remissions in hairy cell leukemia. J Clin Oncol 1991;9:243-6
- Grem J, King SA, Cheson BD. Pentostatin in hairy cell leukemia. Treatment by the exemption mechanism. J Natl Cancer Inst 1989;81:448-53
- Golomb HM, Dodge R, Mick R, Pentostatin treatment of hairy cell leukemia patients who fail initial therapy with recombinant alpha-interferon. A report of CALGB study 8515. Leukemia 1994;8:2037-40
- Catovsky D, Matutes E, Garcia Talavera JG, Long term results with 2´deoxycoformycin in hairy cell leukemia. Leuk Lymphoma 1994;14(Suppl 1):109-13
- Robak T, Błasinska-Morawiec M, Błonski J, 2-chlorodeoxyadenosine (cladribine) in the treatment of hairy cell leukemia and hairy cell leukemia variant: 7-year experience in Poland. Eur J Haematol 1999;62(1):49-56
- Bastie JN, Cazals-Hatem D, Daniel M-T, Five years follow-up after 2-chlorodeoxyadenosine treatment in thirty patients with hairy cell leukemia. Evaluation of minimal residual disease and CD4+ lymphopenia after treatment. Leuk Lymphoma 1999;35:555-65
- Lauria F, Rondelli D, Zinzani PL, Long lasting complete remissions in patients treated with 2-CDA. A 5 year survey. Leukemia 1997;11:629-32
- Estey EM, Kurzrock R, Kantarjian HM, Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CDA). Blood 1992;79:882-7
- Jeh U, Barti R, Dietzfelbinger H, An update: 12 year follow-up of patients with hairy cell leukemia following treatment with 2-chlorodeoxyadenosine. Leukemia 2004;18:1476-81
- Hoffman MA, Janson D, Rose E, Rai KR. Treatment of hairy cell leukemia with cladribine. Response, toxicity and long-term follow-up. J Clin Oncol 1997;15:1138-42
- Tallman MS, Hakimian D, Variakojis D, A single cycle of 2-chlorodeoxyadenosine results in complete remission in the majority of patients with hairy cell leukemia. Blood 1992;9.2203-9
- Piro LD, Ellison JD, Saven A. The Scripps Clinic experience with 2-chlorodeoxyadensine in the treatment of hairy cell leukemia. Leuk Lymph 1994;14:121-5
- Robak T, Jamroziak K, Gora-Taylor J, Cladribine in a weekly versus daily schedule for untreated hairy cell leukemia: final report for the Polish adult Leukemia Group (PALG) of a prspective randomised multicentre trial. Blood 2007;109:3672-5
- Chada P, Rademaker AW, Mendiratta P, Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CDA): long-term follow-up of the Northwestern University experience. Blood 2005;106:241-6
- Arons E, Margulies I, Sorbara L, Minimal residual disease in hairy cell leukemia patients assessed by clone- specific polymerase chain reaction. Clin Cancer Res 2006;12:2804-11
- Noel P. Definition of remission, minimal residual disease, and relapse in hairy cell leukemia bone marrow biopsy histology and immunohistology specimens. Leuk Lymphoma 2011;52(Suppl 2):62-4
- Tallman MS. Implications of minimal residual disease in hairy cell leukemia after cladribine using immuno- histochemistry and immunophenotyping. Leuk Lymphoma 2011;52(Suppl 2):65-8. 644 645
- Lauria F, Lenoci M, Annino L, Efficacy of anti-CD20 monoclonal antibodies (Mabthera) in patients with progressed hairy cell leukaemia. Haematologica 2001;86:1046-50
- Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukaemia. Blood 2003;102:810-13
- Else M, Osuji N, Forconi F, The role of rituximab in combination with pentostatin or cladribine for the treatment of recurrent/refractory hairy cell leukaemia. Cancer 2007;110:2240-7
- Ravandi F, O'Brien S, Jorgensen J, Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood 2011;118:3818-23
- Else M, Dearden CE, Matutes E, Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma 2011;52(Suppl 2):75-8
- Gerrie AS, Zypchen LN, Connors JM. Fludarabine and rituximab for relapsed or refractory hairy cell leukemia. Blood 2012;119:1988-91
- Kreitman RJ. Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. Bio Drugs 2009;23:1-13
- Kreitman RJ, Pastan I. Immunotoxins in the treatment of refractory hairy cell leukemia. Hematol Oncol Clin North Am 2006;20:1137-51
- Kreitman RJ, Arons E, Stetler-Stevenson M, Recombinant immunotoxins and other therapies for relapsed/refractory hairy cell leukemia Leuk Lymphoma. 2011;52(Suppl 2):82-6
- Kreitman RJ, Wilson WH, Bergeron K, Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy cell leukemia. N Engl J Med 2001;345:241-7
- Kreitman RJ, Wilson WH, Robbins D, Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood 1999;94:3340-8
- Kreitman RJ, Squires DR, Stetler-Stevenson M, Phase I trial of recombinant immunotoxin RFB4 (dsFv)-PE38 (BL22) in patients with B-cell malignancies. J.Clin. Oncol 2005;23:6719-29
- Kreitman RJ, Squires DR, Stetler-Stevenson M, Phase II trial of recombinant immunotoxin RFB4 (dsFv)-PE38 (BL22) in patients with hairy cell leukaemia. J Clin Oncol 2009;27:293-2990
- Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011;17(20):6398-405
- Bang S, Nagata S, Onda M, HA22 (R490A) is a recombinant immunotoxin with increased antitumor activity without an increase in animal toxicity. Clin Cancer Res 2005;11:1545-50
- Kreitman RJ, Tallman MS, Coutre S, Phase I dose-escalation study of CAT-8015 (HA22) a CD22 specific targeted immunotoxin in relapsed or refractory hairy cell leukemia (888 abstract). Blood 2009;114:366
- Kreitman RJ, Tallman MS, Robak T, Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol 2012;30(15):1822-8
- Dietrich S, Glimm H, Andrulis M, BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012;366(21):2038-40