
Abstract
Introduction: Mucopolysaccharidosis VI (MPS VI) is an autosomal recessive lysosomal storage disorder caused by a deficiency in the lysosomal enzyme N-acetylgalactosamine 4-sulfatase (arylsulfatase B), leading to an accumulation of glycosaminoglycans within lysosomes. MPS VI patients experience a progressive, chronic and multisystemic disease that causes not only significant morbidity but also early mortality.
Areas covered: Galsulfase, a recombinant human N-acetylgalactosamine 4-sulfatase, rhASB (Naglazyme®), is produced by recombinant DNA technology in a CHO-derived cell line, and was approved in the US (2005) and the EU (2006) for use in MPS VI patients. The authors examine the published pharmacokinetic, safety and efficacy data from the Phase I/II, Phase III, Phase III extension, post-marketing surveillance studies of galsulfase, published case reports and cohort of patients treated with rhASB.
Expert opinion: Galsulfase is generally well tolerated, having an acceptable safety profile. Few infusion reactions have occurred during administration of galsulfase, being generally mild-to-moderate in severity. Improvements in 12-minute walk test, nearly significant improvement in 3-min stair climb and significant reduction in urinary GAG levels were demonstrated in the 24-week, randomized, double-blind Phase III trial that led to the approval of the drug. As in other lysosomal storage diseases, the antibody response to enzyme replacement therapy can differ greatly between patients and may to some extent relate to the genotype. Nevertheless, antibody formation seems to have little impact on clinical outcome in MPS VI patients treated with galsulfase.
1. Introduction
Mucopolysaccharidosis VI (MPS VI) or Maroteaux-Lamy syndrome (OMIM 253200), first characterized by Maroteaux and Lamy in 1963, is a lysosomal storage disease in which deficient activity of the enzyme N-acetylgalactosamine 4-sulfatase (arylsulfatase B) impairs the stepwise degradation of the glycosaminoglycan (GAG) dermatan sulfate. Partially degraded GAGs accumulate within lysosomes in various tissues, causing a multisystemic chronic and progressive disorder with significant functional impairment and early death Citation[1-3].
Clinical features of MPS VI are similar to the other MPS disorders: skeletal dysostosis, coarse face, corneal opacification, visceromegaly, upper airway obstruction and valvular heart disease. Intellectual development is preserved in this disease Citation[3].
Since the advent of enzyme replacement therapy (ERT) for MPS VI with recombinant human N-acetylgalactosamine 4-sulfatase, rhASB (galsulfase, Naglazyme®), several aspects of clinical improvement were reported Citation[4].
In May 2005, in the US, and in July 2006, in Europe, ERT with galsulfase (), a recombinant human rhASB, was approved. It is now available in > 39 countries. Current treatment guidelines suggest the initiation of weekly galsulfase treatment as soon as possible after diagnosis for most patients Citation[3]. Although clinical trial data for the use in patients under the age of 5 years are not available, some case reports from Australia, Japan and a Brazilian cohort of 34 patients under 5 years of age were suggestive of general safety and effectiveness Citation[5-7]. A postmarketing surveillance safety program has been developed and the effectiveness of the use of galsulfase in children who initiated therapy before 6 years of age has been described in data found in a patient registry that collects observational data (see Section 5.4).
Box 1. Drug summary.
2. Overview of the market
Historical treatment for MPS VI was mainly palliative. Based on successes in MPS I-Hurler (OMIM 607014) Citation[8], hematopoietic stem cell transplantation (HSCT) has been performed in small numbers of patients with MPS VI Citation[9,10]. Recently, a review of 45 MPS VI treated with HSCT showed long-term improvements in facial dysmorphism, hepatosplenomegaly, joint mobility and cardiac manifestations Citation[11]; in general, responses to engraftment in terms of biochemical and clinical parameters have been quite satisfactory, although corneal cloudiness and skeletal involvement do progress even with successful engraftment. Since HSCT is associated with substantial morbidity and mortality, is limited by a lack of suitable donors, and no studies have compared the efficacy of HSCT to ERT, further research is warranted to assist clinicians who must evaluate risks and benefits of such treatment on an individual basis for each patient Citation[11].
Another competitor is the soy isoflavone genistein, a natural compound that can inhibit the synthesis of GAGs Citation[12]. Genistein use has been studied in patients with the related disorders MPS IIIA, B, C and D (OMIM 252900, OMIM 252920, OMIM 252930 and OMIM 252940). Nevertheless, an open-label study enrolling 19 patients showed no clinical benefit after 1 year of treatment Citation[13]. Another clinical trial – randomized, placebo-controlled – with 30 MPS III patients demonstrated that genistein led to a mild decreased urinary GAG (uGAG) and plasma heparan sulfate levels; however, no clinical efficacy was detected Citation[14]. Genistein was also used in an open-label study in seven previously untreated attenuated MPS II patients; there were statistically significant improvements in mean active and passive shoulder flexion and abduction Citation[15], but elbow, wrist, hip and knee joint range of motion (ROM) were not improved by treatment. No reports of genistein in MPS VI patients have been found so far. Since MPS VI is not primarily a disorder with brain involvement, most of the studies regarding genistein have been focused on MPS disorders with cognitive decline, such as MPS II and III. It remains to be seen if genistein therapy will benefit peripheral manifestations of the disease in those patients without a primary neurological disorder due to GAGs accumulation in the brain; in such case, genistein therapy could become relevant in the future for the treatment of MPS VI patients.
3. Introduction to galsulfase
Galsulfase is a rhASB. It is a glycoprotein heterogeneously glycosylated with a single-chain protein of 56 kDa, comprising 495 amino acids and containing six asparagine-linked glycosylation sites, four of which carry a bis-mannose-6-phosphate mannose oligosaccharide for specific cellular recognition Citation[16]. Its uptake by cells into lysosomes is most likely mediated by the binding of its bisphosphorylated oligomannose oligosaccharide chains to specific mannose-6-phosphate receptors. Post-translational modification of cysteine-53 produces the catalytic amino acid residue C-formylglycine, which is required for enzyme activity Citation[16].
Galsulfase is produced by recombinant DNA technology in a CHO-derived cell line, using a perfusion process and purified by a series of concentration, column chromatography and filtration steps. The drug product is prepared by sterile filtration and aseptic filling into vials of the Formulated Bulk Drug Substance Citation[16].
Galsulfase is provided as a concentrate for solution for infusion in a single-use vial, which contains a nominal amount of galsulfase 5 mg in 5 ml (concentration 1 mg/ml). Galsulfase is formulated with sodium phosphate monobasic monohydrate, sodium phosphate dibasic heptahydrate, sodium chloride, polysorbate 80 and water for injections. The drug product is a clear to slightly opalescent and colorless to pale yellow solution that has to be diluted in 0.9% sodium chloride solution prior to administration Citation[17,18].
Galsulfase is administered by continuous weekly intravenous (i.v.) infusion, using a 0.2 μm filter, at a recommended dose of 1.0 mg/kg of body weight weekly diluted in 100 ml of 0.9% sodium chloride. The total volume may be administered over 4 h, although a longer infusion time can be used if infusion-related reactions (IRRs) occur, as long as the infusion time does not exceed 24 h. A ramping protocol is suggested based on the final volume of infusion and the weight of the patient; for example, a patient with 20 kg or less should initiate with an infusion rate of 3 ml/h for the first 60 min. Then, if the infusion is well tolerated, the rate may be increased – in steps – up to 32 ml/h Citation[18].
4. Pharmacokinetics
Tissue distribution has been investigated in five of the pharmacology studies in MPS VI-affected cats. rhASB was widely distributed into tissues, with the largest proportion localised to the liver in all studies. There were also significant levels in the spleen, lung, kidney, heart, skin, aorta, cerebrum, cerebellum and lymph nodes in one study, compared to levels in a normal control cat Citation[19].
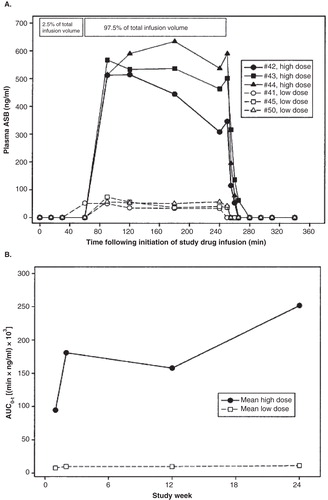
In the Phase I/II study, rhASB plasma concentrations during and 2 h after the first infusion were evaluated (). Maximum observed concentration was 572 ± 60 ng/ml and 75.1 ± 29.2 ng/ml for the high- and low-dose groups, respectively. Antigen was not measurable in the plasma within 10 min after the completion of the enzyme infusion in all patients. rhASB pharmacokinetics appeared to be nonlinear, as reflected by greater than dose-proportionate increases in AUC0-t () Citation[4,20]. AUC0-t increased relative to week 1 values in the high-dose patients, but remained unchanged in the low-dose patients. The area under the plasma concentration-time curve (AUC0-t) for the high-dose group increased from week 1 to week 2, but remained unchanged at weeks 12 – 24. A large difference in mean AUC0-t was observed between the low- and high-dose groups. Pharmacokinetic results at weeks 83, 84 and 96 were similar to those at week 24. The high-dose rhASB led to a more rapid and greater reduction in uGAG concentrations than the lower dose (70 vs 55% at 24 weeks) Citation[4,20].
Figure 1. rhASB plasma concentrations during and 2 h after the first infusion with the area under the plasma concentration-time curve (AUC0-t).
5. Clinical efficacy of galsulfase
5.1 Phase I/II study
This 24-week, randomized, double-blind, Phase I/II study enrolled 7 patients (4 males, 3 females; age 7 – 16 years). Patients were randomized to two dose groups: 0.2 and 1.0 mg/kg. Study drug was given once per week as a 4-h i.v. infusion Citation[4,20].
Seven patients exhibiting disease characteristics varying from moderately to rapidly advancing disease were initially enrolled in the study. One patient dropped out of the study (at week 3) for personal reasons and was replaced Citation[4,20].
In the 6-minute walk test (MWT), patients receiving the 1 mg/kg dose of rhASB had a mean increase of 65 m at week 24 compared to baseline (83% increase); the patients who received 0.2 mg/kg had a mean increase of 23 m (10% increase) Citation[4].
Evaluation of biochemical markers of rhASB activity and urinary excretion of GAGs as well as urinary excretion of dermatan sulfate showed dose-related decreases from baseline levels through week 24, with continued declines through week 96 following transition to the higher dose by the 0.2 mg/kg group. The patients in the 1 mg/kg group had a 70% mean reduction from baseline in uGAGs to a mean level of 100.0 μg/mg creatinine; the patients in the 0.2 mg/kg group had a mean reduction from baseline of 55% to a mean level of 144 μg/mg creatinine. Examination of the time course of the change in urinary excretion of GAGs as a function of weeks on treatment showed a more robust drop in total uGAGs in the 1.0 versus 0.2 mg/kg group by 6 weeks. These data confirm that the higher dose produced a larger change in urinary excretion of both GAGs and dermatan sulfate Citation[4].
All patients showed some improvement in flexion or extension in at least one shoulder at week 24. Liver size as a percent of body weight generally decreased, particularly for 2 patients, one in each dosage group, with the largest livers at baseline. There were no clinically meaningful changes in bone mineral density, height, weight, chest and cervical spine X-rays, ECG, echocardiogram, forced vital capacity, forced expiratory volume in one second, other Childhood Health Assessment Questionnaire (CHAQ) variables and the pinch and grip strength results. There were no clinically meaningful changes in the other efficacy assessments Citation[4].
5.2 Phase II study
This 24-week, open-label, double-blind, Phase II study enrolled 10 patients (7 males, 3 females; age 6 – 22 years). Efficacy, safety and pharmacokinetics of weekly i.v. infusions of 1 mg/kg rhASB were assessed. uGAG levels from the patients ranged from 138.4 to 518.5 μg/mg creatinine at enrollment. All 10 patients were on study through week 72. Its primary efficacy end points were shoulder ROM, stair climb test, 12MWT and uGAG levels Citation[21].
All 10 patients enrolled had improvements in the distance walked at 6 (9/10) and 12 min at week 24 compared to baseline. The mean increase was 64 ± 62 m in 6 min and 155 ± 146 m in 12 min. All 10 patients also showed an increase in the number of stairs climbed in 3 min between baseline and week 24. The mean increase was 48 ± 48 stairs. Also, such improvements could be seen at 6-min time point and 12-min time point after 48 weeks (80 and 138%, respectively) Citation[21]. uGAG levels showed a rapid decline from baseline, with 71% reductions at week 6 and at week 24. Seven patients had an improvement in active shoulder flexion, 9 had improvement in active shoulder extension and 8 had improvements in active lateral rotation at week 24 [Personal Communication]. However, none of these results were clinically significant being < 10 degrees. Similar results were seen with passive ROM.
Decrease in liver and spleen size were seen in the secondary efficacy variable measured, but there were no clinically meaningful changes in the other efficacy variables (measured at week 24 were pulmonary function, physical activity, oxygenation during sleep, bone density, electrocardiogram and echocardiogram) Citation[21].
5.3 Phase III study
This 24-week, Phase III, multi-center, double-blind, placebo-controlled trial enrolled 39 patients. Patients were randomized on a one-to-one basis into a rhASB treatment group or a placebo control group and received a weekly i.v. infusion of either 1.0 mg/kg of rhASB or placebo solution. During the 24-week period, 19 patients received weekly i.v. infusions of rhASB and 20 patients received weekly placebo infusions. One patient in the placebo group dropped out of the trial for reasons unrelated to treatment Citation[22].
The primary efficacy end point (12-min walk test) showed a statistically significant difference in mean distance walked in 12 min between the rhASB and placebo group. The rhASB group walked a mean ± SE of 92 ± 40 m further than the placebo group at week 24 (p value ± 0.025) ().
Table 1. Phase III study: summary of effect on primary and secondary endpoints.
In the secondary end point variables, the rate of stairs climbed per minute was used because the percent of stair climbs in which the patient reached the top of the stairs exceeded the predefined limit of 10% in the data analysis plan. The primary analysis of rate of stair climb (stairs/minute) in all randomized patients showed a difference between the mean change in the rates between rhASB and placebo of 5.7 ± 2.9 stairs/minute (p = 0.053). For both the walk-eligible and ≤ 400 m subsets, the rhASB group climbed a mean of approximately 21 more stairs than the placebo group, p = 0.019 and p = 0.048, respectively ().
After 24 weeks of treatment, patients receiving rhASB experienced a statistically significant reduction (p < 0.001) of GAGs excreted in the urine, compared to patients receiving placebo (). Of 19 rhASB patients, 17 patients, and no placebo patients had a ≥ 50% percent reduction in uGAG levels between baseline and week 24 (p = 0.001). This decline suggests an affect on both lysosomal storage and accumulation of GAG. This initial reduction in uGAG levels was maintained following an additional 24 weeks of treatment in the extension study Citation[22,23].
Tertiary end point variables that assessed joint pain, joint stiffness, endurance and shoulder joint ROM showed little change from baseline over time; however, tertiary variables were not statistically powered to show a difference between the groups. No improvement in cardiac valvular heart disease was observed Citation[22].
5.4 Post-marketing surveillance
The MPS VI Clinical Surveillance Program (MPS VI CSP) is a voluntary, multi-national observational program for patients with MPS VI. It was opened in 2005 to collect observational data from standard clinical and laboratory assessments of patients with MPS VI. Baseline and follow-up data are documented by participating physicians in electronic case report forms. According to a first data analysis, 132 patients with MPS VI were enrolled, most of them from European centers and from US centers Citation[24].
Median age at enrollment was 13 years (range: 0 – 59 years). Mean baseline data showed impaired growth, hepatosplenomegaly and reduced endurance and pulmonary function. Citation[24]. CSP first results seem to confirm the effects of ERT on uGAG levels and endurance that were reported in the clinical trials. In addition, an increase in both height and weight was observed Citation[24]. No improvement or deterioration was seen for cardiac, ophthalmologic or auditory data. Safety data confirmed that ERT with galsulfase is generally well tolerated Citation[24]. The most common findings in the MPS enrolled in the CSP were heart valve disease, reduced visual acuity, impaired hearing and hepatosplenomegaly.
Recently, Harmatz et al. Citation[25] reported a Phase IV study performed in 4 infants with MPS VI, who received weekly infusions of galsulfase 1.0 or 2.0 mg/kg along 52 weeks. The authors concluded that galsulfase at the two dose levels was safe and well tolerated in infants. They speculate that early initiation of galsulfase may prevent or slow progression of some disease manifestations.
6. Safety and tolerability of galsulfase
6.1 Adverse events
The most common adverse events (AEs) in the clinical trials and post-marketing surveillance studies were infusing-drug adverse events (IARs), which were managed by slowing the infusion and/or premedication with antihistamines or steroids Citation[4,20-23]. The incidence of IARs in the published studies varied between 20 and 75%; in Phase III study, the total incidence of AEs, severe AEs and serious AEs (SAEs) in patients in the galsulfase group was similar to that in patients treated with placebo (). Serious or potentially life-threatening IARs were uncommon. Other AEs included limb pain, visual disturbance, chest wall pain, anxiety and dyspepsia Citation[26]. A total of 39 SAEs were reported overall for the 3 studies (2.6% of all events); 27 in rhASB treated patients and 12 in placebo-treated patients. Three SAEs were considered to be related to study drug. Two related SAEs, apnea and urticaria, occurred during study drug infusion, both in patients treated with rhASB. The apneic event, although judged to be possibly related to study drug, may have been precipitated by antihistamine use in the setting of severe upper airway obstruction. Moderate urticaria requiring slowing of the rhASB infusion occurred at week 76.
Table 2. Summary of AEs during study weeks 1 – 48.
The third related SAE, asthma, occurred in an rhASB-treated patient with a long history of steroid-dependent asthma several hours after study drug infusion. A total of 36 SAEs were considered unrelated to study drug administration; none of these occurred during infusion. Twenty-four of these events occurred in patients receiving rhASB. Pneumonia was the most frequently reported unrelated SAE (3 events each for rhASB and placebo) Citation[4,20-23].
Two episodes of increased INR (blood clotting time) were reported for one patient. Single instances of decreased serum albumin, increased alkaline phosphatase, increased phosphorus, decreased potassium, decreased hemoglobin, decreased complement factor, abnormal INR, hyponatremia, hematuria and proteinuria were also reported. None of these events were severe or SAEs Citation[23].
Biphasic anaphylactic reactions have also been observed, which may require prolonged observation. Patients with compromised respiratory function or acute respiratory disease may be at risk of serious acute exacerbation of their respiratory compromise due to IRRs, and these patients require additional monitoring Citation[27].
One single case report of thrombocytopenia in a Turkish MPS VI patient following his third ERT infusion was reversed after decreasing the dose in a subsequent infusion and later returning to the standard dose. The authors suggest it could be related to the antibodies formation against galsulfase although they have not measured those antibodies in their patient Citation[28].
6.2 Antibodies to galsulfase
The development of anti-ASB IgG antibodies was assessed, at a minimum, at 6-week intervals during each of the clinical studies. Initial evidence of antibody development typically appeared following 4 – 8 weeks of treatment. Antibody levels in 14 of 34 patients did not exceed 2.0 OD/µl at any time during the course of their treatment with rhASB. Among patients completing at least 24 weeks of rhASB treatment, 1 of 6 (16.7%) from Phase I/II, 3 of 10 (30%) from Phase II, and 6 of 19 (31.6%) from Phase III developed antibody titres > 10 OD/µl. For patients developing these higher antibody levels, an initial increase to 2.0 OD/µl typically occurred between weeks 6 and 12 of treatment Citation[4,20-22].
In the Phase II study Citation[21], 2 of the 3 patients with high antibody levels had significant reduction in uGAG levels, while the remaining patient did show a smaller reduction in GAG level. The exact mechanism for the variability of the effects of antibody on the ELISA, AUC and uGAG levels is not fully understood, but could be supported by the high genetic variability of the patients (> 40 different genetic mutations in the clinical trials). However, the occurrence of neutralizing antibodies cannot be excluded Citation[29,30].
As in other lysosomal storage diseases, the antibody response to ERT can differ greatly between patients and may to some extent relate to the genotype and residual enzyme activity. Although antibody formation seems to have little impact on clinical outcome, a recent study from The Netherlands demonstrated that the antibody concentration in the blood can reach a level at which it potentially affects the efficacy of ERT by inhibiting uptake of enzyme by the target tissues Citation[31].
7. Access to treatment
Policies about eligibility for galsulfase treatment vary from country to country; these differences typically center around treating patients with more advanced stage of the disease (and thus, irreversible organ damage) who will be less prone to have benefits from ERT or patients with other medical or surgical conditions, which would significantly impair response to or benefit from ERT Citation[32-34].
The availability and accessibility of galsulfase vary considerably between countries/regions and it is not only due to ineligibility of some patients due to advanced stage disease, but specially because of the high costs associated with therapies for orphan diseases Citation[35]. The cost of 1-year treatment with rhASB, for example, is around €150,000 – 450,000, dependent on the patient's weight.
Cost-effectiveness analysis is a valuable tool to increase the transparency of reimbursement decision-making from an economic point of view; nevertheless it does not seem to be the most suitable one for evaluating efficacy and clinical impact of orphan drugs in the treatment of rare diseases. However, at present, the cost-effectiveness of orphan drugs, especially those for very rare diseases, cannot be established with the standard methods used by health technology assessment bodies to inform reimbursement authorities. New tools should be developed to better assess those therapies Citation[36]. Most reimbursement authorities currently accept that it does not make sense to use such standard methods for orphan drugs, at least not those for very rare diseases, based on the rarity of the underlying disease, the unknown costs of not treating these patients and the fact that they are affected by a life-threatening or chronic and serious disease. Moreover, in a condition such as MPS VI, in which cognitive functions are preserved and the treated individual would have clinical benefits, the application of the ‘rule of rescue’ is warranted. This was originally proposed by Jonsen in 1986, referring to the social imperative to rescue identifiable individuals who face avoidable death (or severe disability) Citation[37,38].
8. Conclusion
Three independent clinical trials have evaluated the efficacy and safety of galsulfase: one Phase I/II study, one Phase II study and one Phase III study (). A total of 56 MPS VI patients between 5 and 29 years with a mean age of 12 years were included in these trials, and the majority of cases had a rapidly progressing form of the disease Citation[4,20-23]. An open-label extension study, including all patients who completed these three clinical trials, evaluated the long-term efficacy of galsulfase in endurance and safety and included follow-up data for a period of 97 – 260 weeks Citation[23]. Additional data on the long-term efficacy of galsulfase on pulmonary function were collected during the 97 – 240 weeks of the extension study involving the same patients Citation[39].
Table 3. Summary of study populations.
Recent publications showed impact of ERT on growth, especially in patients who started ERT below 16 years of age Citation[40] and in cardiovascular aspects of the disease, in particular, ventricular septal hypertrophy. Regarding to the cardiovascular aspects, data collected suggest that long-term ERT is effective in reducing intraventricular septal hypertrophy and preventing progression of cardiac valve abnormalities when administered to those < 12 years of age Citation[40].
Taken together, these observations reinforce the need for early diagnosis, reinforcing the data already seen in animal models of the impact of early ERT Citation[41]. Development of new and practical diagnostic tests such as enzyme assay in Dried Blood Spot (DBS) can allow not only for the diagnosis in areas where lysosomal reference laboratories are not available but also for the possibility of newborn screening for the disease Citation[42-44].
9. Expert opinion
The introduction of ERT with galsulfase has been an important milestone in the treatment of MPS VI patients; previously only supportive care and HSCT in selected cases were the available therapies. Enzyme therapy for MPS VI using galsulfase has been shown to result in clinical improvements in endurance (as measured by the 12MWT and 3MSC test) along with a reduction in uGAG levels, using the approved prescribed dose of 1.0 mg/kg of rhASB, administered weekly. In the Phase III trials, patients receiving rhASB walked on average 92 m more in the 12MWT and 5.7 stairs per minute more in the 3MSC than patients receiving placebo. Continued improvement was observed during the extension study. uGAG declined by -227 ± 18 μg/mg more with rhASB than placebo. Proof of therapeutic principle was based on the lowered level of uGAG in all treated patients, which was sustained with on-going treatment. Long-term follow-up will be required to ascertain full clinical benefit on both survival and quality-of-life measures.
Although ERT with galsulfase has been successful in treating some aspects of the disease such as deficits of endurance, pulmonary function, growth and puberty, it has not been able to resolve the symptoms of MPS VI disease occurring in certain regions of the CNS, ophthalmologic system and joints due to the limitations of the blood–brain barrier and comparatively poor vascularization of the joints preventing penetration of enzyme to these regions Citation[27,32,33].
The limited effect of ERT on joint disease and the CNS probably explains why some patients do not show sustained improvement in walk- and stair-climb tests despite an initially positive effect. Other administration routes for ERT, that is, intrathecal and intra-articular, have been studied in order to prevent progression of complications such as spinal cord compression and skeletal dysplasia.
Intrathecal ERT involves the infusion of recombinant enzyme into the spinal fluid Citation[45,46], whereas intraarticular ERT Citation[47,48] involves the direct injection of enzyme into the intra-articular space. Studies in MPS animal models and a few case reports in humans Citation[45] have shown promising results for both techniques, but further studies are warranted.
There is evidence to support the use of galsulfase in the treatment of MPS VI; nevertheless, therapeutic response may be influenced by disease stage and early intervention may lead to better outcomes, as seen in the sibling studies and the case series of young infants treated under 5 years of age Citation[5-7]. Because of the progressive nature of MPS VI, halting disease progression or even slowing the rate of deterioration are beneficial for the patient, reinforcing the positive aspects of starting ERT as soon as possible.
The observation from the other sibling studies (for MPS II and MPS I) also corroborate the need for early diagnosis of MPS patients before irreversible damage is already seen in the patient. In such context, development of high-throughput methods for enzyme analysis using DBSs Citation[49] or the analysis of GAG species in urine samples Citation[50] made feasible the possibility of newborn screening programs for MPS diseases, in particular, for MPS I, II and VI where there is possibility of therapy with bone marrow transplant and/or ERT. At the moment, some pilot programs have been developed that probably will have great impact in the current management of MPS patients since early therapeutic strategies could be offered for such patients Citation[51,52].
Animal studies suggest that the limited effect of ERT on growth might increase if treatment would be started at an earlier age Citation[1-3], but no studies in humans are available as yet. It is possible that certain aspect of the disease may be modified by a longer period of treatment, and additional data is anticipated from the MPS VI CSP. Thus, research is warranted to assess the impact of ERT on individual disease manifestations of MPS VI and its efficacy and safety in very young children Citation[53,54]. At this moment, galsulfase is a therapy that offers fewer risks than HSCT and should be offered to any child suffering from this devastating disorder, keeping in mind that patients with severe advanced disease are less prone to have clinical benefits than patients who started it earlier. It is worthy to note the ERT with galsulfase should be offered in a clinical setting of integrated care, along with physical therapy and medical and surgical management of individual disease complications Citation[55-57].
Declaration of interest
Authors received travel grants, speaker honoraria, investigator fees and unrestricted research and/or educational grants from Actelion, Amicus, BioMarin, Genzyme, Pfizer, Protalix and Shire. The authors declare no conflict of interest and have received no payment in preparation of this manuscript.
Notes
Bibliography
- Maroteaux P, Leveque B, Marie J, Lamy M. A new dysostosis with urinary elimination of chondroitin sulfate B. Presse Med 1963;71:1849-52
- Litjens T, Baker EG, Beckmann KR, et al. Chromosomal localization of ARSB, the gene for human N-acetylgalactosamine-4-sulphatase. Hum Genet 1989;82:67-8
- Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis 2010;5:5
- Harmatz P, Whitley CB, Waber L, et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr 2004;144:574-80
- McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age - a sibling control study. Clin Genet 2009;77(5):492-8
- Furujo M, Kubo T, Kosuga M, Okuyama T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab 2011;104(4):597-602
- Horovitz DDG, Magalhaes TSPC, Acosta A, et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol Genet Metab 2013;109(1):62-9
- Peters C, Balthazor M, Shapiro E, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996;87:4894-902
- Wang C, Hwu W, Lin K. Long-term follow-up of a girl with Maroteaux-Lamy syndrome after bone marrow transplantation. World J Pediatr 2008;4:152-4
- Herskhovitz E, Young E, Rainer J, et al. Bone marrow transplantation for Maroteaux-Lamy syndrome (MPS VI): long-term follow-up. J Inherit Metab Dis 1999;22:50-6
- Turbeville S, Nicely H, Rizzo JD, et al. Clinical outcomes following hematopoietic stem cell transplantation for the treatment ofmucopolysaccharidosis VI. Mol Genet Metab 2011;102(2):111-15
- Friso A, Tomanin R, Salvalaio M, Scarpa M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br J Pharmacol 2010;159:1082-91
- Delgadillo V, O'Callaghan M del M, Artuch R. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis 2011;34:1039-44
- de Ruijter J, Valstar MJ, Narajczyk M, et al. Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann Neurol 2012;71:110-20
- Marucha J, Tylki-Szymanska A, Jakobkiewicz-Banecka J, et al. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am J Med Genet A 2011;155A:2257-62
- Hopwood J, Bate G, Kirkpatrick P. Galsulfase. Nat Rev Drug Discov 2006;5:101-2
- Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125117s111lbl.pdf
- Naglazyme [BioMarin]. Naglazyme prescribing information. 2005. Ref Type: Pamphlet. Available from: http://www.naglazyme.com/hcp/pdfs/DosageandAdminGuide.pdf
- Auclair D, Hopwood JJ, Brooks DA, et al. Replacement therapy in Mucopolysaccharidosis type VI: advantages of early onset of therapy. Mol Genet Metab 2003;78:163-74
- Harmatz P, Kramer WG, Hopwood JJ, et al. Pharmacokinetic profile of recombinant human N-acetylgalactosamine 4-sulphatase enzyme replacement therapy in patients with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): a phase I/II study. Acta Paediatr Suppl 2005;94(447):61-8
- Harmatz P, Ketteridge D, Giugliani R, et al. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 2005;115(6):e681-9
- Harmatz P, Giugliani R, Schwartz I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr 2006;148:533-9
- Harmatz P, Giugliani R, Schwartz IV, et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab 2008;94:469-75
- Hendriksz CJ, Giugliani R, Harmatz P, et al. Design, baseline characteristics, and early findings of the MPS VI (mucopolysaccharidosis VI) Clinical Surveillance Program (CSP). J Inherit Metab Dis 2013;36(2):373-84
- Harmatz PR, Garcia P, Guffon N, et al. Galsulfase (Naglazyme®) therapy in infants with mucopolysaccharidosis VI. J Inherit Metab Dis 2013. [Epub ahead of print]
- Naglazyme, INN-galsulfase, European Medicines Agency. Available from: http://www.ema.europa.eu/ema/
- Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics 2007;120(2):405-18
- Doğan M, Cesur Y, Peker E, et al. Thrombocytopenia associated with galsulfase treatment. Hum Exp Toxicol 2011;30(7):768-71
- White JT, Argento Martell L, Prince WS, et al. Comparison of neutralizing antibody assays for receptor binding and enzyme activity of the enzyme replacement therapeutic Naglazyme (galsulfase). AAPS J 2008;10(3):439-49
- White JT, Martell LA, Van Tuyl A, et al. Development, validation, and clinical implementation of an assay to measure total antibody response to naglazyme (galsulfase). AAPS J 2008;10(2):363-72
- Brands MM, Hoogeveen-Westerveld M, Kroos MA, et al. Mucopolysaccharidosis type VI phenotypes-genotypes and antibody response to galsulfase. Orphanet J Rare Dis 2013;8(1):51
- Giugliani R, Federhen A, Rojas MV, et al. Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol 2010;33(4):589-604
- Waith JE, Vellodi A, Cleary MA, et al. Guidelines for the investigation and management of mucopolysaccharidosis type VI. Available from: http://webarchive.nationalarchives.gov.uk/20130107105354/http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4131929.pdf
- Australian Government Department of Health and Aging. Guidelines for the treatment of Mucopolysaccharidosis Type VI (MPS VI) disease through the Life Saving Drugs Program. Available from: http://www.health.gov.au/internet/main/publishing.nsf/Content/lsdp-info/$File/mps-vi-guidelines-may-2013.pdf
- Hwu WL, Okuyama T, But WM, et al. Current diagnosis and management of mucopolysaccharidosis VI in the Asia-Pacific region. Mol Genet Metab 2012;107(1-2):136-44
- El Dib RP, Pastores GM. A systematic review of new advances in the management of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): focus on galsulfase. Biologics 2009;3:459-68
- Cookson R, McCabe C, Tsuchiya A. Public healthcare resource allocation and the Rule of Rescue. J Med Ethics 2008;34(7):540-4
- Tambuyzer E. Rare diseases, orphan drugs and their regulation: questions and misconceptions. Nat Rev Drug Discov 2010;9(12):921-9
- Harmatz P, Yu ZF, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis 2010;33(1):51-60
- Decker C, Yu ZF, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: growth and pubertal development in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Pediatr Rehabil Med 2010;3(2):89-100
- Braunlin E, Rosenfeld H, Kampmann C, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis 2013;36(2):385-94
- Crawley AC, Niedzielski KH, Isaac EL, et al. Enzyme replacement therapy from birth in a feline model of mucopolysaccharidosis type VI. J Clin Invest 1997;99:651-62
- Wang D, Wood T, Sadilek M, et al. Tandem mass spectrometry for the direct assay of enzymes in dried blood spots: application to newborn screening for mucopolysaccharidosis II (Hunter disease). Clin Chem 2007;53:137-40
- Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis 2006;29:397-404
- Matern D. Newborn screening for lysosomal storage disorders. Acta Paediatr Suppl 2008;97:33-7
- Dickson P, McEntee M, Vogler C, et al. Intrathecal enzyme replacement therapy: successful treatment of brain disease via the cerebrospinal fluid. Mol Genet Metab 2007;91:61-8
- Munoz-Rojas MV, Horovitz DD, Jardim LB, et al. Intrathecal administration of recombinant human N-acetylgalactosamine 4-sulfatase to a MPS VI patient with pachymeningitis cervicalis. Mol Genet Metab 2009;99(4):346-50
- Auclair D, Hein LK, Hopwood JJ, Byers S. Intra-articular enzyme administration for joint disease in feline mucopolysaccharidosis VI: enzyme dose and interval. Pediatr Res 2006;59:538-43
- Wang D, Eadala B, Sadilek M, et al. Tandem mass spectrometric analysis of dried blood spots for screening of mucopolysaccharidosis I in newborns. Clin Chem 2005;51:898-900
- Auray-Blais C, Bhérer P, Gagnon R, et al. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol Genet Metab 2011;102:49-56
- Uribe A, Giugliani R. Selective screening for lysosomal storage diseases with dried blood spots collected on filter paper in 4,700 high-risk colombian subjects. JIMD Rep 2013;11:107-16
- Giugliani R. Newborn screening for lysosomal diseases: current status and potential interface with population medical genetics in Latin America. J Inherit Metab Dis 2012;35(5):871-7
- Auclair D, Hopwood JJ, Lemontt JF, et al. Long-term intra-articular administration of recombinant human N-acetylgalactosamine-4-sulfatase in feline mucopolysaccharidosis VI. Mol Genet Metab 2007;91:352-61
- Giugliani R, Carvalho CG, Herber S, de Camargo Pinto LL. Recent Advances in Treatment Approaches of Mucopolysaccharidosis VI. Curr Pharm Biotechnol 2011;12(6):956-62
- Azevedo AC, Schwartz IV, Kalakun L, et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet 2004;66(3):208-13
- Guarany NR, Schwartz IV, Guarany FC, Giugliani R. Functional capacity evaluation of patients with mucopolysaccharidosis. J Pediatr Rehabil Med 2012;5(1):37-46
- Giugliani R. Mucopolysacccharidoses: from understanding to treatment, a century of discoveries. Genet Mol Biol 2012;35(4 (Suppl)):924-31