Abstract
Introduction: Fragile X syndrome (FXS), the most common inherited form of intellectual disability and autism, is caused by expansion of CGG trinucleotide repeats in FMR1 and marked reduction or absence of the gene product, FMRP. FMRP suppresses synaptic protein synthesis resulting from group 1 metabotropic glutamate receptor (mGluR) activation, a process critical to normal synaptic plasticity. FXS can be characterized as a disorder of synaptic plasticity with physical, cognitive and behavioral manifestations attributable to, at least in part, excessive mGluR activity and downstream effects.
Areas covered: This paper reviews the ‘mGluR theory' of FXS and the targeted drugs investigated in Phase II and III trials based on this theory of pathogenesis. A literature review was conducted using the PubMed database with search terms ‘fragile X syndrome', ‘mGluRs', ‘pharmacotherapy' and specific drug-related terms. Other resources were identified by review of relevant reference lists and consultation with experts in the field.
Expert opinion: While preclinical trials of targeted drugs in animal models of FXS have been encouraging, more studies are needed to determine clinical efficacy in humans. Challenges to clinical trial design and direction for future drug studies, including consideration of NMDA receptor partial agonists and mGluR2/3 agonists, are discussed.
1. Introduction
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability (ID) and autism with approximately 1/2500 – 4000 individuals affected by the full mutation Citation[1-3]. The genetic mutation resulting in the fragile X phenotype was first identified in 1991 on the long arm of the X chromosome at Xq27.3, and the affected gene was designated fragile X mental retardation-1 (FMR1) Citation[4]. FXS results from the unstable expansion of CGG trinucleotide repeats in the 5′ untranslated region of FMR1, leading to hypermethylation of the promoter region and silencing of FMR1 transcription Citation[4]. The full mutation resulting in FXS contains > 200 CGG repeats and is associated with the absence or markedly reduced levels of the gene product, fragile X mental retardation protein (FMRP), an mRNA binding protein Citation[5-7] that regulates translation of proteins important in synaptic plasticity Citation[8-10]. Smaller amplifications, termed premutations (55 – 200 CGG repeats), do not result in the absence of FMRP but rather increased levels of FMR1 mRNA, which is purported to have a direct toxic effect in cells Citation[11-14]. Mosaicism occurs in 20 – 40% of males, indicating the presence of the full mutation in some cells and premutation in others Citation[15]. The number of trinucleotide repeats frequently undergoes expansion in the offspring of female carriers, with the risk of expansion to full mutation directly proportional to the number of carrier CGG repeats Citation[15].
Individuals with the full mutation exhibit a broad array of cognitive, emotional and behavioral impairments, including mild to severe ID, learning disability, hypersensitivity to sensory stimuli, attention deficit hyperactivity disorder (ADHD), anxiety, social phobia, agitation, aggression, self-injury and sleep disturbance Citation[15,16]. Approximately 20 – 30% of individuals with FXS also meet criteria for autistic disorder Citation[17], and many more have features of autism, including repetitive and stereotyped behaviors, poor eye contact and social impairment Citation[18]. The severity of autistic features appears to increase with lower levels of FMRP and is stable over time Citation[17]. ADHD symptoms, particularly hyperactivity, are observed in approximately 75% of younger boys with FXS and typically decline with age Citation[15,18]. Agitation and aggression also follow a developmental course, peaking in early adolescence around the time of puberty and fading for most by young adulthood Citation[18]. The rate of anxiety disorders exceeds 75% in individuals with the full mutation, substantially greater than in other populations with ID Citation[19]. Anxiety is typically manifested as poor eye contact, gaze aversion and excessive shyness but may also drive other common behaviors, including hand flapping, self-injury, aggression and autistic mannerisms Citation[18]. In a study of 97 subjects with FXS, 58% met Diagnostic and Statistical Manual-IV (DSM-IV) criteria for social phobia, 59% for specific phobia and 25% for selective mutism, a particularly rare and severe form of social anxiety Citation[19].
Characteristic physical features in FXS include a large head circumference, long, narrow face, prominent forehead, closely set eyes, large ears, highly arched palate, joint hyperextensibility and macroorchidism Citation[15,16,20]. Seizures are common, affecting 13 – 20% of males and 5% of females with the full mutation Citation[15,16]. Seizures most often occur in childhood, typically resolving before adulthood Citation[16].
In general, the severity of cognitive disability and physical phenotype is inversely proportional to the amount of FMRP produced Citation[21,22]. Autistic behaviors are also more prominent among individuals with lower levels of FMRP Citation[17]. Males with the full mutation typically have mild to moderate ID, with those demonstrating a mosaic pattern less severely affected than those with a fully methylated pattern Citation[15]. Females with FXS are typically less severely affected than males due to higher levels of FMRP production associated with the presence of one normal X chromosome Citation[16]. Approximately 50 – 71% of females with the full mutation have an IQ in the borderline to mild mental retardation range Citation[15,23,24], though others have subtle learning difficulties and no apparent physical manifestations of the syndrome. The degree of clinical expression in females depends in part on the ratio of X chromosome inactivation (XCI) in cells Citation[15,24]. In females who inherit the full mutation and have skewed inactivation of the normal allele, the cognitive, physical and behavioral phenotype is more severe Citation[24,25].
Premutation carriers (55 – 200 CGG repeats) usually have intellectual abilities in the normal range, though they may exhibit mild cognitive and behavioral difficulties, including social anxiety, learning disabilities and occasionally ID and autism Citation[14,26]. As with individuals with the full mutation, the degree of intellectual impairment in male carriers depends on the amount of expressed FMRP Citation[21]. Both males and females with premutations exhibit particular difficulty with arithmetic, suggesting an underlying mechanism for specific cognitive deficits other than FMRP depletion in the premutation state Citation[21,22]. Excess FMR1 mRNA has been identified in male premutation carriers, particularly in those with 100 – 200 CGG repeats, and is likely due to increased rates of transcription as a result of reduced translational efficiency Citation[11,21,22].
Carrier status is also associated with disorders of adult-onset, primarily premature ovarian failure (POF) in women and Fragile X Tremor- Ataxia Syndrome (FXTAS) in men Citation[14,16,27]. Approximately 16% of women with fragile X premutations exhibit POF, which is unrelated to CGG repeat size or deficient FMRP Citation[14,27]. Unlike with the full mutation, skewing of XCI is not associated with the development of POF Citation[28]. FXTAS, developing in 30 – 40% of male carriers in their 50s and beyond, includes intention tremor, gait ataxia, parkinsonism, autonomic dysfunction, executive function deficits and memory loss with progression to dementia in some individuals Citation[14,16,29,30]. The vast majority of carriers who develop late-onset cerebellar ataxia have larger (≥ 70) CGG repeat sizes, however, and the actual prevalence of this syndrome in the general population is likely lower than originally reported Citation[31]. Neuroimaging reveals characteristic white matter lesions in the middle cerebellar peduncles, brain atrophy and periventricular white matter disease Citation[12,14]. Eosinophilic intranuclear inclusions, identified on autopsy throughout the cerebrum and brainstem in a group of premutation carriers, are ubiquitin-positive, suggesting probable intranuclear accumulation of proteins as in other gain-of-function trinucleotide expansion diseases Citation[13,14]. Female premutation carriers develop neurological symptoms much less commonly, an observation that exceeds what would be expected due to X-inactivation effects and suggests additional gender-specific factors Citation[14].
2. Target symptom management
In clinical practice, pharmacotherapy for FXS has historically focused on alleviating the associated emotional and behavioral manifestations of the disorder Citation[32]. A parent survey of more than 1300 children with FXS found that 61% of males and 38% of females were taking medication for at least one symptom, most commonly anxiety and inattention, though parents considered most medications only somewhat effective Citation[32]. Well-designed pharmacological studies for FXS are sparse, and FXS is typically an exclusion criterion in larger trials of individuals with autism Citation[33].
Based on the observation that leukocytes grown in medium deficient in folic acid reveals a fragile site at Xq27.3, several studies have investigated the effects of folic acid treatment in FXS. Though an initial open-label trial and some case reports suggested clinical benefit, no subsequent placebo-controlled trials have shown statistically significant improvement in cognitive or behavioral symptoms with folate treatment Citation[34-40]. A randomized, placebo-controlled, crossover study of 21 males with FXS treated with the metabolically active form of folate, folinic acid, also failed to demonstrate statistically significant benefit on measures of adaptive behaviors, language skills and hyperactivity Citation[41]. However, two subjects withdrew during the crossover arm of the study due to loss of substantial behavioral improvements seen during the first treatment arm; parents had correctly guessed treatment with folinic acid in both cases. A 1996 Cochrane Database review of folic acid in FXS, including review of the five controlled trials between 1986 and 1992, concluded that the studies lacked sufficient power to detect anything but large effects and that no definitive conclusions could be drawn from available data Citation[42]. No actual deficits in folate metabolism have been demonstrated in animal models or humans with FXS, and efforts to investigate folate as a treatment have been abandoned Citation[18].
ADHD symptoms are a common target for pharmacological intervention in FXS, though controlled trials of stimulants and non-stimulant medications are sparse Citation[32]. In 1988, Hagerman et al. reported improvement in hyperactivity and social skills in 10 of 15 children treated with methylphenidate as compared with placebo and dextroamphetamine Citation[43]. An observational study of 12 boys (mean age, 8 years 5 months) with FXS taking either methlyphenidate or dextroamphetamine suggested a response rate of 75% for improved attention, a rate greater than that typically observed in children with autism spectrum disorders (ASD) and idiopathic ID Citation[44]. Academic performance was also enhanced in 75% of subjects during achievement tests with stimulant treatment, though no improvements were seen in excessive motor activity. Of note, the subjects in this small sample were not evaluated for DSM-IV diagnosis of ADHD, and no subjects were scored in the ‘clinically significant' range on the DSM-ADHD rating scale at study entry.
l-Acetylcarnitine (LAC) has been studied as a treatment for hyperactivity in FXS based on the finding that the carnitine ester, involved in N-acteylation of histones at FMR1, caused modest reduction in FMR1 promoter hypermethylation in two of three FXS lymphoblastoid cell lines Citation[45-47]. Decrease in FMR1 methylation status was not associated with transcription reactivation in vitro Citation[45]. In a double-blind, placebo-controlled trial of 63 boys with FXS, LAC was well-tolerated and associated with significant reductions in hyperactivity on the Conners' Global Index for Parents, though not the Conners' Teacher form Citation[47]. Vineland Socialization and Adaptive Behavior ratings also improved significantly for the LAC group compared with placebo; cognitive abilities remained unchanged. Quantitative polymerase chain reaction (PCR) did not detect FMR1 mRNA after 12 months of treatment with LAC, suggesting that observed effects were likely due to its effect on carnitine metabolism.
Similarly, Torrioli et al. reported on valproate, a histone deacetylase inhibitor, as a treatment for ADHD symptoms in a 6-month, open-label study of 10 boys with FXS, finding significant improvement in hyperactivity scores on the Conner's Parent Rating Scale-Revised Short Form Citation[48]. Two subjects withdrew from the study prematurely due to worsened behavior and restlessness.
Melatonin was effective in a randomized, double-blind, placebo-controlled crossover study for decreasing sleep-onset latency and modestly increasing mean night sleep duration in a sample of 12 children with FXS and/or autism Citation[49].
A 12-week, open-label pilot study of aripiprazole in 12 children and young adults with FXS demonstrated significant improvement in irritability, including tantrums, aggression and self-injury, in 10 of 12 (87%) subjects Citation[33]. Adverse effects were generally mild and included drooling, tiredness and insomnia.
3. mGluR theory of FXS
Following the discovery of the genetic defect leading to the fragile X phenotype, a knockout (KO) mouse model was designed to characterize the structure and function of FMRP Citation[50]. Interruption of the Fmr1 gene, the murine homolog to FMR1, resulted in mice lacking Fmr1 protein and exhibiting macroorchidism, mild spatial learning deficits and increased motor activity Citation[50]. KO mice also demonstrated an increased propensity for audiogenic seizures and long, thin, immature dendrites on autopsy, similar to humans with FXS and other forms of ID Citation[51-55]. Drosophila with mutation of the FMR1 homolog dfmr1 also demonstrated dendrites with abnormal synaptic branching and overgrowth Citation[56]. The immature appearance and overgrowth of dendritic spines in humans and animal models suggested altered synaptic plasticity Citation[54] and impairment in the ‘pruning' of extraneous synaptic connections Citation[57].
FMRP has been identified as a widely expressed RNA-binding protein with five functional domains, including two K homology (KH) domains and one RGG box that bind mRNA Citation[5,6]. FMRP preferentially binds via the RGG box to mRNA containing a G-quartet (Gq) structure Citation[10,53,58]. The majority of FMRP is found in the cytoplasm associated with polyribosomes and is likely involved in repressing translation of target mRNAs via this association Citation[59]. Polyribosome-free FMRP, multiple mRNAs and proteins also aggregate to form translationally dormant messenger ribonucleic proteins (mRNPs) Citation[7,60] that associate with cytoskeletal microtubules for transportation to neuronal dendrites Citation[59]. On activation of excitatory, postsynaptic group 1 metabotropic glutamate receptors (mGluRs), translation rapidly ensues, due in part to dissociation of FMRP from target mRNA Citation[61]. One of many consequences of the resultant protein synthesis is long-term depression (LTD) and a reduction in synaptic transmission Citation[62]. If unchecked, excessive LTD can lead to synapse elimination Citation[52,63]. When mGluRs are activated by presynaptic glutamate release, FMRP is also synthesized and inhibits further protein synthesis, creating a negative feedback mechanism for limiting mGluR-dependent LTD Citation[60].
Appropriate inhibition of further protein synthesis by FMRP leads to reversion of synaptic changes within 30 min Citation[52]. In the absence of FMRP, as in FXS, translational inhibition is lifted, and protein synthesis-dependent LTD is exaggerated, thus altering synaptic plasticity Citation[52,64].
More than 500 target mRNAs have been identified Citation[57,65], encoding proteins that themselves suppress translation of synaptic proteins Citation[52], maintain proper neuronal function and mediate neuronal and craniofacial development Citation[10,58,66]. One target protein of interest, for example, is microtubule-associated protein 1B (MAP1B), a protein involved in synapse development Citation[56,67]. MAP1B homologs are overexpressed in mouse and Drosophila KO mutants and are associated with increased neuronal microtubule stability and synaptic overgrowth and branching, respectively Citation[56,67]. These morphological changes in animal models are analogous to the long, thin, immature dendritic spines in individuals with FXS and suggest that upregulated MAP1B may play a role in impaired synapse development and possible cognitive deficits in FXS Citation[67]. Fmr1 KOs also overexpress amyloid precursor protein (APP) and its proteolytic product, Aβ, implicated in the increased susceptibility to seizures observed in FXS, Alzheimer's disease and Down's syndrome Citation[61]. While FMRP is well characterized as an mRNA-binding protein, Brown et al. recently demonstrated that FMRP also directly binds and activates the membrane protein Slack, a sodium-activated potassium channel involved in neuronal firing patterns Citation[68].
Synaptic plasticity is determined by patterns of long-term potentiation (LTP) and LTD, the synaptic processes implicated in learning and memory Citation[52]. While LTP promotes strengthening of synapses, LTD is an activity-dependent synaptic weakening important in synaptic pruning Citation[52]. Fmr1 KO mice show markedly reduced LTP in the cortex and amygdala Citation[69], as well as significantly increased LTD in the hippocampus Citation[70], the latter dependent on activation of group 1 mGluRs Citation[53,70].
mGluR activation and subsequent protein synthesis ultimately leads to internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors, the ionotropic glutamate receptors, from the postsynaptic membrane Citation[63,71]. Removal of excitatory glutamate receptors from the synapse is one proposed mechanism for the expression of mGluR-dependent LTD Citation[63]. AMPAR trafficking at the synaptic membrane plays a role in plasticity, and excessive internalization of AMPARs in the absence of FMRP may underlie impairments in learning and memory seen in FXS Citation[71]. The GluR1 subunits of AMPARs in postsynaptic membranes of amygdala neurons are reduced in Fmr1 KO mice, suggesting that impaired AMPAR expression could be the downstream manifestation of dysregulated LTP Citation[69]. Alternatively, dysregulated LTP could well be the manifestation of impaired AMPAR expression, possibly due to excess mGluR5 signaling during early development. FMRP regulates the translation of proteins STEP and Arc, which are likely responsible for AMPAR endocytosis in LTD Citation[57]. Aberrant internalization of AMPARs is rescued in cultured hippocampal neurons treated with the mGluR5 antagonist 2-methyl-6-phenylethynyl pyridine (MPEP), though not in amygdala neurons Citation[69,71]. The failure of mGluR5 antagonism to normalize AMPAR expression in the amygdala does not necessarily indicate a mechanism for impaired amygdala LTP distinct from impaired hippocampal LTD, however. Dysregulated LTP in the amygdala may be a consequence of overall reduced synaptic connectivity in this region resulting from increased mGluR5-dependent protein synthesis during the development of amygdala circuitry.
Messenger RNAs for several gamma-aminobutyric acid-A (GABAA) receptor subunits in fragile X mouse and Drosophila models are also underexpressed in association with FMRP deficiency Citation[72]. The mechanism by which FMRP regulates GABAAR expression is not fully understood but may involve transport and localization of GABAAR mRNA Citation[72]. GABAARs are the primary inhibitory receptors in the brain, and decreased GABAergic activity in the cortex could contribute to symptoms of anxiety, depression, insomnia and epilepsy, as well as disturbances in learning and memory seen in fragile X patients Citation[18,72]. Despite reduced GABAAR activity, Fmr1 KO mice remain sensitive to the sedative and anxiolytic effects of GABAAR agonists, and the audiogenic seizure phenotype is corrected by treatment with the GABAAR agonists diazepam and ganoxolone. These findings provide the rationale for targeting GABAAR activity in therapeutic drug trials Citation[73].
Group 1 mGluRs consist of mGluR1, primarily expressed in the cerebellum, and mGluR5, expressed in the neocortex, hippocampus and striatum Citation[65]. mGluR5 is of particular interest in FXS given its activity in the neocortex and hippocampus Citation[65]. The group 1 mGluRs are G-protein coupled and transduce excitatory signals via three signaling cascades: activation of phosphoinositol (PI) hydrolysis by phospholipase C (PLC); the phosphoinositide (PI3K)/AKT/mammalian target of rapamycin (mTOR) cascade and the extracellular signal-regulated kinase (ERK) cascade Citation[65]. All three pathways are involved in the regulation of protein synthesis and act together to initiate mRNA translation Citation[65]. FMRP inhibits translation of many of the proteins participating in the signaling cascades, and in Fmr1 KO mice, signaling proteins are overexpressed, further driving protein synthesis Citation[57].
Based on the findings that i) mGluR activation results in protein synthesis and ii) protein synthesis is exaggerated in the absence of FMRP, Bear et al. proposed the ‘mGluR theory' of FXS: that the varied functional consequences of mGluR-dependent protein synthesis can account for the cognitive, behavioral and physical phenotypes that characterize the syndrome Citation[52,74]. Exaggerated LTD in FXS would favor synaptic loss during critical periods of synaptogenesis, accounting for developmental delay and impaired cognitive development Citation[52]. FMR1 is widely expressed throughout the brain, including in most cells with postsynaptic mGluRs, and overactivation of group 1 mGluRs in various regions could explain fragile X symptoms as varied as anxiety, seizures, loose bowel movements, hyperalgesia, obsessive–compulsive symptoms, poor coordination, tactile hypersensitivity and disturbed sleep patterns Citation[52]. Antagonists of group 1 mGluRs reduce protein synthesis triggered by mGluR activation, and it follows that mGluR antagonists could potentially alleviate impairments in the FXS phenotype Citation[52].
The mGluR theory of fragile X is supported by the effects of the non-competitive mGluR5 antagonist MPEP in the treatment of Drosophila KO mutants, rescuing short-term memory, abnormal courtship behavior and mushroom body defects Citation[75,76]. Courtship impairment, indicative of learning and memory deficits, was corrected throughout development and into adulthood, though anatomical defects in the mushroom body, a structure analogous to the mammalian brain, were only rescued if treated early in development Citation[75]. MPEP also suppresses the increased propensity toward seizures and excessive motor activity observed in Fmr1 KO mice Citation[61,77], though chronic mGluR5 inhibition has a smaller effect on macroorchidism. These findings suggest that aspects of the FXS behavioral phenotype may improve with mGluR5 antagonism throughout development, though some anatomical differences are less amenable to correction unless treatment occurs during a critical window in development.
Further support for the mGluR theory comes from the study of a double mutant mouse model with both Fmr1 KO and 50% genetic reduction in mGluR5 expression Citation[78]. Heterozygote crosses showed reduction in audiogenic seizures and normalization of the prepubescent accelerated growth and abnormal dendritic spine density observed in Fmr1 KO mice. Altered ocular dominance plasticity (ODP) in the visual cortex and increased protein synthesis in the hippocampus of Fmr1 KOs were also rescued by reduced mGluR5 expression. Testicular enlargement was not affected by genetic reduction of mGluR5. FMRP and mGluR were thus observed to act in functional opposition, with FMRP acting as a downregulator of synaptic mRNA translation and protein synthesis triggered by mGluR5 activation Citation[65,78]. In the absence of FMRP regulation, unchecked mGluR-dependent protein synthesis results in expression of the fragile X phenotype Citation[65].
Validation of the mGluR theory suggests that medications targeting mGluR5, as well as components of the associated signaling cascades, could be therapeutic in individuals with FXS Citation[79]. Downstream molecules regulated by the mGluR pathway, including individual proteins, GABARs, AMPARs and NMDARs, could also be targets for pharmacological intervention (). While no medications specifically modulating the mGluR pathway have approval from the Food and Drug Administration (FDA) for treatment in FXS, a number of Phase II and III trials have been completed or are underway, as will be described below ().
Figure 1. Illustration of potential targets for drug therapy at a glutamatergic synapse in FXS. When stimulated by presynaptic glutamate release, postsynaptic mGluR1/5 receptors activate intracellular signaling cascades that mobilize translational machinery to translate mRNA into synaptic proteins, examples of which include MMP-9, MAP1B and APP. FMRP inhibits translation of more than 500 target mRNAs in the normal state; in FXS, this negative feedback mechanism is absent or severely reduced, leading to excessive protein synthesis and altered synaptic plasticity. Consequences of increased synaptic protein synthesis include loss of AMPARs and NMDARs from the postsynaptic membrane via endocytosis. Drugs that modulate excitatory glutamatergic and inhibitory GABAergic neurotransmission have the potential to alleviate some phenotypic effects in FXS. Key for labeled mechanisms of action and corresponding drugs: 1) LY2140023 (mGluR2/3 agonist); 2) acamprosate (GABAAR agonist); 3) arbaclofen (GABABR agonist); 4) riluzole (blocks presynaptic glutamate release); 5) riluzole (blocks voltage-dependent sodium channels); 6) riluzole (enhances glutamate reuptake); 7) MPEP, fenobam, AFQ056, RO4917523, STX107, CTEP, acamprosate (mGluR5 antagonists); 8) CX516 (ampakine); 9) memantine (weak NMDA antagonist); d-serine, d-cycloserine, GLYX-13 (glycine site NMDAR partial agonists); 10) lithium, CX516 (increase BDNF); 11) lithium (GSK3 inhibition); 12) minocycline (MMP-9 inhibition).
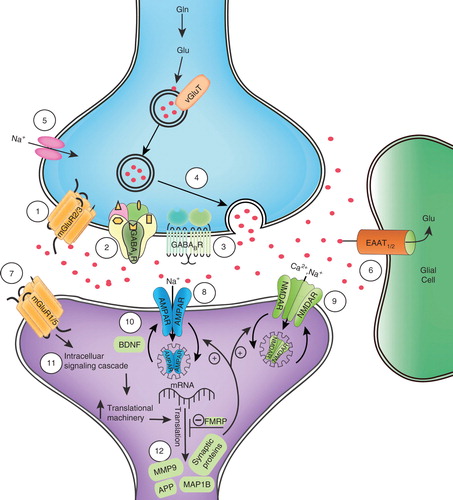
Table 1. Comparison of Phase II and III clinical trials in FXS.
4. mGluR5 antagonists
Extracellular antagonists of group 1 mGluRs, particularly mGluR5, have garnered much excitement for therapeutic potential based on the finding that 50% knockdown of mGluR5 activity in Fmr1 KO mice and mGluR5 blockade with MPEP in animal models can correct aspects of the fragile X cognitive and behavioral phenotype, including propensity toward seizures, increased motor activity, memory impairment, repetitive behavior, sensorimotor processing impairment and accelerated prepubescent growth Citation[61,75,77,78,80,81]. MPEP has also been shown to correct deficits in presynaptic glutamate release in the amygdala of Fmr1 KO mice, suggesting a possible therapeutic role for symptoms of anxiety and hyperarousal in FXS Citation[69]. Antagonists of mGluR5 have been identified as better therapeutic targets than mGluR1 antagonists, as mGluR1 is widely expressed in the cerebellum, and blockade is associated with motor impairment Citation[57,80]. mGluR1 antagonists also appear less potent than mGluR5 antagonists in altering the fragile X phenotype Citation[80]. In a recent study, the non-competitive mGluR1 antagonist JNJ16259685 diminished marble burying, a measure of repetitive behavior, in Fmr1 KO mice but failed to significantly affect motor activity, motor coordination or susceptibility to seizures, as MPEP did Citation[80]. MPEP is the prototypical mGluR5 antagonist but is not specific for mGluR5 and also inhibits NMDARs at high concentrations, leading to significant side effects Citation[82]. Consequently, MPEP has not been studied in human subjects as attention has turned to more selective mGluR5 antagonists.
4.1 Fenobam
Like MPEP, fenobam (N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea) is a selective and potent non-competitive mGluR5 antagonist Citation[83] that was studied as a novel, non-benzodiazepine anxiolytic in the 1980s Citation[84]. While fenobam demonstrated efficacy and safety for anxiety, similar to diazepam, in a 5-week, double-blind, placebo-controlled study of 29 outpatients Citation[84], another Phase II trial reported no anxiolytic effect and psychostimulant-like side effects, including hallucinations, vertigo, paresthesias and insomnia, and further development of fenobam as an anxiolytic was discontinued Citation[83]. Porter more recently found that fenobam and MPEP bind the same allosteric modulatory site of the mGluR5 receptor and have similar mechanisms of action Citation[83]. With emergent interest in the role of mGluR5 activity in a number of neuropsychiatric disorders, fenobam is again being investigated as a potential therapeutic agent for anxiety Citation[83], as well as FXS Citation[81,85]. In rodent models of anxiety, fenobam had a potent anxiolytic effect Citation[83]. Fmr1 KO mice treated with fenobam also demonstrate rescue of the immature dendritic spine morphology in hippocampal neurons Citation[81], as well as impaired procedural memory formation and avoidance behavior Citation[86]. Of note, however, wild-type (WT) mice treated with fenobam showed severe impairment in motor learning, highlighting the importance of accurate diagnosis in treating intellectually impaired individuals with mGluR5 antagonists Citation[86].
A pilot open-label, single dose trial of fenobam in adults with FXS was conducted to evaluate safety and pharmacokinetic properties in human subjects Citation[85]. Outcome measures included performance on a prepulse inhibition (PPI) task and the Carolina Fragile X Project Continuous Performance Test (FXCPT) commission scores, chosen to assess the phenotypic features of sensory gating, attention and response inhibition. Twelve verbal subjects (six men and six women) were administered a single oral dose of fenobam – 50, 100 or 150 mg – with the first subject of each gender receiving 50 mg, the second 100 mg and all subsequent subjects 150 mg. Dosing was based on previous trials for anxiety, with 150 mg representing the middle dose range in which central nervous system (CNS) side effects were observed. Subjects completed the PPI and FXCPT tasks 60 min later; side effects and fenobam plasma concentrations were assessed over the course of 6 h. Fenobam concentration in plasma peaked at 2 – 3 h after dosing, though changes in PPI were determined earlier at 1 h post-dose, possibly underestimating any therapeutic effect. The pharmacokinetic properties of fenobam show wide intersubject variability and suggest that effective dosing may be highly variable as well. Fifty percent of subjects (four men and two women) met the predetermined response criterion for improvement in sensory processing, defined as 20% improvement from baseline in the PPI, a proportion significantly greater than would be expected by chance alone. No significant effect for attention and inhibition was observed on the FXCPT due to low rates of commission errors at baseline and ceiling effects. A single dose of fenobam was well tolerated, with three subjects experiencing mild sedation, and nine subjects showing ‘calmed' behavior with improvement in symptoms of impaired eye contact, impaired social interaction, anxiety and hyperactivity. This observed clinical improvement indicates that different behavioral outcome measures are needed to fully characterize the effect of treatment. Conclusions could not be drawn about CNS side effects that emerged in previous studies with chronic dosing in adult subjects with anxiety disorders. As in other open-label studies, results should be interpreted cautiously, as a large placebo effect and observer bias may overestimate the apparent efficacy of fenobam. Randomized, double-blind, placebo-controlled trials are the gold standard in clinical trial design and minimize the influence of such biases, and rigorous controlled trials would be required to draw more definitive conclusions regarding therapeutic effect.
4.2 AFQ056
AFQ056 is a selective, non-competitive mGluR5 antagonist with a short half-life that is currently in clinical trials for the treatment of l-DOPA-induced dyskinesia in Parkinson's disease, Huntington's disease and FXS Citation[82]. AFQ056 has been shown to rescue deficits in PPI and the elongated dendritic spine morphology in cultured hippocampal neurons from Fmr1 KO mice without causing adverse effects in WT mice Citation[82]. Jacquemont et al. recently reported on a Phase II randomized, double-blind, crossover study of AFQ056 in 30 male subjects with FXS aged 18 – 35 years with the Aberrant Behavior Checklist-Community Edition (ABC-C) sum score as a primary outcome measure Citation[87]. As a group, subjects did not show overall improvement on the ABC-C or a number of secondary outcome measures after 19 – 20 days of treatment, with the exception of a modest reduction in repetitive behavior. However, a subgroup of subjects (n = 7) with full methylation of the FMR1 promoter and no detectable FMR1 mRNA improved significantly on the ABC-C (p < 0.001), Including subscales for Stereotypy, Hyperactivity and Inappropriate Speech, while those with partial promoter methylation (n = 18) showed no response. The subpopulation with full methylation also showed improvement on secondary measures of clinician-rated improvement, including stereotypic behavior, restricted interests and autistic behaviors, but not overall adaptive functioning. Twenty-four of 30 subjects (80%) reported mild to moderate adverse events, most commonly fatigue and headache. Results from a follow-up study suggest that full methylation status may predict AFQ056 response, though as expected, AFQ056 does not have any effect on degree of FMR1 methylation or transcription in vitro Citation[88].
Phase II randomized, double-blind, placebo-controlled trials of AFQ056 in both adolescents and adults with FXS are underway to assess safety, tolerability and efficacy; 2-year open-label extension studies are also planned Citation[89-92]. Novartis is also sponsoring a Phase I open-label study of pharmacokinetics, safety and tolerability of single and multiple oral doses of AFQ056 in children aged 3 – 11 years with FXS Citation[93].
4.3 Acamprosate
Acamprosate (calcium acetylhomotaurine) is a drug with putative mGluR5 antagonism approved by the FDA for the maintenance of abstinence from alcohol in individuals with alcohol dependence Citation[94]. Characterization in animal models indicates that acamprosate may also act as a weak NMDA receptor antagonist and GABAAR agonist and have anti-oxidant effects Citation[94]. In an open-label, pilot study of three males aged 18 – 23 years with full mutation FXS and autistic disorder, all subjects demonstrated significant improvement in expressive and pragmatic use of language, though change in non-verbal communication, including eye gaze, was not observed Citation[94]. Two of the three subjects developed dose-dependent nausea and/or emesis, and one subject reported sedation. Observations suggest a positive therapeutic effect, though the small sample size and open-label nature of the study limit any definitive conclusions. While acamprosate is widely available with documented safety in humans, disadvantages include poor oral bioavailability, the need for high and frequent dosing and significantly less potency at mGluR5 in comparison with the prototypical mGluR5 antagonist MPEP Citation[94]. Activity at NMDA and GABAARs rather than at mGluR5 receptors may be responsible for its observed therapeutic effect Citation[94]. A Phase III open-label trial of acamprosate (NCT01300923) for the treatment of behavioral, cognitive and social deficits in children aged 5 – 17 with FXS is currently underway at Indiana University Citation[95].
4.4 mGluR5 antagonists in development
RO4917523 (Hoffman-LaRoche, South San Francisco, CA, USA) is an mGluR5 antagonist that has recently been investigated for efficacy in treatment-resistant depression and as an adjunctive treatment for adults with major depressive disorder (MDD) and incomplete response to an antidepressant (NCT00809562; NCT01437657) Citation[96]. Phase II trials in male adults with FXS are currently underway to assess safety, tolerability, pharmacokinetics and efficacy with daily dosing over 6 – 12 weeks (NCT01015430; NCT01517698) Citation[96]. Another mGluR5 antagonist, STX107 (Seaside Therapeutics, Cambridge, MA, USA), has completed Phase I single-dose trials in healthy male adult volunteers (NCT00965432), and Phase II trials are planned to assess tolerability in adult males with FXS (NCT01325740) Citation[97]. No data are publicly available at the time of this report.
2-Chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP) is a recently developed, highly selective mGluR5 antagonist with potency 30- to 100-fold greater than MPEP and fenobam in vivo Citation[98]. CTEP confers the advantages of bioavailability approaching 100%, as well as a long half-life of 18 h in mice, enabling sustained receptor blockade with dosing once every 48 h Citation[98]. In young adult Fmr1 KO mice, CTEP has been shown to rescue biochemical, structural and behavioral aspects of the mutant phenotype, including learning and memory impairment, hypersensitivity to an auditory stimulus, susceptibility to seizure, motor hyperactivity, macroorchidism (partial rescue), abnormal dendritic spine density in the visual cortex, increased hippocampal LTD, increased synaptic protein synthesis and increased signaling through ERK and mTOR signaling pathways Citation[99]. Amelioration of multiple fragile X features in young adulthood, after the phenotype is fully established, is particularly notable and implies that correction of an imbalance in synaptic signaling can substantially improve outcome Citation[99]. No trials of CTEP in human subjects have been reported.
5. Other glutamatergic agents
5.1 Riluzole
Riluzole is a drug with unknown exact mechanism of action that is believed to modulate glutamatergic and GABAergic neurotransmission via inhibition of presynaptic glutamate release, enhanced glutamate reuptake, potentiation of postsynaptic GABAAR activity and blockage of GABA reuptake Citation[100]. Riluzole is FDA-approved for the treatment of amyotrophic lateral sclerosis (ALS), and preliminary studies suggest efficacy for treatment-resistant depression and obsessive–compulsive disorder (OCD) Citation[100]. Riluzole was studied in six young adult males (aged 19 – 24 years) with FXS, autism and moderate ID, with primary outcome measures including the ABC subscales and Children's Yale-Brown Obsessive Compulsive Scale modified for Pervasive Developmental Disorders (CY-BOCS-PDD), the latter intended to capture any change in repetitive, compulsive behaviors Citation[100]. One of six subjects (16%) responded to treatment with a reduction in repetitive behaviors and irritability; there was no significant change on any secondary measures. Despite a lack of clinical response, ERK activation, known to be delayed in FXS and considered a potential biomarker of treatment response, decreased significantly in all patients. Liver function tests increased asymptomatically in five of six (83%) subjects.
5.2 Memantine
Memantine is a non-competitive NMDA receptor antagonist with weak receptor affinity that causes blockade at low synaptic glutamate levels and release of blockade at high glutamate levels and may have a neuroprotective effect Citation[101]. Overactivity of the excitatory glutamate system and underactivity of inhibitory GABAergic pathways have been proposed as causative factors in autism, and memantine may function to ameliorate this imbalance Citation[102]. Approved for the treatment of moderate to severe Alzheimer's dementia and off-label use of memantine for social deficits, cognitive and behavioral symptoms in ASD have been reported, with greatest improvement seen in the areas of language and social behavior Citation[101]. Memantine has been shown to promote maturation of dendritic spines and stimulate excitatory synapse formation in cultured cerebellar granule cells from Fmr1 KO mice Citation[102]. Erickson et al. conducted a chart review of six young adult patients (aged 13 – 22 years) with FXS and a comorbid diagnosis of ASD treated with memantine for a mean duration of 34.7 weeks. Four of six subjects (67%) showed significant clinical improvement as measured by the Clinical Global Impressions-Improvement (CGI-I) score, though specific symptom rating scale scores did not change Citation[101]. Two of six subjects (33%) developed treatment-limiting irritability. Of those tolerating memantine, the greatest gains were reported in social behavior, inattention and irritability. In a separate case report, one 65-year-old female premutation carrier with FXTAS demonstrated a 50% reduction in FXTAS symptoms, including head and hand tremor, ataxia, neuropathy, depressed mood and anxiety with the addition of memantine to venlafaxine Citation[103].
6. Drugs affecting intracellular signaling pathways and target proteins
6.1 Minocycline
Minocycline, a tetracycline derivative antibiotic that inhibits matrix metalloproteinase-9 (MMP-9) and may reduce CNS inflammation, has been studied as a treatment for stroke, multiple sclerosis (MS), amyotrophic lateralizing sclerosis (ALS), Huntington's disease, Parkinson's disease, Alzheimer's disease and autism Citation[104,105]. MMP-9 is involved in normal synaptic structure and function via its role in remodeling the extracellular matrix and is overexpressed in response to mGluR activation Citation[106,107]. Elevated levels of MMP-9 are found in Fmr1 KO hippocampal neurons and have been linked to immature dendritic spine morphology Citation[104]. Treatment of Fmr1 KO mice with minocycline restores mature dendritic spine formation, leads to reduced anxiety and more strategic exploratory behavior in maze tasks, and increases mating-related ultrasonic vocalizations, a measure of social communication, to levels comparable with WT controls Citation[104,108]. Minocycline has also been found to restore normal synaptic structure in Drosophila dfmr1 mutants Citation[107].
Paribello et al. conducted a Phase II open-label study of minocycline in 20 adolescent and young adult subjects with FXS treated with 100 mg or 200 mg daily for 8 weeks Citation[106]. Minocycline significantly improved ABC-C Irritability subscale scores, CGI-I scores and the visual Analog Scale for Behavior (VAS). Improvements were also seen in ABC subscales for Stereotypy, Hyperactivity and Inappropriate Speech. Two subjects asymptomatically seroconverted to a positive ANA. Other side effects were mild and included dizziness, diarrhea, drowsiness and headache. Children were not included in the study, as minocycline can cause permanent tooth discoloration during tooth development in childhood. As with other open-label studies, results must be interpreted cautiously. A randomized, placebo-controlled trial of minocycline in children under age 13 with FXS was recently completed Citation[109], though results are not yet reported. A survey of side effects in 50 individuals with FXS treated with minocycline revealed that gastrointestinal (GI) symptoms were most common, including loss of appetite, GI upset and diarrhea Citation[110].
6.2 Lithium
Lithium, traditionally used as a mood-stabilizing agent in bipolar disorder, has several known mechanisms of action, including inhibition of the metabolic regulatory enzyme glycogen synthase kinase-3 (GSK3), which is overactive in the absence of FMRP Citation[111-113]. Treatment with MPEP in Fmr1 KO mice also results in reduced GSK3 activity, suggesting that mGluR5 antagonists and direct GSK3 inhibitors affect a common signaling pathway and lending support to GSK3 as a potential therapeutic target Citation[112,114]. GSK3 regulates a variety of neuronal substrates, including transcription factors, cell signaling cascades and structural proteins involved in cell architecture, such as (MAP1B) microtubule-associated protein 1B Citation[113]. GSK3 promotes activation of MAP1B, and lithium's inhibitory effects on this system could theoretically correct aspects of the fragile X phenotype linked to upregulated MAP1B activity in Drosophila Citation[112]. Lithium has also been shown to increase brain-derived neurotrophic factor (BDNF), a regulator of learning, memory and synaptic plasticity, in Fmr1 KO mice Citation[114]. Fmr1 KO mice treated with lithium show amelioration of increased audiogenic seizures, hyperactivity, social preference differences, anxiety-related behaviors in social interactions, learning and memory deficits, abnormal dendritic spine length and density, mGluR-mediated LTD in hippocampal neurons and increased cerebral protein synthesis in the limbic system and hypothalamus Citation[112-117]. Restoration of normal hippocampal LTD persisted in FMR1 KO mice with long-term treatment of lithium into adulthood, suggesting benefits for treatment throughout the lifespan Citation[116].
Following evidence of improvement in cognitive and behavioral abnormalities in fragile X animal models treated with lithium Citation[75,112,114], Berry-Kravis et al. conducted an open-label pilot study of lithium in 15 subjects with FXS, aged 6 – 23 years Citation[111]. Though improvement in ABC-C Irritability subscale scores, the primary outcome measure, was not significant, subjects were rated as significantly improved in ABC-Hyperactivity and Inappropriate Speech subscale scores, CGI, VAS and Vineland Adaptive Behavior Scales (VABS)-Maladaptive Behavior subscale scores. Overall improvement fell in the mild to moderate range. Eleven of 15 subjects reported polyuria and/or polydipsia, and 4 subjects developed elevated thyroid-stimulating hormone (TSH). ERK activity, a potential biomarker for changes in cell signaling, normalized for the 11 subjects tested following treatment with lithium. The authors noted that lithium's diverse biochemical activities and general mood-stabilizing properties may account for some of the observed improvements. Lack of a placebo comparison group and double blinding in this open-label study increases the likelihood that results were influenced by observation and placebo bias and should be interpreted cautiously.
7. GABA receptor agonists
7.1 Arbaclofen
Arbaclofen (STX209; Seaside Therapeutics), the R-enantiomer of baclofen, is a GABABR agonist that inhibits presynaptic release of glutamate, thereby indirectly reducing downstream signaling of mGluR5 Citation[118]. The therapeutic effect of GABAergic agents may also extend from reduction of the imbalance between excitatory and inhibitory neurotransmission in FXS. Fmr1 mutant mice show reduced audiogenic seizures during acute treatment with arbaclofen, though tolerance to anticonvulsant effects develops rapidly with treatment extended beyond a single dose Citation[119-121]. In Fmr1 KO mice, arbaclofen has also been shown to reduce excessive protein synthesis, decrease dendritic spine density to WT values and correct aberrant AMPAR endocytosis Citation[121]. The restoration of normal spine density occurred after weaning and the peak period of new spine development, suggesting that arbaclofen can correct an established phenotype in FXS mouse models. Berry-Kravis et al. recently reported results of arbaclofen in a Phase II randomized, double-blind, placebo-controlled crossover study of 63 patients, aged 6 – 40 years, with FXS Citation[122]. Overall, subjects did not show statistically significant improvement on the primary outcome measure, the ABC-C Irritability subscale. Global improvement as measured by the CGI-I showed a trend toward improvement but also did not reach significance. With post hoc subset analysis of the 27 subjects presenting with the most severe ratings of social withdrawal, arbaclofen was associated with significant improvement on global measures, as well as the Vineland II-Socialization raw score and the ABC-Social Avoidance subscale, newly developed for use in FXS. In further post hoc analysis with the ABC-Social Avoidance subscale, significant improvements in social interaction were observed for the broader study population. Arbaclofen was well tolerated with no serious adverse events; headaches and sedation were most commonly reported. Phase III randomized, placebo-controlled trials of arbaclofen in children, adolescents and adults with FXS across 20 sites are currently underway; long-term, open-label extension trials are also planned Citation[123,124]. Seaside Therapeutics has also reported positive results from an open-label Phase IIa study of arbaclofen in 32 children with ASD, including significant global improvement on CGI-I and significant improvement on the ABC-C Irritability and Social Withdrawal subscales Citation[123].
8. Ampakines
8.1 CX516
Excessive internalization of AMPA receptors from the postsynaptic membrane in the absence of FMR1 has been associated with exaggerated LTD, reduced LTP and resultant deficits in learning and memory in FXS Citation[63,71]. Positive modulators of AMPA receptors (‘ampakines') prolong depolarization and excitatory postsynaptic potentials (EPSPs), primarily by delaying deactivation of the AMPA receptor Citation[125-127]. In turn, enhanced depolarization of the postsynaptic membrane unblocks voltage-sensitive NMDA receptors, which may ultimately account for enhanced induction of LTP Citation[127]. As LTP is thought to be involved in memory encoding, drugs that positively modulate AMPAR activity and thus indirectly facilitate LTP could potentially strengthen some types of memory and improve memory impairment Citation[126,128]. Ampakines have also been shown to increase production of BDNF, which may offer protection from excitotoxic neuronal damage and promote synaptic maturation Citation[127].
CX516 (1-(quinoxalin-6-ylcarbonyl)piperidine; Cortex Pharmaceuticals Irvine, CA, USA) and related ampakines have been shown to improve memory encoding in rats in a number of behavioral paradigms in association with increased hippocampal neuronal firing Citation[128-130]. CX516 also enhanced delayed recall of nonsense syllables in elderly subjects Citation[131] and improved performance on measures of attention and memory in adults with schizophrenia concomitantly treated with stable doses of clozapine Citation[132]. Based on these promising results, CX516 was investigated in a Phase II, randomized, placebo-controlled trial in 49 adults with FXS and ID Citation[133]. While CX516 was well tolerated with no serious adverse events other than a substantial rash in one patient, those taking CX516 did not demonstrate any improvement compared with placebo on a variety of cognitive and behavioral measures during the 4-week investigation period. However, the relatively brief period of monitoring may have precluded detection of more gradually developing cognitive benefits. In a study of elderly subjects, blood concentrations of CX516 were noted to drop by more than 50% between tests administered at 75 and 135 min after drug ingestion Citation[131]. Furthermore, CX516 is a relatively weak ampakine, with primate studies suggesting that a dosage three to four times that used may have been needed to observe a significant effect Citation[133]. The study was somewhat limited by choice of cognitive measures, some of which proved too difficult or complex to provide meaningful results for this population with an average Full Scale Intelligence Quotient (FSIQ) of 43.
9. Oxytocin
Oxytocin (OT) is a neuropeptide synthesized in the hypothalamus that promotes uterine contractions during parturition and milk let-down, as well as prosocial behaviors, including bonding, attachment and sexual behavior. OT also has anxiolytic and stress-reduction effects, and receptors for OT are found throughout the limbic system. Deficits in social interaction associated with autism may be related to aberrant functioning in the OT and related vasopressin systems, and several studies have reported that administration of synthetic OT can improve social behavior Citation[134-136] and repetitive behaviors Citation[137] in individuals with ASD.
Hall et al. recently reported results of a small, randomized, double-blind, placebo-controlled, single-dose trial of intranasal OT for the treatment of social anxiety in 10 young adult males with FXS Citation[138]. A small but significant increase in eye gaze frequency during a structured social challenge task was observed with a dose of 24 IU, and salivary cortisol levels decreased at 48 IU. OT was well tolerated with no adverse effects or changes in heart rate. The authors propose that OT may be effective in reducing social anxiety in FXS, potentially by reducing amygdala reactivity to perceived social threat, decreasing hypothalamic–pituitary–adrenal (HPA) axis activation, and promoting social motivation.
10. Acetylcholinesterase inhibitors
10.1 Donepezil
Aberrant activity in cholinergic pathways may contribute to cognitive impairments frequently observed in FXS, including executive dysfunction and deficits in learning and memory Citation[139]. Kesler et al. identified a significantly lower right choline/creatine ratio in the dorsolateral prefrontal cortex (DLPFC) using 1H magnetic resonance spectroscopy in nine males with FXS and a trend toward significantly reduced absolute choline levels Citation[139]. In a 6-week, open-label trial of the acetylcholinesterase inhibitor donepezil, subjects showed improvement in working memory, mental flexibility, hyperactivity, irritability and inattention Citation[139]. A randomized controlled trial of donepezil in young adults with FXS is currently underway at Stanford University (NCT01120626) Citation[140].
11. Conclusions
FXS is the most common single-gene cause of ID and autism with characteristic cognitive, physical and behavioral manifestations. Severe reduction or absence of FMRP results in loss of translational inhibition and overexpression of proteins involved in synaptic plasticity, purported to account for these pathognomonic signs and symptoms. Targeted drug treatments in animal models and humans antagonize group 1 mGluR activity, downstream signaling cascades or upregulated target proteins in an attempt to restore normal synaptic plasticity. Related targets for intervention include presynaptic GABAR and postsynaptic NMDAR and AMPAR activity. Large, well-designed placebo-controlled clinical trials in FXS are lacking and are needed to clarify early findings. The results have important implications for a number of neuropsychiatric disorders that may share clinical features and affected signaling pathways, including autism and a subset of ID.
12. Expert opinion
FXS is a single-gene disorder with neurobiological deficits that are increasingly well understood. In the absence of FMRP acting as a translational brake at excitatory synapses, group 1 mGluRs are overactive, resulting in excessive protein synthesis, enhanced LTD and loss of AMPARs and NMDARs from the synaptic membrane. Like schizophrenia and autism, FXS has been characterized as a ‘synapsopathy', a developmental disorder of synaptic development and plasticity Citation[79], with associated imbalance between the excitatory glutamatergic and inhibitory GABAergic systems. The proposed mechanism of disease in FXS affords an exciting opportunity for a targeted treatment approach to a neurodevelopmental disorder with psychiatric manifestations, as the pharmacological treatment of mental illness has traditionally relied on a ‘top down', serendipitous approach to drug development, resulting in symptom relief but less often correction of the underlying deficit. Targeted therapeutics in FXS thus have important implications for many other disorders with overlapping symptom clusters, including autism, ID, ADHD and anxiety disorders, as some of these disorders may share affected pathways. Autistic symptoms are particularly common in FXS, and many of the drugs in FXS Phase II and III trials, including OT, memantine and arbaclofen, show promise for the treatment of core symptoms of ASD, as well. While multiple genetic variations have been linked to the development of autism, a subset of individuals with ASD may have a disturbance of glutamatergic transmission similar to fragile X that is also amenable to therapeutics targeting the mGluR5 pathway.
In animal models of FXS, data supporting the mGluR theory and drugs that correct mGluR overexpression are robust, with many studies reporting phenotypic ‘rescue' and behavior that is indiscernible from WT. In human trials, results of targeted treatment trials have been encouraging but somewhat less striking. Some novel therapeutics, such as the ampakine CX516, appeared promising in preclinical trials but failed to demonstrate efficacy in clinical trials. The differences observed between animal and human subjects may reflect the milder fragile X phenotype observed in Fmr1 KO mice, which show less impairment in learning and memory than their human counterparts. Models of neuropsychiatric symptoms in animals are also unlikely to fully represent the complexity of human behavior and experience, and differences in pharmacokinetics, metabolism and dosing protocols between animals and humans could impact outcomes. Future development of targeted treatments should also consider potential unwanted effects based on their neurobiological mechanism of action. Drugs inhibiting overexpressed synaptic proteins, for example, could have off-target effects and cellular toxicity due to participation of these proteins in multiple cellular signaling pathways Citation[57].
In preclinical trials, mGluR5 antagonists have raised excitement for their ability to not only improve maladaptive behaviors but to reverse aspects of the physical phenotype, such as dendrite morphology, and to correct these abnormalities into adulthood. Still, aberrant brain development is present from the earliest stages of life, and some aspects of the phenotype are unlikely to improve with late intervention. One can assume that the most favorable outcomes would be obtained with early childhood intervention, and the choice of target population in future clinical trials should be carefully considered Citation[79]. Drugs that alter glutamate transmission and synaptic protein synthesis may improve cognitive functioning in individuals with FXS but lead to cognitive impairment in healthy volunteers without similar deficits, and this differential effect should also be considered during analysis of Phase I trials for newly developed drugs Citation[79].
Clinical trials in FXS have been hampered by methodological challenges. The fragile X population is relatively small, and recruitment of subjects to adequately power randomized controlled trials is difficult, even in multi-site studies. The majority of completed trials have used an open-label study design that carries significant potential for bias, particularly placebo and observer bias, and increased risk of type II error. Furthermore, many potential subjects with FXS have significant emotional and behavioral symptoms and are excluded from participation in Phase II and III drug trials because they are already taking psychotropic medications. Individuals with FXS have historically been excluded from larger studies of autism, and outcome measures that have been developed for more common neuropsychiatric disorders may lose validity when applied to the specific fragile X phenotype. FXS is uniquely associated with a diverse assortment of physical, psychological and cognitive symptoms, and because it has been unclear which features novel drugs may affect, choosing appropriate outcome measures has been challenging. While some studies have primarily focused on cognitive changes, others have chosen to track specific behavioral measures, such as social relatedness or irritability. Others have included biochemical markers of altered synaptic activity, though change in these markers has not always correlated with significant clinical effect. Failure to capture change in a particular area of functioning due to inadequate outcome measures could represent a missed therapeutic opportunity.
The ABC-C is a behavioral outcome measure with five subscales that has been validated and extensively utilized in ID and autism populations, and a recent exploratory and confirmatory factor analysis examined its psychometric properties in 630 children and young adults with FXS Citation[141]. The findings support modification of the original five factors (Irritability; Lethargy/Withdrawal; Stereotypy; Hyperactivity; Inappropriate Speech), as well as the addition of a new sixth factor, Social Avoidance, to improve sensitivity and specificity in the fragile X population. Further studies are needed to develop new behavioral outcome measures that specifically reflect the FXS phenotype, as well as cognitive measures that are appropriately matched to ability level and can accurately reflect change with intervention.
A number of multi-site randomized, controlled trials to further explore the efficacy of novel medications in FXS are underway, including minocycline, arbaclofen, donepezil and the mGluR5 antagonists AQ056, R04917523 and STX107. A major consequence of group 1 mGluR overactivity is loss of ionotropic NMDARs and AMPARs from the postsynaptic membrane, and future targeted treatment trials should also include drugs with the potential to modulate glutamatergic neurotransmission at these sites. Both reduced and enhanced NMDAR functioning has been associated with ASD-like phenotypes in mice, suggesting that maintaining balanced levels of NDMAR activity is critical Citation[142]. NMDAR partial agonists, including d-cycloserine and the related compound d-serine, bind to the regulatory glycine binding site on NMDARs and enhance activation in the presence of low concentrations of glycine site agonists but inhibit receptor/ion channel complex activity when agonist concentration is high Citation[143]. In a pilot study of d-cycloserine in 10 subjects with autistic disorder, 40% showed meaningful overall improvement, particularly in symptoms of social withdrawal Citation[144]. Mice with ASD-like behaviors and mutation in the Shank2 gene show markedly decreased NMDAR function and impaired social behaviors, both of which significantly improve with d-cycloserine Citation[142]. Human trials of d-cycloserine in other putative hypoglutamatergic neuropsychiatric disorders, such as schizophrenia, have been less promising, possibly due to d-cycloserine's relatively low affinity for the glycine site Citation[145]. d-Serine, however, is a full agonist at the glycine binding site and may have a more potent therapeutic effect; trials of d-serine in the treatment of prodromal schizophrenia are currently underway (NCT00826202) Citation[146]. GLYX-13 is a more recently developed NMDAR glycine site partial agonist that both enhances LTP and suppresses LTD in the hippocampus in vitro, suggesting promise for impairments in learning and memory in FXS and other neuropsychiatric disorders Citation[147]. Compared with d-cycloserine, GLYX-13 is a more potent glycine site agonist and has greater selectivity for NR2B-subunit-containing NMDARs, which may explain its more selective enhancement of LTP Citation[147]. GLYX-13 has been shown to improve pro-social vocalizations in mouse models of autism Citation[143]. Phase I clinical trials have been completed, and a Phase II trial of GLYX-13 as an add-on treatment in MDD is planned Citation[148]. In the design of future in vivo studies of GLYX-13, consideration should be given to its differential effects depending on drug concentration. At lower concentrations, it appears to enhance LTP and block LTD, though at higher concentrations, it can impair LTP and lose its effect on LTD Citation[147].
An alternative mechanism for reducing activation of postsynaptic mGluR5s, other than direct antagonism, is stimulation of mGluR2s and mGluR3s, presynaptic autoreceptors that modulate release of glutamate into the synaptic cleft. LY2140023 (pomaglumetad methionil) is a selective mGluR2/3 receptor agonist that demonstrated initial efficacy for reducing psychotic symptoms in schizophrenia similar to currently available antipsychotic drugs, though a larger Phase II clinical trial demonstrated a large placebo response and failed to replicate previous findings Citation[149,150]. LY2140023 appeared well tolerated with the notable exception of increased seizure activity. However, in a subsequent 24-week, open-label, active-controlled study comparing LY2140023 and standard of care treatment with atypical antipsychotics, no seizures in the LY2140023 group were reported Citation[149]. Risk of seizure activity should be further characterized before considering trials in individuals with FXS, many of whom have a concomitant seizure disorder.
Characterization of the neurobiological defects and targeted drug development in FXS affords an exciting opportunity for the application of translational medicine in the treatment of neuropsychiatric disorders. FXS is the most common known genetic cause of autism and ID, and studies in FXS therefore have potentially important implications for the broader understanding and treatment of these disorders. Future clinical trials are needed to clarify the efficacy of existing targeted drugs for different symptom domains, explore the utility of recently developed drugs that impact glutamatergic neurotransmission in novel ways and define outcome measures that promote consistency and allow comparison across trials.
Article highlights.
Fragile X syndrome (FXS) is a single-gene disorder with characteristic cognitive, physical and behavioral features that overlap many more common neurodevelopmental disorders, including intellectual disability (ID) and autism spectrum disorder (ASD), and serves as an important neurobiological model for developing targeted drug treatments.
The proposed disease mechanism in FXS involves overactivity of group 1 metabotropic glutamate receptors (mGluRs) and increased synaptic protein synthesis in the absence of translational inhibition normally provided by Fragile X Mental Retardation Protein-1 (FMRP), leading to impairments in long-term depression (LTD) and synaptic plasticity.
Modulators of glutamatergic and GABAergic neurotransmission have the potential to ameliorate behavioral symptoms and possibly improve cognitive deficits in FXS.
Newly developed N-methyl-d-aspartate receptor (NMDAR) partial agonists and mGluR2/3 agonists are drugs that have not been studied in FXS to date and warrant further investigation.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Acknowledgments
The authors would like to thank Ben Kelmendi, MD, of Yale University Department of Psychiatry, for graciously designing the illustration in this manuscript.
Notes
This box summarizes key points contained in the article.
Bibliography
- Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet 1996;64:196-7
- Pesso R, Berkenstadt M, Cuckle H, Screening for fragile X syndrome in women of reproductive age. Prenat Diagn 2000;20:611-14
- Hagerman PJ. The fragile X prevalence paradox. J Med Genet 2008;45:498-9
- Verkerk AJMH, Pieretti M, Sutcliffe JS, Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991;65:905-14
- Ashley CT Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science 1993;262:563-6
- Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 1993;74:291-8
- Feng Y, Absher D, Eberhart DE, FMRP Associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell 1997;1:109-18
- Verheij C, Bakker CE, de Graaff E, Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 1993;363:722-4
- Li Z, Zhang Y, Ku L, The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res 2001;29:2276
- Darnell JC, Jensen KB, Jin P, Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 2001;107:489-99
- Tassone F, Hagerman RJ, Taylor AK, Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Gent 2000;66:6-15
- Brunberg JA, Jacquemont S, Hagerman RJ, Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol 2002;23:1757-66
- Greco CM, Hagerman RJ, Tassone F, Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002;125:1760-71
- Hagerman PJ, Hagerman RJ. Fragile X-associated Tremor/Ataxia Syndrome (FXTAS). Ment Retard Devel Disabil Res Rev 2004;10:25-30
- Tsiouris JA, Brown WT. Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs 2004;18:687-703
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Advances in the treatment of fragile X syndrome. Pediatrics 2009;123:378-90
- Hatton DD, Sideris J, Skinner M, Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A 2006;140A:1804-13
- Tranfaglia MR. The psychiatric presentation of fragile X: evolution of the diagnosis and treatment of the psychiatric comorbidities of fragile X syndrome. Dev Neurosci 2011;33:337-48
- Cordeiro L, Ballinger E, Hagerman R, Hessl D. Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J Neurodev Disord 2011;3:57-67
- Brown WT, Jenkins EC, Krawczun MS, The fragile X syndrome. Ann NY Acad Sci 1986;477:129-50
- Loesch DZ, Huggins RM, Bui QM, Effect of fragile X status categories and FMRP deficits on cognitive profiles estimated by robust pedigree analysis. Am J Med Genet A 2003;122A:13-23
- Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Devel Disabil Res Rev 2004;10:31-41
- Hagerman RJ, Jackson C, Amiri K, Girls with fragile X syndrome: physical and neurocognitive status and outcome. Pediatrics 1992;89:395-400
- De Vries BB, Wiegers AM, Smits AP, Mental status of females with an FMR1 gene full mutation. Am J Hum Genet 1996;58:1025
- Martinez R, Bonilla-Henao V, Jimenez A, Skewed X inactivation of the normal allele in fully mutated female carriers determines the levels of FMRP in blood and the fragile X phenotype. Mol Diagn 2005;9:157-62
- Tassone F, Hagerman RJ, Taylor AK, Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet 2000;91:144-52
- Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Fragile X premutation is a significant risk factor for premature ovarian failure: the international collaborative POF in fragile X study—preliminary data. Am J Med Genet 1999;83:322-5
- Spath MA, Nillesen WN, Smits APT, X chromosome inactivation does not define the development of premature ovarian failure in fragile X premutation carriers. Am J Med Genet A 2010;152A:387-93
- Hagerman RJ, Leehey M, Heinrichs W, Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001;57:127-30
- Jacquemont S, Hagerman RJ, Leehey M, Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet 2003;72:869-78
- Jacquemont S, Leehey MA, Hagerman RJ, Size bias of fragile X premutation alleles in late onset-onset movement disorders. J Med Genet 2006;43:804-9
- Bailey DB, Raspa M, Bishop E, Medication utilization for targeted symptoms in children and adults with fragile X syndrome. J Dev Behav Pediatr 2012;33:62-9
- Erickson CA, Stigler KA, Wink LK, A prospective open-label study of aripiprazole in fragile X syndrome. Psychopharmacology 2011;216:85-90
- Lejeune J, Rethore MO, de Blois MC, Trial of folic acid treatment in fragile X syndrome. Ann Genet 1984;27:230-2
- Brown WT, Cohen IL, Fisch GS, High dose folic acid treatment of fragile (X) males. Am J Med Genet 1986;23:263-71
- Hagerman RJ, Jackson AW, Levitas A, Oral folic acid versus placebo in the treatment of males with the fragile X syndrome. Am J Med Genet 1986;23:241-62
- Wells TE, Madison LS. Assessment of behavior change in a fragile-X syndrome male treated with folic acid. Am J Med Genet 1986;23:291-6
- Gillberg C, Wahlstrom J, Johansson R, Folic acid as an adjunct in the treatment of children with the autism fragile-X syndrome (AFRAX). Dev Med Child Neurol 1986;28:624-7
- Froster-Iskenius U, Bodeker K, Oepen T, Folic acid treatment in males and females with fragile-(X)-syndrome. Am J Med Genet 1986;23:273-89
- Fisch GS, Cohen IL, Gross AC, Folic acid treatment of fragile X males: a further study. Am J Med Genet 1988;30:393-9
- Strom CM, Brusca RM, Pizzi WJ. Double-blind, placebo-controlled crossover study of folinic acid (Leucovorin®) for the treatment of fragile X syndrome. Am J Med Genet 1992;44:676-82
- Rueda J-R, Ballesteros J, Guillen V, Folic acid for fragile X syndrome. Cochrane Database Syst Rev John Wiley & Sons, Ltd, 2011(5):CD008476
- Hagerman RJ, Murphy MA, Wittenberger MD. A controlled trial of stimulant medication in children with the fragile X syndrome. Am J Med Genet 1988;30:377-92
- Roberts J, Miranda M, Boccia M, Treatment effects of stimulant medication in young boys with fragile X syndrome. J Neurodevel Disord 2011;3:175-84
- Pascale E, Battiloro E, Reale GC, Modulation of methylation in the FMR1 promoter region after long term treatment with L-carnitine and acetyl-L-carnitine. J Med Genet 2003;40:e76-6
- Torrioli MG, Vernacotola S, Mariotti P, Double-blind, placebo-controlled study of L-acetylcarnitine for the treatment of hyperactive behavior in fragile X syndrome. Am J Med Genet 1999;87:366-8
- Torrioli MG, Vernacotola S, Peruzzi L, A double-blind, parallel, multicenter comparison of L-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in fragile X syndrome boys. Am J Med Genet A 2008;146:803-12
- Torrioli MG, Vernacotola S, Setini C, Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A 2010;152:1420-7
- Wirojanan J, Jacquemont S, Diaz R, The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med 2009;5:145
- Bakker CE, Verheij C, Willemsen R, Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 1994;78:23-33
- Comery TA, Harris JB, Willems PJ, Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci 1997;94:5401-4
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci 2004;27:370-7
- Kooy RF. Of mice and the fragile X syndrome. Trends Genet 2003;19:148-54
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci 2001;21:5139-46
- Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci 2002;99:1639-44
- Zhang YQ, Bailey AM, Matthies HJG, Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 2001;107:591-603
- Berry-Kravis E, Knox A, Hervey C. Targeted treatments for fragile X syndrome. J Neurodevel Disord 2011;3:193-210
- Brown V, Jin P, Ceman S, Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001;107:477-87
- Wang H, Dictenberg JB, Ku L, Dynamic association of the fragile X mental retardation protein as a messenger ribonucleoprotein between microtubules and polyribosomes. Mol Biol Cell 2008;19:105-14
- Weiler IJ, Irwin SA, Klintsova AY, Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci 1997;94:5395
- Westmark CJ, Westmark PR, Malter JS. MPEP reduces seizure severity in Fmr-1 KO mice over expressing human Abeta. Int J Clin Exp Pathol 2010;3:56-68
- Huber KM. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 2000;288:1254-6
- Snyder EM, Philpot BD, Huber KM, Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci 2001;4:1079-85
- Qin M, Kang J, Burlin TV, Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the Fmr1 null mouse. J Neurosci 2005;25:5087-95
- Dolen G, Carpenter RL, Ocain TD, Bear MF. Mechanism-based approaches to treating fragile X. Pharmacol Ther 2010;127:78-93
- Bittel DC, Kibiryeva N, Butler MG. Whole genome microarray analysis of gene expression in subjects with fragile X syndrome. Genet Med 2007;9:464-72
- Lu R, Wang H, Liang Z, The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci 2004;101:15201
- Brown MR, Kronengold J, Gazula V-R, Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci 2010;13:819-21
- Suvrathan A, Hoeffer CA, Wong H, Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci 2010;107:11591-6
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci 2002;99:7746-50
- Nakamoto M, Nalavadi V, Epstein MP, Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci 2007;104:15537
- D'Hulst C, De Geest N, Reeve SP, Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res 2006;1121:238-45
- Heulens I, D'Hulst C, Van Dam D, Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res 2012;229(1):244-9
- Bear MF. Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav 2005;4:393-8
- McBride SMJ, Choi CH, Wang Y, Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 2005;45:753-64
- Dolen G, Bear MF. Courting a Cure for Fragile X. Neuron 2005;45:642-4
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005;49:1053-66
- Dolen G, Osterweil E, Rao BSS, Correction of fragile X syndrome in mice. Neuron 2007;56:955-62
- Bear MF, Dolen G, Osterweil E, Nagarajan N. Fragile X: translation in action. Neuropsychopharmacology 2008;33:84-7
- Thomas A, Bui N, Perkins J, Yuva-Paylor L. Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berl) 2012;219:47-58
- de Vrij FMS, Levenga J, van der Linde HC, Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 2008;31:127-32
- Levenga J, Hayashi S, de Vrij F, AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis 2011;42:311-17
- Porter RHP. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther 2005;229(1):244-9
- Pecknold JC, McClure DJ, Appeltauer L, Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacol 1982;2:129-32
- Berry-Kravis E, Hessl D, Coffey S, A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet 2009;46:266-71
- Vinueza Veloz MF, Buijsen RAM, Willemsen R, The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav 2012;11:325-31
- Jacquemont S, Curie A, des Portes V, Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential responses to the mGluR5 antagonist AFQ056. Sci Transl Med 2011;3:64ra1
- Tabolacci E, Pirozzi F, Gomez-Mancilla B, The mGluR5 antagonist AFQ056 does not affect methylation and transcription of the mutant FMR1 gene in vitro. BMC Med Genet 2012;13:13-17
- Novartis. Safety and efficacy of AFQ056 in adult patients with fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01253629?term=AFQ056+and+fragile+x&rank=4 [Last accessed 30 September 2012]
- Novartis. Safety and efficacy of AFQ056 in adolescent patients with fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01357239?term=AFQ056+and+fragile+x&rank=1 [Last accessed 30 September 2012]
- Novartis. Long-term, safety, tolerability and efficacy study of AFQ056 in adult patients with fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01348087?term=AFQ056+and+fragile+x&rank=2 [Last accessed 30 September 2012]
- Novartis. Long-term, safety and tolerability study of AFQ056 in adolescent patients with fragile X syndrome (open-label). NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01433354?term=AFQ056+and+fragile+x&rank=3 [Last accessed 30 September 2012]
- Novartis. Clinical study to assess the pharmacokinetics, safety and tolerability of single and multiple oral doses of AFQ056 in children with fragile X syndrome (FXS). NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01482143?term=AFQ056+and+fragile+x&rank=5 [Last accessed 30 September 2012]
- Erickson CA, Mullett JE, McDougle Christopher J. Brief Report: acamprosate in fragile X syndrome. J Autism Devel Disord 2010;40:1412-16
- Indiana University. Acamprosate in youth with fragile X syndrome. NIH: clinicalTrials.gov. 2011. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01300923?term=acamprosate+fragile+x&rank=1 [Last accessed 30 September 2012]
- NIH. Search of: RO4917523 - list results - ClinicalTrials.gov. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/results?term=RO4917523&Search=Search [Last accessed 30 September 2012]
- NIH. Search of: STX107- list results- ClinicalTrials.gov. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/results?term=stx107&Search=Search [Last accessed 30 September 2012]
- Lindemann L, Jaeschke G, Michalon A, CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J Pharmacol Exp Ther 2011;339:474-86
- Michalon A, Sidorov M, Ballard TM, Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 2012;74:49-56
- Erickson CA, Weng N, Weiler IJ, Open-label riluzole in fragile X syndrome. Brain Res 2011;1380:264-70
- Erickson CA, Mullett JE, McDougle Christopher J. Open-label memantine in fragile X syndrome. J Autism Devel Disord 2009;39:1629-35
- Wei H, Dobkin C, Sheikh AM, The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS One 2012;7:e36981
- Ortigas MC, Bourgeois JA, Schneider A, Improving Fragile X-Associated Tremor/Ataxia Syndrome symptoms with memantine and venlafaxine. J Clin Psychopharmacol 2010;30:642-4
- Bilousova TV, Dansie L, Ngo M, Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 2008;46:94-102
- Siller SS, Broadie K. Matrix Metalloproteinases and minocycline: therapeutic avenues for fragile X syndrome. Neural Plast 2012;2012:1-9
- Paribello C, Tao L, Folino A, Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol 2010;10:91
- Siller SS, Broadie K. Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech 2011;4:673-85
- Rotschafer SE, Trujillo MS, Dansie LE, Minocycline treatment reverses ultrasonic vocalization production deficit in a mouse model of Fragile X Syndrome. Brain Res 2012;1439:7-14
- University of California, Davis. Trial of minocycline to treat children with fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01053156?term=fragile+x+syndrome+and+minocycline&rank=1 [Last accessed 30 September 2012]
- Utari A, Chonchaiya W, Rivera SM, Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am J Intellect Dev Disabil 2010;115:433-43
- Berry-Kravis E, Sumis A, Hervey C, Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Devel Behav Pediatr 2008;29:293
- Min WW, Yuskaitis CJ, Yan Q, Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 2009;56:463-72
- Liu Z-H, Chuang D-M, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 2012;14:618-30
- Yuskaitis CJ, Mines MA, King MK, Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of Fragile X syndrome. Biochem Pharmacol 2010;79:632-46
- Mines MA, Yuskaitis CJ, King MK, GSK3 Influences Social Preference and Anxiety-Related Behaviors during Social Interaction in a Mouse Model of Fragile X Syndrome and Autism. PLoS ONE 2010;5:e9706
- Choi CH, Schoenfeld BP, Bell AJ, Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res 2011;1380:106-19
- Liu Z-H, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis 2012;45:1145-52
- Hopkins CR. ACS chemical neuroscience molecule spotlight on STX209 (arbaclofen). ACS Chem Neurosci 2011;2:381-1
- Pacey LKK, Heximer SP, Hampson DR. Increased GABAB receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol 2009;76:18-24
- Pacey LKK, Tharmalingam S, Hampson DR. Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J Pharmacol Exp Ther 2011;338:897-905
- Henderson C, Wijetunge L, Kinoshita MN, Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med 2012;4:152ra128
- Berry-Kravis EM, Hessl D, Rathmell B, Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med 2012;4:152ra127
- Seaside Therapeutics. News: seaside Therapeutics reports positive data from phase 2 study of STX209 in autism spectrum disorders. Seaside Therapeutics. 2010. Available from: http://www.seasidetherapeutics.com/news [Last accessed 30 September 2012]
- Seaside Therapeutics. A fixed-dose study of the efficacy, safety, and tolerability of STX209 (arbaclofen) administered for the treatment of social withdrawal in children with fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01325220?term=STX209&rank=3 [Last accessed 30 September 2012]
- Arai A, Kessler M, Xiao P, A centrally active drug that modulates AMPA receptor gated currents. Brain Res 1994;638:343-6
- Arai A, Kessler M, Rogers G, Effects of a memory-enhancing drug on DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor currents and synaptic transmission in hippocampus. J Pharmacol Exp Ther 1996;278:627-38
- Lynch G. AMPA receptor modulators as cognitive enhancers. Curr Opin Pharmacol 2004;4:4-11
- Staubli U, Rogers G, Lynch G. Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci 1994;91:777-81
- Hampson RE, Rogers G, Lynch G, Facilitative effects of the ampakine CX516 on short-term memory in rats: enhancement of delayed-nonmatch-to-sample performance. J Neurosci 1998;18:2740-7
- Hampson RE, Rogers G, Lynch G, Deadwyler SA. Facilitative effects of the ampakine CX516 on short-term memory in rats: correlations with hippocampal neuronal activity. J Neurosci 1998;18:2748-63
- Lynch G, Granger R, Ambros-Ingerson J, Evidence that a positive modulator of AMPA-type glutamate receptors improves delayed recall in aged humans. Exp Neurol 1997;145:89-92
- Goff DC, Leahy L, Berman I, A placebo-controlled pilot study of the ampakine CX516 added to clozapine in schizophrenia. J Clin Psychopharmacol 2001;21:484-7
- Berry-Kravis E, Krause SE, Block SS, Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol 2006;16:525-40
- Hollander E, Bartz J, Chaplin W, Oxytocin increases retention of social cognition in autism. Biol Psychiatry 2007;61:498-503
- Andari E, Duhamel J-R, Zalla T, Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci 2010;107:4389-94
- Guastella AJ, Einfeld SL, Gray KM, Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol Psychiatry 2010;67:692-4
- Hollander E, Novotny S, Hanratty M, Oxytocin infusion reduces repetitive behaviors in adults with Autistic and Asperger's Disorders. Neuropsychopharmacology 2003;28:193-8
- Hall SS, Lightbody AA, McCarthy BE, Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology 2012;37:509-18
- Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in fragile X syndrome and potential intervention: a preliminary 1H MRS study. Am J Med Genet A 2009;149:403-7
- Stanford University. Randomized controlled study of donepezil in fragile X syndrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01120626?term=fragile+x+and+donepezil&rank=2 [Last accessed 30 September 2012]
- Sansone S, Widaman K, Hall S, Psychometric study of the Aberrant Behavior Checklist in fragile X syndrome and implications for targeted treatment. J Autism Dev Disord 2012;42:1377-92
- Won H, Lee H-R, Gee HY, Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012;486:261-5
- Moskal JR, Burgdorf J, Kroes RA, A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci Biobehav Rev 2011;35:1982-8
- Posey DJ, Kem DL, Swiezy NB, A pilot study of D-cycloserine in subjects with autistic disorder. Am J Psychiatry 2004;161:2115-17
- Goff DC, Herz L, Posever T, A six-month, placebo-controlled trial of D-cycloserine co-administered with conventional antipsychotics in schizophrenia patients. Psychopharmacology (Berl) 2005;179:144-50
- Nathan Kline Institute for Psychiatric Research. D-serine for the schizophrenia prodrome. NIH: clinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00826202?term=D-serine+schizophrenia&rank=3 [Last accessed 30 September 2012]
- Zhang X, Sullivan JA, Moskal JR, Stanton PK. A NMDA receptor glycine site partial agonist, GLYX-13, simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hippocampus. Neuropharmacology 2008;55:1238-50
- Naurex, Inc. Phase 2, double-blind, placebo controlled, randomized withdrawal, parallel efficacy and safety study of GLYX-13 in subjects with inadequate/partial response to antidepressants during the current episode of Major Depressive Disorder. NIH: ClinicalTrials.gov. 2012. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01684163?term=GLYX-13&rank=1 [Last accessed: 30 September 2012]
- Kinon BJ, Gomez J-C. Clinical development of pomaglumetad methionil: a non-dopaminergic treatment for schizophrenia [Internet]. Neuropharmacology 2012; http://dx.doi.org.ezp-prod1.hul.harvard.edu/10.1016/neuropharm.2012.06.002
- Kinon BJ, Zhang L, Millen BA, A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol 2011;31:349-55