Abstract
Introduction: Thirty to forty percent of orphan drugs approved are used in the management of malignancy. This reflects the revolution in molecular oncology and biotechnology witnessed over the last 30 years. The identification of molecular pathways intrinsic to the origin of cancer has resulted in finding targets for anticancer agents and molecular biomarkers predictive of drug response. The clinical application of biomarkers in oncology has resulted in the identification of rare cancers within common cancer types. Orphan drug development plays a key role in sustaining the era of personalized cancer medicine.
Areas covered: The development of crizotinib and its companion Vysis ALK Break Apart FISH Probe Kit is presented in this paper to highlight how the field of medical oncology is integrating advances in molecular biology to deliver the goal of personalized cancer care. The molecular basis of ALK-positive lung cancer and the evidence that supported crizotinib in successfully receiving an accelerated, orphan drug marketing approval status are also discussed.
Expert opinion: The international oncology community strives to improve cancer outcomes. The revolution in molecular oncology has enabled the transition toward an era of targeted therapy and personalized medicine. Oncologists actively encourage pharmaceutical companies to develop novel anticancer agents in conjunction with a companion diagnostic test. This identifies a subgroup of patients most likely to respond to a drug thereby limiting unnecessary toxicity and cost. This together with the financial incentives established by the Orphan Drug Act will fuel continued research in oncology orphan drug development in association with predictive biomarkers.
1. Introduction
Diseases that arise in patient populations representing at the maximum 6 – 8% of the world population are defined as orphan or rare diseases. In the USA, an orphan disease is defined by the Food and Drug Administration (FDA) as one affecting fewer than 200,000 people. Symptoms of rare diseases may occur early, at birth or in childhood, including cystic fibrosis or familial adenomatous polyposis. However, more than 50% of rare diseases become apparent during adulthood, such as renal cell carcinoma, glioma and acute myeloid leukemia. Currently, there are over 5000 rare diseases listed in the world with 5 new rare diseases being described every week in the medical literature. Four out of five rare diseases have a genetic basis. Other rare diseases are the result of infections and allergies, or are due to degenerative and proliferative causes. An orphan drug is a medicinal product that has been developed specifically to treat a rare medical condition, the condition itself being referred to as an ‘orphan disease'. A total of 362 drugs have been given orphan drug marketing approval since 1983. Orphan drugs have been approved for every pathologic area.However, over time there has been a definite shift in the development of orphan drugs to target certain key pathologic areas including oncology, hematology, gastrointestinal (GI) inborn error, rheumatology and neurology. Between 1983 and 1988, orphan drugs targeting cancers did not feature in the top five pathologic areas for orphan drug approvals. Whereas today 30 – 40% of orphan drugs approved are used in the management of malignancy. This increase in oncology drugs successfully receiving orphan drug marketing approval status reflects the revolution in molecular oncology and biotechnology witnessed over the last 30 years. Several factors are recognized as key elements in the successful development of an orphan drug (). Therefore, orphan drug development is increasingly a crucial component in drug development in oncology. As previously outlined, the management of cancer is becoming customized to the individual patient and the specific tumor biology. The identification of molecular pathways intrinsic to the origin of cancer has resulted in finding molecular biomarkers and targets for anticancer agents. Through research in the area of molecular oncology, the oncology community has come to realize the enormity of heterogeneity present in all cancer subtypes. Indeed, it is now realized that one cancer subtype, for example breast cancer, encompasses a number of different cancers each with their own unique pathologic, prognostic and pharmacodynamic characteristics () Citation[1].
Figure 1. Factors influencing successful orphan drug development.
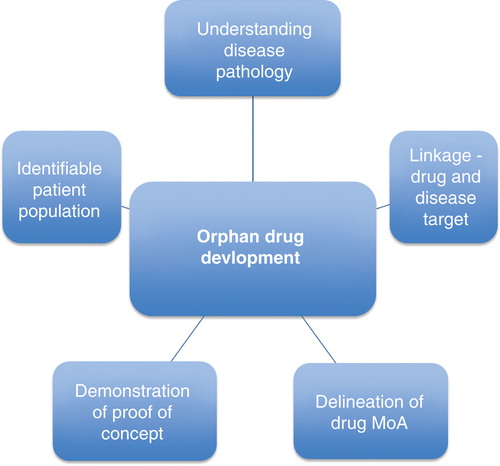
Figure 2. Intrinsic breast cancer subtype DNA microarray.
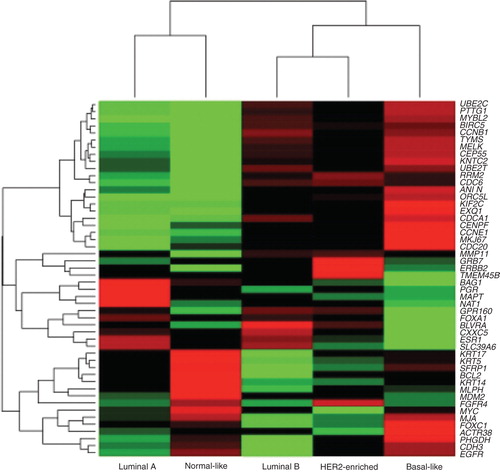
The clinical application of biomarkers in oncology has resulted in the identification of rare cancers within common cancer types (), for example, human epidermal growth factor receptor (HER2)-positive breast cancer, triple negative breast cancer, estrogen receptor-positive breast cancer, EGFR-mutated advanced non-small cell lung cancer (NSCLC), KRAS wild–type (WD) and mutated metastatic colorectal cancer and BRAF (v-raf murine sarcoma viral oncogene homolog B1)-mutated advanced melanoma. It may be more accurate to describe the rare cancers identified by molecular biomarkers within common cancer types as adopted as opposed to orphan diseases.
Table 1. Examples of predictive biomarkers for drug response.
Predictive biomarkers can identify distinct subgroups, within common cancer types, that are sensitive to specific targeted agents (). Thus, representing potential targets for orphan drug development. In NSCLC, 16.6% of Caucasian and 30 – 50% of Asian patients with lung adenocarcinoma carry a mutation in EGFR Citation[2,3] which confers sensitivity to tyrosine kinase inhibitors (TKIs) including erlotinib and gefitinib Citation[4]. Similarly, in melanoma the presence of a BRAF mutation is seen in 60% of tumors Citation[5]. As a result, these tumors exhibit enhanced BRAF kinase activity and increased phosphorylation of downstream targets, particularly MEK. Vemurafenib (PLX4032) has marked antitumor effects against melanoma expressing the BRAF V600E mutation but not against tumors with WD BRAF Citation[6]. However, although the response to BRAF inhibition is often profound it appears to be temporary due to the development of resistance. Understanding the mechanisms leading to resistance has allowed the development of other therapeutic approaches, for example, the combined inhibition of MEK and RAF may prove useful in patients whose resistance is due to mutations affecting RAS proteins Citation[7]. The BRAF story highlights the point that in the future we will need multiple agents targeting different pathways simultaneously to overcome the limitations that resistance to targeted agents confers on a tumor. There is also considerable interest in identifying types of c-Kit mutations in patients with GI stromal tumors for both prognostic and predictive information, and possibly more appropriate drug dosing Citation[8,9].
This article uses the development of crizotinib, an orphan drug, and its companion Vysis ALK Break Apart FISH (fluorescent in situ hybridization) Probe Kit to highlight how the field of medical oncology is integrating advances in molecular biology to deliver the goal of personalized medicine to individuals with cancer.
2. Personalized cancer medicine: targeted treatment
The revolution in molecular oncology and biotechnology has created a paradigm shift toward personalized cancer medicine Citation[10]. We are moving away from the empirical strategy of ‘one drug fits all' toward a customized cancer treatment where individual tumor biomarker expression profiles drive management Citation[10,11].
Understanding the concept of oncogene addiction has led to a rationalized approach to therapeutic selectivity of tumor cells over normal cells currently seen in personalized cancer medicine. This concept refers to the idea that cancer cells are dependent on the molecular pathways that drive their malignant pathways. If a molecular abnormality crucial to the development of the cancer cell is targeted there is potential to exert greater anticancer effect with significantly less toxicity to normal tissue than the broadly antiproliferative cytotoxic drugs that dominate current therapy Citation[12].
The term ‘biomarker' refers to a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes or pharmacological responses to a specified therapeutic intervention Citation[13]. Biomarkers in oncology can be prognostic, pharmacodynamic or predictive Citation[14]. A prognostic biomarker anticipates the likely outcome of an illness. In the setting of adjuvant cancer, a prognostic biomarker gives information regarding disease recurrence and in the metastatic setting time to progression or survival. The National Institute of Health (NIH) Consensus Conference suggests that a clinically useful prognostic marker must be a proven independent, significant factor that is easy to determine and interpret and has therapeutic consequences Citation[15]. An example of prognostic biomarkers available today in the setting of adjuvant breast cancer include the Oncotype DX test Citation[16], mammaprint, Rotterdam signature and breast cancer gene expression ratio Citation[17]. Pharmacodynamic biomarkers determine or measure the effect of a drug on the disease Citation[18]; for example, the degree of change on a substrate regulated by an enzymatic drug target (such as phosphorylation after inhibition of a protein kinase) Citation[19]. Predictive biomarkers help to foretell the likelihood of a cancer responding to a particular drug. Pharmaceutical companies are being actively encouraged to identify such biomarkers as companion diagnostic tests for the drugs they develop. Use of predictive biomarkers attempts to subselect the population most likely to respond to a drug, thereby minimizing unnecessary toxicity and reducing cost. Predictive biomarkers that are now commonly employed by a practicing oncologist include: estrogen and progesterone receptor protein expression (ER, PR), HER2/neu overexpression, EGFR, KRAS and BRAF mutation detection ().
2.1 ELM4-ALK-positive NSCLC: the crizotinib story
Lung cancer is a leading cause of cancer deaths worldwide Citation[20,21]. Based on GLOBOCAN 2008 estimates, lung cancer accounts for 13% (1.6 million) of the total cancer cases and 18% (1.4 million) of the cancer-related deaths in 2008 Citation[22]. NSCLC accounts for 80% of lung cancers diagnosed. Unfortunately, 30 – 40% of patients present with advanced stage disease Citation[23], when chemotherapy regimens fail to impact significantly on survival Citation[24]. The median survival for patients with clinical stage IV disease is 6 months Citation[25]. Poor outcomes and the plateau reached by cytotoxic chemotherapy in terms of efficacy encouraged research into new anticancer agents that target proteins that are selectively expressed by and/or undergo genomic alterations in cancer cells. As outlined previously, specific genetic lesions that drive the proliferation of cancer cells render some cancers sensitive to therapeutic inhibitors targeting the mutated pathway. Testing for these genetic mutations and directing therapies against these targets can lead to improved patient outcomes.
Chromosomal translocation has long been linked to cancer causation Citation[26]. This genetic abnormality is implicated in approximately 20% of cancers Citation[27]. Fusion genes are common in lymphoid, hematopoietic and connective tissue cancers; however, they are believed to be rare in solid tumors, with the exception of prostatic carcinoma Citation[28,29]. EML4-ALK consists of a fusion of the echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) genes resulting from the chromosome inversion inv(2)(p21;p23) Citation[30]. Soda et al. generated a retroviral cDNA expression library from a lung adenocarcinoma specimen resected from a 62-year-old male smoker. To build this library, they used SMART method (clontech) for preferential amplification of full-length cDNAs from clinical specimens. One of the amplified cDNAs contained an open reading frame for a predictive protein. The amino-terminal portion of which was identical to that of human EML4. The carboxy-terminal portion was identical to the intracellular domain of human ALK. This suggested that the cDNA was a derivative from a fusion product of EML4 and ALK. EML4 is a member of the family of echinoderm microtubule-associated protein-like proteins Citation[31]. ELM4 consists of an N-terminal basic region, a hydrophobic echinoderm microtubule-associated protein-like protein (HELP) domain Citation[32] and WD repeats Citation[33]. In the predictive gene fusion protein, the N-terminal half of EML4 is fused to the intracellular juxtamembrane region of ALK. This results in constitutive dimerization of the kinase domain of ALK and a consequent increase in its catalytic activity.
The ELM4-ALK fusion gene protein has previously been identified in other tumors, including anaplastic large cell lymphoma Citation[34], neuroblastoma Citation[35] and inflammatory myofibroblastic tumor Citation[36]. ALK fusion gene rearrangements have also been identified in non-clear cell renal carcinoma, breast and colorectal cancer Citation[37]. ELM4-ALK-activating mutations in NSCLC occur for the most part in the absence of gene mutations in KRAS/EGFR mutations Citation[38,39]. In the majority of cases when such mutations are found they help to identify distinct disease subgroups. The presence of the activating, oncogenic, fusion gene of ELM4-ALK is uncommon and present in around 5% of patients with NSCLC. This is equivalent to over 70,000 patients worldwide. Thus, creating a small subgroup of patients with NSCLC that may be susceptible to and respond to ALK TKIs Citation[40]. This constitutes a rare disease. NSCLC harboring the ELM4-ALK fusion gene tend to occur in younger patients, most often non-smokers, with a predilection for lung adenocarcinomas Citation[41] however, Wong et al. has observed its presence in various histologic types of lung cancer Citation[42]. ALK gene translocations have been detected in anaplastic large cell lymphomas for many years and a variety of diagnostic methods have been validated. However, in NSCLC there is no standard method for identifying an EML4-ALK fusion translocation. Methods currently being evaluated include polymerase chain reaction (PCR), immunohistochemistry (IHC) and FISH Citation[43]. In NSCLC, alternative fusion partners for ALK have been identified including KIF5B and KLC1 Citation[44,45].
Soda et al. observed the oncogenic nature of the ELM4-ALK fusion gene in lung cancer Citation[46]. Preclinical studies showed that when ALK inhibitors were directed at EML4-ALK-positive lung cancer cell lines, apoptosis and down-regulation of cell survival pathways occurred Citation[47]. ELM4-ALK-positive lung cancers do not respond to EGFR TKIs Citation[48] and unlike EGFR-mutated lung cancer uncertainty remains as to whether ALK-mutated lung cancers may have enhanced sensitivity to chemotherapy. Currently, there is one agent targeting EML4-ALK fusion gene translocation that has been clinically approved.
3. Crizotinib (PF-02341066)
PF-02341066 (crizotinib) is an oral, bioavailable, small-molecule, multi-targeted TKI shown to inhibit the proliferation and growth of EML4-ALK-positive tumors including NSCLC () Citation[49]. Several clinical studies have now shown the efficacy of the ALK inhibitor in ALK-positive advanced NSCLC. Kwak et al. reported an overall response rate (ORR) of 57% for patients receiving 250 mg of crizotinib daily, in at least the second-line setting Citation[50]. At the data cutoff time, 77% of patients continued to receive crizotinib, and the estimated probability of 6-month progression-free survival (PFS) was 72%. Crinò et al. presented the ‘Initial Phase II results with crizotinib in advanced ALK-positive NSCLC: PROFILE 1005', an open-label study examining the efficacy of crizotinib for previously treated patients with advanced ALK-rearranged NSCLC, at the American Society of Clinical Oncology (ASCO) in 2011. A total of 136 patients with ALK-rearranged advanced NSCLC, from 12 countries, who had progressed after at least one line of chemotherapy were enrolled to receive crizotinib. The results were promising with 83% experiencing tumor shrinkage and 54% had a partial response by RECIST criteria. Crizotinib was well tolerated and quality of life was maintained Citation[51].
Table 2. Clinical trials studying crizotinib in ALK-positive advanced NSCLC.
There are also two Phase III trials in progress. The first, PROFILE 1007, randomized patients to compare crizotinib with docetaxel or pemetrexed in the second-line setting for advanced ALK-positive NSCLC. In June 2012, Pfizer reported that this study has met its primary end point demonstrating that crizotinib significantly improves PFS over single-agent chemotherapy in the second-line setting Citation[52].
PROFILE 1014 is the second Phase III trial in progress Citation[53] studying crizotinib in the first-line setting for ALK-rearranged, advanced NSCLC. This study randomizes patients to receive the standard dose crizotinib or doublet, platinum/pemetrexed-based chemotherapy (carboplatin/pemetrexed or cisplatin/pemetrexed). The international oncology community eagerly awaits the results of these trials.
Chromosomal rearrangements involving the ROS1 receptor tyrosine kinase gene have recently been identified in NSCLC. Early data suggest that crizotinib may have meaningful activity in this group. ROS1 rearrangements may define another orphan disease in which criztonib is a treatment option Citation[54].
4. Crizotinib: orphan drug market approval and associated companion diagnostic test
On 26 August 2011, the US FDA approved crizotinib (Xalkori, Pfizer) for the management of locally advanced and metastatic NSCLCs that express the ELM4-ALK fusion gene. Significantly, crizotinib approval was in conjunction with approval of a companion diagnostic test that detects the abnormal ALK fusion gene, a first-of-a-kind genetic test called the Vysis ALK Break Apart FISH Probe Kit Citation[55]. Use of a companion diagnostic test to identify patients with NSCLC that harbor an abnormal ALK fusion gene essentially allows us to create a new disease entity that represents approximately 5% of all patients with advanced NSCLC. Consequently, crizotinib received orphan drug marketing approval. Crizotinib was reviewed under the FDA's priority review program. This program provides for an expedited 6-month review of drugs that may offer major advances in treatment or that provide a treatment when no adequate therapy exists. The FDA through an accelerated approval program approved crizotinib. This program empowers the agency to approve drugs in a timely fashion, developed to treat serious diseases, on the basis of clinical data showing that the drug is safe and has an effect on an end point that is likely to predict a clinically meaningful outcome for the patient. The program was created with the aim to give patients earlier access to promising new drugs. Accelerated approval of a drug is on the premise that further studies would follow to confirm the drug's clinical benefit.
The Vysis ALK Break Apart FISH Probe Kit is test used in NSCLC to detect rearrangements involving the ALK gene via FISH to identify patients eligible for treatment with crizotinib. The Vysis ALK Break Apart FISH Probe Kit assay requires four kits:
Vysis ALK Break Apart FISH Probe Kit
Vysis Paraffin Pretreatment IV and Post Hybridization Wash Buffer Kit
ProbeChek ALK Negative Control Slides
ProbeChek ALK Positive Control Slides
The safety and efficacy of the Vysis ALK Break Apart FISH Probe Kit was determined by conducting a number of studies including: concentration optimization, hybridization quality evaluation, establishment of the normal cutoff, probe localization, FISH success rate, analytical sensitivity, analytical specificity, microbial contamination, robustness, kit shelf-life stability, reproducibility and repeatability testing.
The analytical sensitivity and specificity were defined as the percentage of chromosome targets with expected normal signal pattern and the percentage of signals that hybridize only with the correct locus, respectively. Analytical sensitivity and specificity for each probe was found to be 100% (95% confidence interval (CI) 98.5 – 100.0%) and 100% (95% CI 97.0 – 100.0%), respectively.
The FDA approved the Vysis ALK Break Apart FISH Probe Kit on the basis of preclinical and clinical evidence supporting that it was safe, efficacious (data from the PROFILE 1005 study was used to support this finding) and reproducible Citation[56].
Alternative methods for detecting ALK gene rearrangements include reverse transcriptase polymerase chain reaction (RT-PCR), IHC and next-generation DNA sequencing (NGS). However, none of these are standard techniques for the identification of ALK rearrangements, and have not been validated in clinical trials to identify responders to ALK-targeted therapies. FISH was the gold standard test used to identify ALK-mutated NSCLC in clinical trials that showed efficacy for ALK inhibition. However, ongoing work continues in relation to the development of IHC along with RT-PCR and NGS and some of these look promising.
Both crizotinib and the companion Vysis ALK Break Apart FISH Probe Kit were approved ahead of the drug's 30 September 2011 FDA review goal date and the companion diagnostics' 28 September 2011 review goal date Citation[55].
5. The economic paradox
The small population affected by an orphan disease often discourages pharmaceutical industry from pursuing research and drug development for these illnesses Citation[57]. Effective therapy is traditionally not deemed worth the research and development expenditures to bring a product to market. There has been a paradigm shift toward a personalized approach to medicine over recent years. Orphan drug development plays a key role in sustaining the era of personalized medicine.
In 2008, the worldwide cost of cancer due to premature death and disability (not including direct medical costs) has been estimated to be US$895 billion Citation[58]. The international, oncology community is now, more than ever, aware of the rising cost of cancer care and acknowledges its responsibility in the need to develop realistic solutions to curb this cost Citation[59]. Advances in biomolecular technology, in particular predictive biomarkers, are heralded as a way to reduce the cost of drug expenditure. Predictive biomarkers identify a population most likely to respond to a drug, thereby reducing unnecessary cost and toxicity. New anticancer drugs are very expensive and the oncology community actively encourages the development of companion diagnostic tests to identify the population most likely to experience clinical benefit. Utilization of predictive biomarkers as seen with the ALK-positive NSCLC story often lead to the identification of orphan diseases. There has been a tendency for the pharmaceutical industry to avoid drug development for rare diseases. With respect to the field of oncology, new expensive drugs are less likely to be approved unless they have an associated diagnostic test to identify the target population, which in doing so creates a rare, orphan disease.
The passage of the Orphan Drug Act in 1983 created incentives designed to encourage investment in research and drug development for rare diseases Citation[60]. First, the Act instructed the FDA to establish protocols to facilitate orphan drug approvals. Second, Congress created a 50% tax credit for clinical trial costs for orphan drug development. Finally, the FDA approved that drugs with orphan drug designation would benefit from 7-year market exclusivity. These incentives, together with the fact that orphan drugs have fewer competitors and lower promotional and distribution costs have led the pharmaceutical industry to expand orphan drug development in the oncology setting. Newly approved oncology drugs are more likely to have a disproportionately high share of FDA priority review ratings and orphan drug designations at approval Citation[61].
6. Conclusion
We are living in an exciting time of research and drug development. The improved understanding of molecular oncology and ongoing advances in biotechnology will ensure continued development of targeted agents to fuel the personalized era of cancer medicine. Innovative oncology drugs will increasingly be approved in conjunction with predictive, drug-response specific biomarkers. Thus, creating orphan diseases, adopted within common cancers, which act as therapeutic targets for novel anticancer agents.
7. Expert opinion
Cancer is the second most common cause of death in the Western world, where the lifetime risk of developing cancer is approximately 40% Citation[62]. Improving cancer care treatment and outcomes remains an important goal for the international oncology community. Decades of cancer research have increased the basic understanding of the molecular pathways driving cancer cell growth, proliferation and survival. This in turn has led to significant advances in the treatment of individuals with cancer. Increasingly, the clinical heterogeneity observed in pathologically and anatomically similar cancers can be explained by distinct molecular features. This article explores the transition in the management of cancer as a result of important developments in the molecular understanding of cancer as we move away from the empiric, ‘one drug fits all' and toward ‘customized' or ‘personalized' treatment of cancer. For the individual with cancer selecting a treatment based on biological rational offers increased chance of response and limits exposure to drugs and their associated toxicity for minimal clinical benefit. For industry and regulators this approach represents financial sense. Before expensive novel biological therapies are approved, it is inevitable that drug approval agencies will require an associated companion diagnostic test identifying the subgroup expected to gain most from a specific drug. This shift will require academia, pharmaceutical and biotechnology sectors to develop processes where predictive biomarkers are developed, integrated and validated alongside the drug development pathway. Assessment of efficacy of drugs targeting small subgroups of patients with a particular cancer (e.g., ALK mutation-positive NSCLC) represents a significant challenge. To robustly assess the efficacy of targeted drugs in the small subgroups expected to respond, novel approaches to clinical trial design will be required. Trials with adaptive features represent one option. These are studies that can change according to examination of accumulated data at an interim point in the clinical trial. There is a prospectively planned opportunity to modify one or more specified aspects of the study design and hypotheses based on analysis of data from subjects in the study. An example of this new approach is the ‘Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging and Molecular Analysis 2′ (I-SPY 2 trial), which has an adaptive clinical trial design Citation[63]. These types of studies will require many individuals to undergo screening in order to identify a small number with the biomarker in question, unlike traditional clinical trials design where thousands of patients are accrued and randomized, these smaller scale studies often exploiting the neo-adjuvant setting provide unique challenges Citation[64,65].
The expanding knowledge regarding tumor biology and molecular pathways that drive proliferation of cancer allow us now to recategorize common cancers with similar anatomical and morphological features into distinct subgroups reflecting the underlying molecular heterogeneity. The identification of molecular biomarkers that allow us to create these subpopulations with unique and often rare forms of common cancers also represent potential therapeutic targets. Increasingly, oncology drugs are meeting the criteria for orphan drug designation approval. New innovative anticancer drugs are expensive to manufacture. The incentives received following orphan drug designation approval significantly reduce the cost of research and drug development. Therefore, orphan drug designation is something now actively sought by pharmaceutical companies with respect to oncology drug development. Through 2010, 362 orphan designations resulted in approval, with oncology representing the largest clinical subcategory Citation[66].
A novel cancer therapeutic now rarely costs less than US$5000 per month. With treatment courses costing as much as US$100,000, it is reasonable to expect a greater return on such investments Citation[67]. One method to ensure a greater return is to limit administration of a new drug to those most likely to respond. Companion diagnostic tests now allow us to select patients that will most likely yield a clinically significant response to a particular targeted agent. This helps to reduce the cost of cancer care by avoiding inappropriate patient selection for novel agents and reducing unnecessary toxicity. Both the medical oncology community and the pharmaceutical industry encourage oncology-related drug development together with a companion diagnostic test. Examples include crizotinib, for ALK-positive advanced NSCLC, and vemurafenib, a drug approved by the FDA in 2011 for the management of metastatic melanoma expressing a BRAF mutation Citation[6]. Many more drugs with a companion diagnostic test like crizotinib and vemurafenib will receive orphan drug marketing approval in the coming years.
There is an emerging concern regarding the safety and efficacy of orphan drugs that received accelerated marketing approval. For example, gemtuzumab ozogamicin, an orphan drug approved in 2000 for acute myeloid leukemia, was removed from the market in 2010 when a confirmatory trial failed to show a survival benefit. In fact, it reported a greater number of deaths in the gemtuzumab ozogamicin arm Citation[68]. In contrast to this, crizotinib has already been shown to improve PFS in the second-line setting compared with single-agent chemotherapy in the treatment of advanced NSCLC. The nature of drug development and research for orphan diseases is such that the number of patients eligible for clinical trial enrollment is small. Kesselheim et al. compared clinical trial design features of all orphan and non-orphan oncology drugs approved by the FDA between 2004 and 2010. Compared with the pivotal trials used to approve non-orphan drugs, those used to approve orphan drugs were more likely to enroll a significantly smaller number of study participants, have a non-randomized and unblinded study design and use surrogate end points, such as disease response rather than overall survival, to assess efficacy Citation[69]. We cannot lose sight of the fact that accelerated approval regulations, for certain drugs, were established by the FDA to promote earlier access to therapeutics for life-threatening diseases Citation[70]. In 2011, the FDA announced a commitment to comprehensive and timely follow-up testing and post-marketing surveillance of cancer drugs granted accelerated approval Citation[71]. The application of this promise to orphan drugs would hopefully go a long way to alleviate some of the concerns that have emerged regarding safety and efficacy.
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Notes
Bibliography
- Perou CM, Sorlie T, Eisen MB, Molecular portraits of human breast tumours. Nature 2000;406(6797):747-52
- Rosell R, Moran T, Queralt C, Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361(10):958-67
- Mok TS, Wu YL, Thongprasert S, Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361(10):947-57
- Keedy VL, Temin S, Somerfield MR, American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011;29(15):2121-7
- Davies H, Bignell GR, Cox C, Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949-54
- Chapman PB, Hauschild A, Robert C, Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507-16
- Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med 2011;364(8):772-4
- Group GSTM-A. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol 2010;28(7):1247-53
- Kim TW, Lee H, Kang Y-K, Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer Res 2004;10(9):3076-81
- Duffy MJ, Crown J. A personalized approach to cancer treatment: how biomarkers can help. Clin Chem 2008;54(11):1770-9
- Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev Drug Discov 2010;9(5):363-6
- Reid A, Baird R, Workman P. Emerging molecular technologies: drugs interfering with signal transduction pathways. Chapter 17. Principles of Molecular oncology. 3rd edition. Springer; 2008
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001. 69(3):89-95
- Park JW, Kerbel RS, Kelloff GJ, Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res 2004;10(11):3885-96
- NIH consensus conference. Treatment of early-stage breast cancer. JAMA 1991;265:391-5
- Paik S, Shak S, Tang G, A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351(27):2817-26
- Harris L, Fritsche H, Mennel R, American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol 2007;25(33):5287-312
- Sarker D, Workman P. Pharmacodynamic biomarkers for molecular cancer therapeutics. Adv Cancer Res 2007;96:213-68
- Ratain MJ, Schilsky RL, Conley BA, Egorin MJ. Pharmacodynamics in cancer therapy. J Clin Oncol 1990;8(10):1739-53
- Thun MJ, DeLancey JO, Center MM, The global burden of cancer: priorities for prevention. Carcinogenesis 2010;31(1):100-10
- Jemal A, Siegel R, Ward E, Cancer statistics, 2006. CA Cancer J Clin 2006;56(2):106-30
- Jemal A, Bray F, Center MM, Global cancer statistics. CA Cancer J Clin 2011;61(2):69-90
- Lu C, Onn A, Vaporciyan AA, "78: Cancer of the Lung". Holland-Frei Cancer Medicine. 8th edition. People's Medical Publishing House; 2010; ISBN: 9781607950141
- Schiller JH, Harrington D, Belani CP, Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002;346(2):92-8
- Goldstraw P, Crowley J, Chansky K, The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol 2007;2(8):706-14
- Aplan PD. Causes of oncogenic chromosomal translocation. Trends Genet 2006;22(1):46-55
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007;7(4):233-45
- Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Evidence of recurrent gene fusions in common epithelial tumors. Trends Mol Med 2006;12(11):529-36
- Tomlins SA, Rhodes DR, Perner S, Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310(5748):644-8
- Soda M, Choi YL, Enomoto M, Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448(7153):561-6
- Pollmann M, Parwaresch R, Adam-Klages S, Human EML4, a novel member of the EMAP family, is essential for microtubule formation. Exp Cell Res 2006;312(17):3241-51
- Eichenmuller B, Everley P, Palange J, The human EMAP-like protein-70 (ELP70) is a microtubule destabilizer that localizes to the mitotic apparatus. J Biol Chem 2002(2):1301-9
- Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci 1999;24(5):181-5
- Kutok JL, Aster JC. Molecular biology of anaplastic lymphoma kinase-positive anaplastic large-cell lymphoma. J Clin Oncol 2002;20(17):3691-702
- George RE, Sanda T, Hanna M, Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455(7215):975-8
- Pulford K, Lamant L, Espinos E, The emerging normal and disease-related roles of anaplastic lymphoma kinase. Cell Mol Life Sci 2004;61(23):2939-53
- Debelenko LV, Raimondi SC, Daw N, Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 2011;24(3):430-42
- Inamura K, Takeuchi K, Togashi Y, EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol 2008;3(1):13-17
- Kuo Y-W, Wu S-G, Ho C-C, Shih J-Y. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol 2010;5(12):2039-40
- McDermott U, Iafrate AJ, Gray NS, Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008;68(9):3389-95
- Koh Y, Kim DW, Kim TM, Clinicopathologic characteristics and outcomes of patients with anaplastic lymphoma kinase-positive advanced pulmonary adenocarcinoma. suggestion for an effective screening strategy for these tumors. J Thorac Oncol 2011;6(5):905-12
- Wong DW, Leung EL, So KK, The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115(8):1723-33
- Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46(10):1773-80
- Wong DW, Leung EL, Wong SK, A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer 2011;117(12):2709-18
- Togashi Y, Soda M, Sakata S, KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012;7(2):e31323
- Soda M, Takada S, Takeuchi K, A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA 2008;105(50):19893-7
- Koivunen JP, Mermel C, Zejnullahu K, EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008;14(13):4275-83
- Shaw AT, Yeap BY, Mino-Kenudson M, Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27(26):4247-53
- Christensen JG, Zou HY, Arango ME, Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther 2007;6(12 Pt 1):3314-22
- Kwak EL, Bang YJ, Camidge DR, Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363(18):1693-703
- Crino L, Kim D, Riely GJ, Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol 2011;29(Suppl):abstract 7514
- Pfizer Announces Positive Results From Phase 3 Study PROFILE 1007 Evaluating XALKORI® (crizotinib) In Previously Treated Patients With ALK-Positive Advanced Non-Small Cell Lung Cancer. 2012. Available from: http://www.pfizer.com/news/press_releases/pfizer_press_release.j
- Shaw AT, Yasothan U, Kirkpatrick P. Crizotinib. Nat Rev Drug Discov 2011;10:897-8
- Bergethon K, Shaw AT, Ou SH, ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30(8):863-70
- FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. 2011. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856html
- Vysis ALK Break Apart FISH Probe Kit; Vysis Paraffin Pretreatment IV and Post Hybridization Wash Buffer Kit; ProbeChek ALK Negative Control Slides; and ProbeChek ALK Positive Control Slides - P110012. Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cftopic/pma/pma.cfm?num=p110012
- Brewer GJ. Drug development for orphan diseases in the context of personalized medicine. Transl Res 2009;154(6):314-22
- Ferlay J, Shin HR, Bray F, Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127(12):2893-917
- Sullivan R, Peppercorn J, Sikora K, Delivering affordable cancer care in high-income countries. Lancet Oncol 2011;12(10):933-80
- Shulman SR, Manocchia M. The US orphan drug programme 1983 – 1995. Pharmacoeconomics 1997;12(3):312-26
- DiMasi JA, Grabowski HG. Economics of new oncology drug development. J Clin Oncol 2007;25(2):209-16
- American Cancer Society. Lifetime Risk of Developing or Dying From Cancer. Available from: http:/www.cancer.org/Cancer/CancerBasics/lifetime- probability-of-developing-or-dying-from-cancer 2010
- Barker AD, Sigman CC, Kelloff GJ, I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther 2009;86(1):97-100
- Prowell TM, Pazdur R. Pathological complete response and accelerated drug approval in early breast cancer. N Engl J Med 2012;366(26):2438-41
- Schott AF, Hayes DF. Defining the benefits of neoadjuvant chemotherapy for breast cancer. J Clin Oncol 2012;30(15):1747-9
- Wellman-Labadie O, Zhou Y. The US Orphan Drug Act: rare disease research stimulator or commercial opportunity? Health Policy 2010;95(2-3):216-28
- Fojo T, Grady C. How much is life worth: cetuximab, non-small cell lung cancer, and the $440 billion question. J Natl Cancer Inst 2009;101(15):1044-8
- US Food and Drug Administration. Mylotarg (gemtuzumab ozogamicin): market withdrawal. 2010. Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlerts forHumanMedicalProducts/ucm216458.htm [Accessed 21 April 2011]
- Kesselheim AS, Myers JA, Avorn J. Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA 2011;305(22):2320-6
- Richey EA, Lyons EA, Nebeker JR, Accelerated approval of cancer drugs: improved access to therapeutic breakthroughs or early release of unsafe and ineffective drugs? J Clin Oncol 2009;27(26):4398-405
- Johnson JR, Ning YM, Farrell A, Accelerated approval of oncology products: the food and drug administration experience. J Natl Cancer Inst 2011;103(8):636-44
- Parker JS, Mullins M, Cheang MCU, Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27(8):1160-7