
Abstract
Introduction: At least 40,000 people per year worldwide are born with β-thalassemia. Patients with β-thalassemia major are reliant on regular red blood cell transfusions for survival from a young age. For those with β-thalassemia intermedia or hemoglobin (Hb) E/β-thalassemia, symptoms range from mild clinical presentation to a more severe phenotype and patients are not necessarily transfusion-dependent.
Areas covered: Here, β-thalassemia treatment strategies including transfusion, splenectomy, fetal hemoglobin induction, hematopoietic stem-cell transplantation, in addition to potential future treatment options, are reviewed. Approaches for the monitoring and management of complications are also described.
Expert opinion: The most important advances in the treatment of transfusion-dependent β-thalassemia major patients since the advent of iron chelation therapy are the introduction of oral iron chelators in addition to technologies for the direct measurement of iron in organs. For non-transfusion-dependent patients with β-thalassemia intermedia or HbE/β-thalassemia, recent studies have highlighted the significance of iron overload-related complications and the increase in incidence with advancing age, prompting the development of much-needed clinical treatment guidelines. Future research should focus on improving the treatment of β-thalassemia major patients to further extend survival and quality of life, and continued identification of β-thalassemia intermedia or HbE/β-thalassemia patients who may benefit from transfusion and iron chelation therapy.
1. Introduction to β-thalassemia
Annually, over 40,000 people worldwide are born affected by β-thalassemia, though it has been suggested that this is likely to be an underestimate Citation[1]. Incidence is highest in the Mediterranean, the Middle East, North America and South East Asia (particularly India, Thailand and Indonesia; this region accounts for ∼ 50% of affected births) and incidence is increasing worldwide as a result of migration Citation[2,3].
β-Thalassemias are characterized by a reduction or deficiency of β-globin chains and a subsequent imbalance in globin chains of the hemoglobin (Hb) molecule, which leads to impaired erythropoiesis. A wide variety of different mutations (> 200) have been documented that affect the β-globin gene, for which patients may be either homozygous or heterozygous. Phenotypic effects, therefore, range widely from slight impairment to the complete inhibition of β-globin chain synthesis Citation[4].
β-Thalassemia therefore comprises a number of different conditions, including β-thalassemia major in addition to β-thalassemia intermedia and HbE/β-thalassemia. Among these, β-thalassemia major and the most severe form of HbE/β-thalassemia are frequently referred to as transfusion-dependent thalassemia (TDT). In contrast, β-thalassemia intermedia and the mild and moderate forms of HbE/β-thalassemia are among several thalassemia types that are grouped as non-TDT (NTDT) ().
Figure 1. Transfusion dependence distinguishes β-thalassemias and their severity.
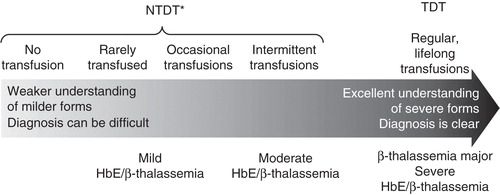
Patients with β-thalassemia major have symptoms at the severe end of the clinical spectrum and are reliant on regular red blood cell transfusions for survival from a young age. Each year, ∼ 23,000 people are born affected by β-thalassemia major Citation[1,3]. In contrast, β-thalassemia intermedia occurs less frequently, although accurate global incidence has not yet been established Citation[5]. In this case, the scale of severity varies significantly, ranging from mild clinical presentation with almost normal growth, to a more severe phenotype characterized by early onset anemia and symptoms including growth and development retardation and skeletal deformities. Some patients can also become transfusion-dependent during their lifetime Citation[6]. Of note, though β-thalassemia major describes patients with a clinically severe form of the disease, whereas β-thalassemia intermedia describes patients with less severe symptoms, the underlying molecular and pathophysiological basis for the two diseases is not necessarily distinct Citation[4].
HbE/β-thalassemia is the most common form of thalassemia in North Eastern/Eastern India, Bangladesh and South East Asia Citation[2], and occurs as a result of co-inheritance of HbE (caused by substitution in the β-globin gene, leading to the production of structurally abnormal Hb) and a β-thalassemia allele. Clinically, HbE/β-thalassemia is a highly heterogeneous disease. Typically, anemia with splenomegaly develops from 6 to 12 months of age, with impaired growth during the first decade of the patient’s life Citation[7]. Clinical symptoms among patients with mild and moderate forms of the disease vary widely; patients may be transfusion-dependent or require occasional or intermittent transfusion. In contrast, patients with the severe form are often transfusion-dependent and are generally managed in the same way as β-thalassemia major patients.
2. Treatment strategies
2.1 Transfusion
As mentioned, β-thalassemia major patients are dependent on transfusions for survival. For other patients, transfusion may occur rarely, occasionally or intermittently (). For example, occasional or intermittent transfusion therapy may be appropriate for β-thalassemia patients who are not transfusion-dependent as it provides healthy erythrocytes and reduces the occurrence of ineffective erythropoiesis Citation[8,9]. Of note, such NTDT patients should also be closely monitored for triggers of regular transfusion, as delayed transfusion can lead to growth and pubertal delay, thalassemic facies and hypersplenism Citation[6].
Table 1. Clinical requirements for occasional, intermittent or chronic transfusions in patients with β-thalassemia.
Clinicians should follow transfusion practices and policies recommended in relevant regional guidelines as these often vary depending on local considerations Citation[6,10-15]. Particular attention should be given to hemovigilance practices Citation[16] in order to minimize issues relating to appropriate blood screening, such as transmission of infectious diseases. A record should be kept of the patient’s transfusion history as the rate of transfusional iron loading may be important for subsequent iron chelator dose selection.
Furthermore, a number of considerations should be taken into account when initiating and continuing transfusion in patients with β-thalassemia intermedia or mild/moderate HbE/β-thalassemia. Caution should be exercised so that the patient is not subjected to frequent transfusions unnecessarily. Hb levels can fluctuate in non-transfusion-dependent patients, and therefore, upon diagnosis, patients should be followed carefully over several months before a treatment decision is made. Quality of life must also be taken into account as patients can survive and even thrive with an Hb level around 7 g/dl (particularly those with HbE/β-thalassemia) Citation[17-19]. Finally, it is important that patients are regularly reassessed, following initiation of transfusion, to determine whether continued transfusion is necessary.
A number of complications can occur as a result of transfusion. These include iron overload and related complications, such as cardiac, liver and endocrine problems. In addition, alloimmunization may occur, whereby the recipient mounts an immune response to donor antigens, resulting in various clinical consequences. Of note, this is more likely in non-transfusion-dependent patients (as transfusion is often initiated later in life), and in pregnant women and splenectomized patients. Alloimmunization can be minimized with good blood matching procedures Citation[20,21].
2.2 Splenectomy
Hypersplenism may occur as a result of large numbers of cells being pooled and destroyed in the spleen’s reticulo-endothelial system and hemodilution because of an increased plasma volume. Therefore, spleen size should be carefully monitored in all patients with β-thalassemia.
Many patients with β-thalassemia major require splenectomy. This should be performed in specific, defined clinical circumstances Citation[12] including splenic enlargement accompanied by left upper quadrant pain or early satiety, or leucopenia or thrombocytopenia due to hypersplenism. However, good clinical management may delay or prevent hypersplenism, reducing the need for splenectomy Citation[22].
In contrast, among patients with β-thalassemia intermedia or mild/moderate HbE/β-thalassemia, splenectomy should not be the first management option if others are available as it is associated with multiple adverse outcomes Citation[6,23-25]. As abnormalities of platelets and red blood cells in these patients become more prominent following splenectomy, patients are at an increased risk of thrombotic and vascular events, with an associated increased mortality rate Citation[26,27]. Splenectomy should be avoided in NTDT patients younger than 5 years of age, and in general should be reserved for very specific patients, including those with worsening anemia leading to poor growth and development; where transfusion and iron chelation are not possible or available; and in cases of hypersplenism or splenomegaly/massive splenomegaly.
For all patients, should splenectomy be performed, laparoscopic splenectomy is preferable to open surgery, and preventative measures – such as vaccination, prophylactic antibiotics and coagulation prophylaxis – should be taken to minimize the risks Citation[6,12,28].
2.3 HbF induction
Inducing agents such as hydroxyurea may be used in patients with β-thalassemia to increase the production of γ-globin (a β-globin-like molecule), which binds to α-chains to produce HbF, addressing the imbalance in globin chains. This, in turn, reduces the occurrence of ineffective erythropoiesis, decreases hemolysis and increases total Hb. There is, unfortunately, a lack of randomized clinical trials investigating the efficacy of hydroxyurea treatment. Though data are available from a large number of single-arm trials or retrospective analyses of hydroxyurea therapy, patient numbers are small and results have not been consistently reproduced (for a review of all of these studies, see reference Citation[29]). However, the clinical outcomes for patients in these studies are summarized as they represent an indication of potential efficacy.
In patients with β-thalassemia major, the proportion of patients who are no longer dependent on transfusion after treatment varies greatly between studies from around 25 to 80%. Around 20 – 50% of patients exhibit a ‘partial response’ whereby transfusion requirements are reduced Citation[29]. Improvements in transfusion requirements are also associated with a reduction in iron overload and hemolytic indices.
In β-thalassemia intermedia, responses vary greatly and study end points differ according to the severity of the disease before treatment. In a small study of β-thalassemia intermedia patients who were previously transfusion-dependent as a result of the severity of the disease, eight of nine patients showed a good response (> 70% reduction in transfusion requirements) Citation[30]. In studies of non-transfusion-dependent β-thalassemia intermedia patients, Hb increases of > 10 g/dl were observed in around 60 – 70% of patients, though this was not always maintained during a follow-up of more than 12 months Citation[31,32]. Hydroxyurea treatment is associated with a decreased incidence of many of the morbidities associated with this disease. In a study of 584 patients in the OPTIMAL CARE study, hydroxyurea treatment was associated with a reduction in pulmonary hypertension, leg ulcers, hypothyroidism, osteoporosis and extramedullary hematopoietic tumors, although there was an increased risk of hypogonadism Citation[33].
Fewer studies have been performed in patients with HbE/β-thalassemia, and again end points tend to vary between studies. A positive response (such as elimination of transfusion requirement or a significant increase in Hb) has been observed in around 35 – 40% of patients Citation[34,35], and improvement in anemia is generally associated with improvement in quality-of-life-related parameters Citation[29].
Where there is no immediate need for transfusion or splenectomy, and no indication that Hb levels will drop suddenly, it is suggested that a modulator of HbF is trialed. If Hb levels do drop suddenly, then transfusion prior to splenectomy might be more appropriate.
2.4 Hematopoietic stem-cell transplantation
Hematopoietic stem-cell transplantation (HSCT) is the only treatment that can be considered a cure for β-thalassemia Citation[36], and enhanced conditioning regimens and donor selection procedures have improved outcomes of this treatment in recent years Citation[37]. In such a procedure, the donor is ideally a human leukocyte antigen (HLA)-identical sibling of the patient, though ∼ 60% of patients lack such a suitable family donor. In these cases, transplant from matched unrelated donors may be possible Citation[37].
In patients with β-thalassemia major, the outcomes of HSCT vary according to a variety of factors. Patients are classified as Class 1, 2 or 3 based on iron overload, liver fibrosis and hepatomegaly status, and the highest survival rates are observed among Class 1 and 2 patients. It is also preferable that HSCT is performed at a young age, during the patient’s childhood, as survival rates ≥ 88% and thalassemia-free survival ≥ 83% are observed among these patients Citation[12,38]. Of note, HSCT is not generally performed for patients with less severe disease (i.e., β-thalassemia intermedia or HbE/β-thalassemia) as a result of a high risk of complications.
HSCT is a challenging procedure associated with several risks and complications. Graft-versus-host disease (GVHD) is the major cause of morbidity and mortality following allogeneic HSCT Citation[39]. Risk factors include older age of the recipient, use of a female donor for a male recipient, alloimmunization of the donor and HLA mismatching Citation[40]. In order to minimize the risk of GVHD, immunosuppressant conditioning should take place before and after HSCT Citation[37].
Veno-occlusive disease (VOD) may also occur in the first month after treatment, caused by hepatocyte and sinusoidal endothelial vessel damage following HSCT Citation[41]. Risk factors include patient age < 6.7 years, the type of VOD prophylaxis and the use of busulfan-containing conditioning regimens Citation[41]. Symptoms include pain in the upper right abdomen, weight gain, jaundice and ascites. In severe cases, VOD may lead to liver failure, hepato-renal syndrome, portal hypertension and eventual death from multiorgan failure Citation[41]. It is recommended that patients are monitored to allow prompt identification of VOD-induced inversion of portal flow as these patients might benefit from treatment with heparin and recombinant tissue plasminogen activator Citation[41].
Iron overload should also be considered in patients undergoing HSCT as excess iron can accumulate before and after HSCT. Levels may remain high for several years, even among patients who become transfusion-independent. Iron overload can affect survival, increasing the risks of infection and GVHD, and acting as a negative prognostic factor. Therefore, patients should be assessed and treated as appropriate for iron overload both pre- and post-HSCT Citation[42].
2.5 Future treatment options
Several new molecules and treatment strategies are currently in development, some of which show some promise for the treatment of β-thalassemia. Gene therapy has been trialed in several exploratory studies, with the aim of transferring β-globin in stem cells to reduce the α–β imbalance in erythroid cells, eventually leading to transfusion independence Citation[43]. In one exploratory clinical trial, an adult patient with severe HbE/β-thalassemia dependent on monthly transfusions since early childhood became transfusion-independent for 21 months following gene therapy Citation[44]. In this patient, Hb levels of 9 – 10 g/dl have been maintained, of which one-third contain vector-encoded β-globin.
Janus kinase 2 (Jak2) inhibitors have also been investigated for the treatment of β-thalassemia as they may limit the overproduction of immature erythroid cells in thalassemia, potentially reversing extramedullary hematopoiesis Citation[45]. Based on the available preclinical evidence, it is anticipated that Jak2 inhibitors might reduce the occurrence of splenomegaly, transfusion requirements and possibly iron overload in patients with β-thalassemia intermedia, though clinical data are not yet available Citation[46].
Finally, techniques for the correction of anemia without transfusions have been explored, including sotatercept (angiotensin-converting enzyme [ACE]-011) and ACE-536, modified activin type IIa or IIb receptor fusion proteins that inhibit signaling induced by other members of the transforming growth factor β super family, promoting maturation of terminally differentiating erythroblasts Citation[47,48]. In a murine model of β-thalassemia, the murine ortholog of ACE-536 (RAP-536) attenuated ineffective erythropoiesis, ameliorated anemia and improved associated comorbidities Citation[47], and in healthy volunteers ACE-536 produced a dose-dependent increase in red blood cells and Hb levels Citation[49]. Phase II trials of sotatercept are currently underway, though interim results are promising with a ≥ 2 g/dl increase in hemoglobin occurring in 33% of patients in the highest dose cohort, and generally good tolerability Citation[48].
3. Management strategies (complications)
3.1 Iron overload and chelation
In patients with transfusion-dependent β-thalassemia major, iron overload occurs as a result of accumulation of iron from transfusions and, to a lesser extent, increased intestinal absorption. Conversely, iron overload in patients with β-thalassemia intermedia or mild/moderate HbE/β-thalassemia is due to increased intestinal absorption secondary to ineffective erythropoiesis and, to a much lesser extent, accumulation of iron from transfusions Citation[50].
Iron overload is associated with an increased likelihood of complications. In β-thalassemia major patients, if left untreated, iron overload is fatal early in life, usually as a result of cardiac failure Citation[51]. Of note, access to cardiac MRI techniques has led to a significant reduction in mortality as a result of cardiac iron overload in recent years Citation[3,52]. However, as the management of cardiac disorders is improving, liver damage is coming to the forefront, and deaths from hepatic complications are increasing relative to other iron overload-related conditions Citation[53]. Among patients with β-thalassemia intermedia or mild/moderate HbE/β-thalassemia, there is generally an absence of cardiac siderosis irrespective of liver iron concentration (LIC), and the most common complications include osteoporosis, extramedullary hematopoiesis, hypogonadism and cholelithiasis Citation[33].
Iron load should be regularly monitored in patients with β-thalassemia and iron load values used to guide treatment decisions, including initiation and cessation of chelation and dose escalation Citation[54]. MRI is considered the gold standard; however, associated facilities and the required technical expertise are not always available. Serum ferritin assessment may be a practical and useful alternative for β-thalassemia major patients in particular Citation[55-57]. However, caution is advised as serum ferritin may underestimate LIC in patients with β-thalassemia intermedia or mild/moderate HbE/β-thalassemia compared with those with β-thalassemia major Citation[58,59]. Furthermore, serum ferritin may also fluctuate in response to infection/inflammation. For NTDT patients, data from the THALASSA study show that serum ferritin thresholds of 800, 2000 and 300 ng/ml may be useful to guide treatment decisions in terms of initiating chelation therapy, escalating dose and interrupting therapy, respectively, in the absence of access to liver MRI techniques Citation[6,60].
The introduction of regular transfusion and chelation regimens has led to improved survival in β-thalassemia major patients, as well as a substantial reduction in iron-related complications. In a study of 977 such patients treated with transfusion and deferoxamine (DFO), lower iron burden was associated with a lower probability of heart failure and hypogonadism and with prolonged survival Citation[61]. Increased survival without cardiac disease was observed in transfused patients in whom iron overload had been reduced as a result of chelation Citation[62].
It has also been acknowledged that optimal outcomes are achieved when chelation is tailored according to individual patient requirements, including transfusional iron loading Citation[57], as well as patient factors that may influence adherence Citation[63]. In regularly transfused patients, details of the rate of transfusional iron loading can guide chelation therapy decisions. Where this information is unavailable, this is estimated based on the assumption that one donor unit contains 200 mg iron Citation[12].
Several iron chelation agents are available. DFO was the first available chelator and has been in regular use since the 1980s Citation[64]. In transfusion-dependent patients with β-thalassemia major, DFO is effective in reducing liver iron overload. Among patients with baseline LIC between 7 and 14 mg Fe/g dry weight (dw), LIC was reduced by 1.9 ± 2.93 mg Fe/g dw, and among those with LIC > 14 mg Fe/g dw, the value was reduced by 6.4 ± 6.93 mg Fe/g dw Citation[55]. Furthermore, myocardial iron is improved; treatment with doses equivalent to 35 mg/kg/day for 7 days per week over ∼1 year led to changes in myocardial T2* from 13.3 to 15.0 ms Citation[65], and over 2 years from 11.6 to 14.2 ms Citation[66]. In β-thalassemia major patients with acute decompensated heart failure, continuous 24 h per day intravenous chelation with DFO is recommended, supplemented with deferiprone Citation[67]. Among 14 patients with β-thalassemia intermedia treated with DFO, a significant increase in urinary iron excretion was observed Citation[68]. However, growth retardation and bone changes (e.g., metaphyseal dysplasia) are common with doses > 60 mg/kg/day, especially those who begin chelation in the first 3 years of life Citation[69]. The risk of such complications may be reduced at doses ≤ 40 mg/kg, and pediatric patients should be monitored for body weight and growth every 3 months Citation[64].
Treatment with DFO requires frequent and prolonged subcutaneous administration, which can lead to concerns with patient compliance and quality of life. The introduction of oral chelators, together with routine MRI assessments of liver and cardiac R2 iron load, has substantially improved clinical outcomes for patients Citation[70].
Deferiprone is a bidentate oral iron chelator introduced in 1999 Citation[71]. Deferiprone improves LIC and cardiac iron load in β-thalassemia major patients and allows for combination chelation strategies with DFO Citation[65,72]. A very small number of patients with β-thalassemia intermedia have been treated with deferiprone in clinical trials, though iron overload was reduced Citation[73,74]. In 30 non-transfusion-dependent HbE/β-thalassemia patients with iron overload, serum ferritin, non-transferrin-bound iron and malondialdehyde decreased significantly (p < 0.05) after 1 year Citation[75]. Agranulocytosis and neutropenia are the most serious adverse events associated with deferiprone Citation[76-78]; patient absolute neutrophil count should be determined before starting treatment and monitored on a weekly basis, and therapy stopped if neutropenia develops (absolute neutrophil count < 1.5 × 109/l) Citation[71].
Deferasirox is the first once-daily oral iron chelator. Among β-thalassemia major patients, treatment with deferasirox 40 mg/kg/day resulted in LIC decreases from 30.6 ± 18.0 mg Fe/g dw at baseline to 14.4 ± 16.6 mg Fe/g dw after 2 years Citation[66,79]. Combination therapy of deferasirox and DFO has also been assessed, with serum ferritin decreases of 2174 from 5551 ng/ml at baseline and cardiac T2* increases from 7.03 to 8.24 ms after 1 year Citation[80]. Furthermore, as the only iron chelator to have gained approval for the treatment of iron burden in non-transfusion-dependent patients, deferasirox is indicated for the treatment of chronic iron overload in patients ≥ 10 years of age with β-thalassemia intermedia and HbE/β-thalassemia Citation[81]. In the THALASSA study, significant decreases in LIC and serum ferritin were observed over 12 months with deferasirox (least squares mean ± SD LIC decreased by 1.95 ± 0.50 mg Fe/g dw [from baseline 13.11 ± 7.29 mg Fe/g dw] with deferasirox 5 mg/kg/day, and by 3.80 ± 0.48 mg Fe/g dw [from baseline 14.56 ± 7.92 mg Fe/g dw] with deferasirox 10 mg/kg/day) and reductions continued over 2 years of treatment with similar safety profile Citation[82]. Furthermore, chelation with deferasirox can continue until iron reaches near-normal levels Citation[83,84]. Patients undergoing treatment with deferasirox do require regular, close monitoring as rare cases of acute renal failure, hepatic injury or gastrointestinal hemorrhages have been reported, mostly in elderly patients with multiple comorbidities and those with advanced hematologic malignancies Citation[81,85,86].
3.2 Other complications
A number of other complications occur in patients with β-thalassemia, involving several different organ systems (, ). Historically, in patients with β-thalassemia major, the most common complications have been cardiac-related. However, as mentioned above, with increased use of cardiac MRI techniques and effective chelation, liver complications are becoming more prominent. Infections (bacterial, viral or fungal) are also a significant cause of mortality and morbidity, particularly in patients with immune abnormalities, severe anemia, splenectomy or iron overload Citation[24,53,61,87], and chelation therapy with DFO has been associated with opportunistic infections by microorganisms that utilize the iron bound by the chelator Citation[88]. In addition, transmission of viruses such as hepatitis viruses or human immunodeficiency virus may occur where transfusion screening practices are not optimal Citation[89,90].
Table 2. Management options for specific β-thalassemia complications.
Figure 2. Major complications observed in patients with either transfusion-dependent or non-transfusion-dependent β-thalassemia. Patients with β-thalassemia intermedia or HbE/β-thalassemia exhibit a unique profile of complications compared with those with β-thalassemia major, although several complications including osteoporosis, endocrinopathies and hepatic complications, are observed across all patient types.
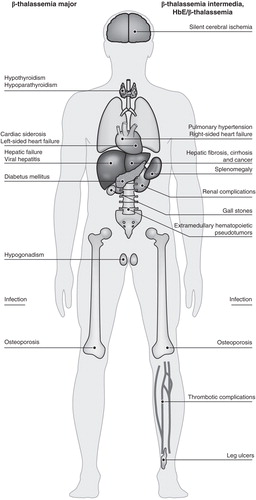
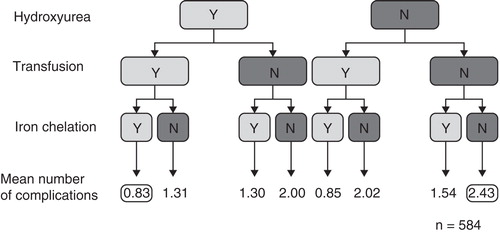
Patients with β-thalassemia intermedia or HbE/β-thalassemia exhibit a different profile of disease- and treatment-related complications (). The OPTIMAL CARE study was a retrospective review of 584 patients with β-thalassemia intermedia at six comprehensive care centers (Lebanon, Italy, Iran, Egypt, UAE and Oman), performed to assess the rate of complications in relation to currently practiced treatment options Citation[33]. This demonstrated that the most common disease-related complications were osteoporosis (23%), extramedullary hematopoiesis (21%), hypogonadism (17%) and cholelithiasis (17%), followed by thrombosis (14%), pulmonary hypertension (11%), abnormal liver function (10%) and leg ulcers (8%), whereas hypothyroidism, heart failure and diabetes mellitus were less frequently observed (all < 6%). The highest rates of complications were seen in patients who did not receive any treatment (), and among those who received transfusion, the incidence of complications was lower in those who also received chelation, compared with those who did not. However, older age and splenectomy were also independently associated with an increased risk of most complications.
Figure 3. Flow chart depicting the mean number of complications that occurred in patients with β-thalassemia intermedia undergoing different management schemes. Numbers reported are mean incidences for the 584 patients included in the retrospective OPTIMAL CARE study, according to the management strategy applied that may have involved hydroxyurea, red blood cell transfusion and iron chelation.
Regular patient monitoring is important in order for the clinician to identify and appropriately manage all complications, thus achieving the optimal outcome for the patient, though the financial resources, expertise and facilities required for assessments should also be taken into consideration. provides a comprehensive summary of β-thalassemia complications and appropriate management strategies. details the recommended frequency and relative expense of observation strategies for monitoring potential complications. In addition, monitoring may alter throughout the patient’s life, for example, growth and sexual development should be monitored most frequently in pediatric patients and monitoring in relation to iron overload may become more important in later life (from 8 to 10 years of age), as iron load increases with time.
Table 3. Frequency and relative expense of observation strategies for monitoring iron overload and other complications in β-thalassemias.
4. Expert opinion
Since the introduction of transfusion and iron chelation therapy with DFO in the 1970s and 1980s, the lives of patients with β-thalassemia major have been transformed, demonstrating that prolonged life is possible with effective iron removal. In recent years, the most important advances in the treatment of patients with β-thalassemia major have been the introduction of oral iron chelators and the availability of technologies that allow the direct measurement of iron in various organs, such as the heart and the liver. Therefore, physicians are now able to monitor and adjust iron chelation therapy more effectively based on more than just serum ferritin levels alone (e.g., see data from patients in the UK Citation[91]). As a result, further improvements in patient survival and quality of life are anticipated. In addition, the development of effective antibiotics and vaccinations along with recommendations for optimal screening practices prior to transfusion have had a positive impact on patients with thalassemia, though the prevention and treatment of infection does remain a challenge.
For patients with β-thalassemia intermedia, the OPTIMAL CARE study outlined various associated clinical complications related to iron overload (such as liver disease and liver cancer), and demonstrated how incidence increases with age. These observations highlighted the need for iron chelation therapy in these patients and prompted the development of guidelines for the clinical management of patients with non-transfusion-dependent β-thalassemia Citation[6]. The guidelines, along with data from randomized clinical trials in this patient group, can help inform treatment decision making. Data from the first randomized, placebo-controlled study evaluating iron chelation therapy in patients with β-thalassemia intermedia, HbE/β-thalassemia and other NTDT conditions (the THALASSA study) not only demonstrated the efficacy and safety of deferasirox in this patient population Citation[82,83], but also confirmed significant iron burden requiring chelation.
Further research is needed into the treatment and management of all β-thalassemia patients. There is still room for improvement in the conventional treatment of patients with β-thalassemia major to further extend survival and quality of life. For patients with β-thalassemia intermedia and HbE/β-thalassemia, efforts should be focused on the identification and recognition of patients who may benefit from transfusion therapy complemented by iron chelation therapy. As studies have highlighted the impact of iron accumulation in the absence of regular transfusion, and the increased rate of complications with advancing age, the importance of effective monitoring and early treatment intervention have become very clear. Clinicians would benefit from increased availability of educational resources and tools for the identification of such patients, as well as to guide their treatment.
Currently, gene therapy is proving to be an interesting approach in the treatment and possible cure of β-thalassemias. Few patients have received gene therapy to date; one trial in France is ongoing with results for one patient published Citation[44] and the remainder still under evaluation; and two Phase I/II trials are currently recruiting patients in the US and France, with the first patient transplanted in France in December 2013 Citation[92-95]. Another interesting and promising approach is the induction of erythropoiesis by molecules such as ACE-536 and sotatercept; the final results of ongoing Phase II trials are awaited.
In conclusion, it is suggested that the most important developments in the treatment of thalassemia patients have been an increased appreciation of the clinical significance of iron overload – in both transfusion-dependent and non-transfusion-dependent patients with β-thalassemia – along with the introduction of techniques for the direct measurement of iron load and oral iron chelators. It is proposed that future research should focus on the continued improvement and refinement of treatment options for transfusion-dependent patients and the identification of patients who require prompt and effective clinical management in spite of a lack of dependence on regular blood transfusion.
Article highlights.
Currently available treatment strategies for patients with β-thalassemia include red blood cell transfusion, splenectomy, fetal hemoglobin induction and hematopoietic stem-cell transplantation.
Several potential future treatment options are also undergoing clinical trials, including gene therapy, Janus kinase 2 inhibitors and techniques for the correction of anemia without transfusions including angiotensin-converting enzyme-536, a modified activin type IIb receptor fusion protein.
Iron overload occurs in patients with β-thalassemia, regardless of their transfusion status, and is associated with an increased likelihood of complications. Though access to cardiac MRI techniques has led to a reduction in mortality due to cardiac iron overload, liver damage is now coming to the forefront.
A number of other complications may also occur, though they vary between patients with β-thalassemia major and those with β-thalassemia intermedia or hemoglobin E/β-thalassemia. Cardiac and hepatic complications may occur in addition to endocrinopathies, pulmonary hypertension, extramedullary hematopoietic pseudotumors, osteoporosis, thromboembolic events and leg ulcers. Regular patient monitoring is essential to facilitate effective management.
Tailored iron chelation regimens have led to improved survival in β-thalassemia major patients, as well as a substantial reduction in iron-related complications.
Future research should focus on the continued improvement and refinement of treatment options for transfusion-dependent patients, in addition to the identification of patients who require prompt and effective clinical management in spite of a lack of dependence on regular blood transfusion.
Declaration of interest
MD Cappellini is receiving honoraria as an invited speaker by Novartis Pharmaceuticals, Genzyme and Shire; V Viprakasit received research grant support and lecture fees from Novartis Pharmaceuticals and research grant support from GPO-L-ONE, Thailand, FerroKin Biosciences and National Research University (NRU), Thailand; AT Taher reports receiving research funding and honoraria from Novartis Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Writing assistance was utilized in the production of this manuscript and funded by Novartis Pharmaceuticals.
Acknowledgements
The authors thank Bethan Hahn of Mudskipper Business Ltd for medical editorial assistance.
Notes
This box summarizes key points contained in the article.
Bibliography
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010;115(22):4331-6
- Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of beta-thalassemias and hemoglobin E disorders. Expert Rev Hematol 2010;3(1):103-17
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008;86(6):480-7
- Thein SL. The molecular basis of beta-thalassemia. Cold Spring Harb Perspect Med 2013;3(5):a011700
- Weatherall DJ, Clegg JB. The thalassaemia syndromes. Blackwell Science, Oxford; 2001
- Guidelines for the management of non transfusion dependent thalassaemia (NTDT). Thalassaemia International Federation. 2013. Available from: http://www.thalassaemia.org.cy/wp-content/uploads/pdf/educational-programmes/Publications/Non-Transfusion%20Dependent%20Thalassaemias%20%282013%29/NTDT%20ENGLISH.pdf
- Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med 2012;2(8):a011734
- Cazzola M, Finch CA. Evaluation of erythroid marrow function in anemic patients. Haematologica 1987;72(3):195-200
- Cazzola M, Pootrakul P, Huebers HA, et al. Erythroid marrow function in anemic patients. Blood 1987;69(1):296-301
- Angelucci E, Barosi G, Camaschella C, et al. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica 2008;93(5):741-52
- Musallam KM, Angastiniotis M, Eleftheriou A, et al. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol 2013;130(2):64-73
- Guidelines for the clinical management of thalassaemia, 2nd Revised Edition. Thalassaemia International Federation. 2008. Available from: http://www.thalassaemia.org.cy/wp-content/uploads/pdf/educational-programmes/Publications/Guidelines%20%282008%29/Thalassaemia%20Guidelines%20ENGLISH.pdf
- Standards of Care Guidelines for Thalassemia. Vichinsky E, Levine L. 2012. Available from: http://hemonc.cho.org/thalassemia/documents/SOCGuidelines2012.pdf
- Guidelines for the clinical care of patients with thalassemia in Canada. Thalassemia Foundation of Canada. 2009. Available from: http://www.thalassemia.ca/wp-content/uploads/Thalassemia-Guidelines_LR.pdf
- Standards for the clinical care of Children and adults with thalassaemia in the UK. United Kingdom Thalassaemia Society. 2008. Available from: http://sct.screening.nhs.uk/cms.php?folder=2493
- de Vries RR, Faber JC, Strengers PF. Haemovigilance: an effective tool for improving transfusion practice. Vox Sang 2011;100(1):60-7
- Olivieri NF, Muraca GM, O’Donnell A, et al. Studies in haemoglobin E beta-thalassaemia. Br J Haematol 2008;141(3):388-97
- Premawardhena A, Fisher CA, Olivieri NF, et al. Haemoglobin E beta thalassaemia in Sri Lanka. Lancet 2005;366(9495):1467-70
- Olivieri N. Treatment strategies for hemoglobin E beta-thalassemia. Blood Rev 2012;26(Suppl 1):S28-30
- Matteocci A, Pierelli L. Red blood cell alloimmunization in sickle cell disease and in thalassaemia: current status, future perspectives and potential role of molecular typing. Vox Sang 2013;106(3):197-208
- Chou ST, Liem RI, Thompson AA. Challenges of alloimmunization in patients with haemoglobinopathies. Br J Haematol 2012;159(4):394-404
- Piga A, Serra M, Longo F, et al. Changing patterns of splenectomy in transfusion-dependent thalassemia patients. Am J Hematol 2011;86(9):808-10
- Singer ST, Kuypers FA, Styles L, et al. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol 2006;81(9):670-5
- Vento S, Cainelli F, Cesario F. Infections and thalassaemia. Lancet Infect Dis 2006;6(4):226-33
- Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol 2012;158(1):16-29
- Cappellini MD, Robbiolo L, Bottasso BM, et al. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol 2000;111(2):467-73
- Taher A, Isma’eel H, Mehio G, et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost 2006;96(4):488-91
- Alexakis N, Dardamanis D, Albanopoulos K, et al. Incidence, risk factors, and outcome of portal vein thrombosis after laparoscopic-assisted splenectomy in beta-thalassemia patients: a prospective exploratory study. J Laparoendosc Adv Surg Tech A 2013;23(2):123-8
- Musallam KM, Taher AT, Cappellini MD, et al. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood 2013;121(12):2199-212
- Bradai M, Pissard S, Abad MT, et al. Decreased transfusion needs associated with hydroxyurea therapy in Algerian patients with thalassemia major or intermedia. Transfusion 2007;47(10):1830-6
- Mancuso A, Maggio A, Renda D, et al. Treatment with hydroxycarbamide for intermedia thalassaemia: decrease of efficacy in some patients during long-term follow up. Br J Haematol 2006;133(1):105-6
- Rigano P, Pecoraro A, Calzolari R, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. Br J Haematol 2010;151(5):509-15
- Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood 2010;115(10):1886-92
- Italia KY, Jijina FF, Merchant R, et al. Effect of hydroxyurea on the transfusion requirements in patients with severe HbE-beta-thalassaemia: a genotypic and phenotypic study. J Clin Pathol 2010;63(2):147-50
- Singer ST, Kuypers FA, Olivieri NF, et al. Fetal haemoglobin augmentation in E/beta(0) thalassaemia: clinical and haematological outcome. Br J Haematol 2005;131(3):378-88
- Thomas ED, Buckner CD, Sanders JE, et al. Marrow transplantation for thalassaemia. Lancet 1982;2(8292):227-9
- Lucarelli G, Isgro A, Sodani P, et al. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med 2012;2(5):a011825
- Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev 2008;22(2):53-63
- Lee SJ, Klein JP, Barrett AJ, et al. Severity of chronic graft-versus-host disease: association with treatment-related mortality and relapse. Blood 2002;100(2):406-14
- Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood 2011;117(11):3214-19
- Cesaro S, Pillon M, Talenti E, et al. A prospective survey on incidence, risk factors and therapy of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Haematologica 2005;90(10):1396-404
- Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematol Am Soc Hematol Educ Program 2010;2010:456-62
- Yannaki E, Karponi G, Zervou F, et al. Hematopoietic stem cell mobilization for gene therapy: superior mobilization by the combination of granulocyte-colony stimulating factor plus plerixafor in patients with beta-thalassemia major. Hum Gene Ther 2013;24(10):852-60
- Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010;467(7313):318-22
- Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol 2009;16(3):187-94
- Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev 2012;26(Suppl 1):S12-15
- Suragani RNVS, Li R, Cawley S, et al. ACE-536 improves ineffective erythropoiesis, anemia and co-morbidities in beta-thalassemia. Blood 2012;120(21):abstract 248
- Cappellini MD, Porter J, Origa R, et al. A Phase 2a, open-label, dose-finding study to determine the safety and tolerability of sotatercept (ACE-011) in adults with beta-thalassemia: interim results. Blood 2013;122(21):abstract 3448
- Attie KM, Boyd IE, Wilson DM, et al. ACE-536 increases hemoglobin in healthy postmenopausal women: a Phase 1, randomized, double-blind, placebo-controlled, multiple ascending dose study. Blood 2013;122(21):abstract 3194
- Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011;118(16):4321-30
- Zurlo MG, De Stefano P, Borgna-Pignatti C, et al. Survival and causes of death in thalassaemia major. Lancet 1989;2(8653):27-30
- Chouliaras G, Berdoukas V, Ladis V, et al. Impact of magnetic resonance imaging on cardiac mortality in thalassemia major. J Magn Reson Imaging 2011;34(1):56-9
- Voskaridou E, Ladis V, Kattamis A, et al. A national registry of haemoglobinopathies in Greece: deducted demographics, trends in mortality and affected births. Ann Hematol 2012;91(9):1451-8
- Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med 2000;343(5):327-31
- Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006;107(9):3455-62
- Olivieri NF, Brittenham GM, Matsui D, et al. Iron-chelation therapy with oral deferiprone in patients with thalassemia major. N Engl J Med 1995;332(14):918-22
- Cappellini MD, Porter JB, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin trends: the prospective multicenter EPIC study of deferasirox in 1744 patients with various transfusion-dependent anemias. Haematologica 2010;95(4):557-66
- Musallam KM, Taher AT. Iron-chelating therapy for transfusional iron overload. N Engl J Med 2011;364(15):1476
- Taher A, El Rassi F, Isma’eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica 2008;93(10):1584-6
- Taher A, Porter J, Viprakasit V, et al. Serum ferritin for predicting clinically relevant LIC thresholds to guide management of patients with non-transfusion-dependent thalassemia treated with deferasirox: THALASSA study extension analysis. Haematologica 2013;98(Suppl 1):abstract S1171
- Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004;89(10):1187-93
- Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med 1994;331(9):574-8
- Trachtenberg FL, Mednick L, Kwiatkowski JL, et al. Beliefs about chelation among thalassemia patients. Health Qual Life Outcomes 2012;10:148
- Desferal® (deferoxamine) basic prescribing information. Novartis Pharmaceuticals. 2011. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/desferal.pdf
- Pennell DJ, Berdoukas V, Karagiorga M, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006;107(9):3738-44
- Pennell DJ, Porter JB, Piga A, et al. Deferasirox compared with deferoxamine for the removal of cardiac iron in patients with beta-thalassemia major: 2-year data from the CORDELIA extension. Blood 2013;122(21):abstract 1018
- Pennell DJ, Udelson JE, Arai AE, et al. Cardiovascular function and treatment in beta-thalassemia major: a consensus statement from the American Heart Association. Circulation 2013;128(3):281-308
- Cossu P, Toccafondi C, Vardeu F, et al. Iron overload and desferrioxamine chelation therapy in beta-thalassemia intermedia. Eur J Pediatr 1981;137(3):267-71
- Brill PW, Winchester P, Giardina PJ, et al. Deferoxamine-induced bone dysplasia in patients with thalassemia major. AJR Am J Roentgenol 1991;156(3):561-5
- Nichols-Vinueza DX, White MT, Powell AJ, et al. MRI-guided iron assessment and oral chelator use improve iron status in thalassemia major patients: A six-year single center retrospective cohort study. Blood 2013;122(21):abstract 563
- Ferriprox summary of product characteristics. Apotex. 2011. Available from: http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000236/WC500022050.pdf
- Tanner MA, Galanello R, Dessi C, et al. Combined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunction. J Cardiovasc Magn Reson 2008;10:12
- Olivieri NF, Koren G, Matsui D, et al. Reduction of tissue iron stores and normalization of serum ferritin during treatment with the oral iron chelator L1 in thalassemia intermedia. Blood 1992;79(10):2741-8
- Rombos Y, Tzanetea R, Konstantopoulos K, et al. Chelation therapy in patients with thalassemia using the orally active iron chelator deferiprone (L1). Haematologica 2000;85(2):115-17
- Akrawinthawong K, Chaowalit N, Chatuparisuth T, et al. Effectiveness of deferiprone in transfusion-independent beta-thalassemia/HbE patients. Hematology (Am Soc Hematol Educ Program) 2011;16(2):113-22
- Ceci A, Baiardi P, Felisi M, et al. The safety and effectiveness of deferiprone in a large-scale, 3-year study in Italian patients. Br J Haematol 2002;118(1):330-6
- Cohen AR, Galanello R, Piga A, et al. Safety and effectiveness of long-term therapy with the oral iron chelator deferiprone. Blood 2003;102(5):1583-7
- Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood 2003;102(1):17-24
- Deugnier Y, Turlin B, Ropert M, et al. Improvement in liver pathology of patients with beta-thalassemia treated with deferasirox for at least 3 years. Gastroenterology 2011;141(4):1202-11
- Aydinok Y, Kattamis A, Cappellini MD, et al. Deferasirox–deferoxamine combination therapy reduces cardiac iron with rapid liver iron removal in patients with severe transfusional iron overload (HYPERION). Blood 2013;122(21):abstract 2257
- EXJADE® (deferasirox) US Prescribing Information. Novartis Pharmaceuticals. 2013. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/exjade.pdf
- Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol 2013;92(11):1485-93
- Taher AT, Porter J, Viprakasit V, et al. Deferasirox significantly reduces iron overload in non-transfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012;120(5):970-7
- Taher A, Porter J, Viprakasit V, et al. The safety profile of deferasirox remains consistent as non-transfusion-dependent thalassemia patients approach the target liver iron concentration of <3 mg Fe/g dw for interrupting chelation. Haematologica 2013;98(Suppl 1):abstract P391
- Grange S, Bertrand DM, Guerrot D, et al. Acute renal failure and Fanconi syndrome due to deferasirox. Nephrol Dial Transplant 2010;25(7):2376-8
- Yew CT, Talaulikar GS, Falk MC, et al. Acute interstitial nephritis secondary to deferasirox causing acute renal injury needing short-term dialysis. Nephrology (Carlton) 2010;15(3):377
- Mokhtar GM, Gadallah M, El Sherif NH, et al. Morbidities and mortality in transfusion-dependent Beta-thalassemia patients (single-center experience). Pediatr Hematol Oncol 2013;30(2):93-103
- Boelaert JR, de Locht M, Van Cutsem J, et al. Mucormycosis during deferoxamine therapy is a siderophore-mediated infection. In vitro and in vivo animal studies. J Clin Invest 1993;91(5):1979-86
- Cunningham MJ, Macklin EA, Neufeld EJ, et al. Complications of beta-thalassemia major in North America. Blood 2004;104(1):34-9
- Mallat ME, Sharara AI. Treatment and prevention of hepatitis B and C in thalassemia. Hemoglobin 2009;33(Suppl 1):S139-44
- Modell B, Khan M, Darlison M, et al. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2008;10:42
- Sadelain M. Recent advances in globin gene transfer for the treatment of beta-thalassemia and sickle cell anemia. Curr Opin Hematol 2006;13(3):142-8
- A study evaluating the safety and efficacy of the LentiGlobin® BB305 drug product in beta-thalassemia major subjects. NCT01745120. 2013. Available from: http://www.clinicaltrials.gov/show/NCT01745120
- Beta-thalassemia product development. Bluebirdbio. 2014. Available from: http://www.bluebirdbio.com/product-thalassemia.php
- Learn about the Northstar Study. Bluebirdbio. 2014. Available from: http://northstarstudy.com/
- Taher AT, Musallam KM, Karimi M, et al. Contemporary approaches to treatment of beta-thalassemia intermedia. Blood Rev 2012;26(Suppl 1):S24-7
- Lobo C, Angulo IL, Aparicio LR, et al. Retrospective epidemiological study of Latin American patients with transfusional hemosiderosis: the first Latin American epidemiological study in iron overload–the RELATH study. Hematology (Am Soc Hematol Educ Program) 2011;16(5):265-73
- Maakaron JE, Cappellini MD, Graziadei G, et al. Hepatocellular carcinoma in hepatitis-negative patients with thalassemia intermedia: a closer look at the role of siderosis. Ann Hepatol 2013;12(1):142-6
- European Association of the Study of the Liver. 2011 European Association of the Study of the Liver hepatitis C virus clinical practice guidelines. Liver Int 2012;32(Suppl 1):2-8
- Ramachandran P, Fraser A, Agarwal K, et al. UK consensus guidelines for the use of the protease inhibitors boceprevir and telaprevir in genotype 1 chronic hepatitis C infected patients. Aliment Pharmacol Ther 2012;35(6):647-62
- Wanachiwanawin W. Infections in E-beta thalassemia. J Pediatr Hematol Oncol 2000;22(6):581-7
- Borgna-Pignatti C, Carnelli V, Caruso V, et al. Thromboembolic events in beta thalassemia major: an Italian multicenter study. Acta Haematol 1998;99(2):76-9
- Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost 2010;8(10):2152-8
- Karimi M, Khanlari M, Rachmilewitz EA. Cerebrovascular accident in beta-thalassemia major (beta-TM) and beta-thalassemia intermedia (beta-TI). Am J Hematol 2008;83(1):77-9
- Succar J, Musallam KM, Taher AT. Thalassemia and venous thromboembolism. Mediterr J Hematol Infect Dis 2011;3(1):e2011025
- Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis 2006;37(1):12-20
- Borgna-Pignatti C, Gamberini MR. Complications of thalassemia major and their treatment. Expert Rev Hematol 2011;4(3):353-66
- Dore F, Cianciulli P, Rovasio S, et al. Incidence and clinical study of ectopic erythropoiesis in adult patients with thalassemia intermedia. Ann Ital Med Int 1992;7(3):137-40
- Shin KH, Sharma S, Gregoritch SJ, et al. Combined radiotherapeutic and surgical management of a spinal cord compression by extramedullary hematopoiesis in a patient with hemoglobin E beta-thalassemia. Acta Haematol 1994;91(3):154-7
- Haidar R, Mhaidli H, Taher AT. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur Spine J 2010;19(6):871-8
- Origa R, Fiumana E, Gamberini MR, et al. Osteoporosis in beta-thalassemia: Clinical and genetic aspects. Ann NY Acad Sci 2005;1054:451-6
- National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 1979;28(12):1039-57
- Kosaryan M, Mahdavi MR, Aliasgharian A, et al. Credibility of measurement of fructosamine and hemoglobin A1C in estimating blood glucose level of diabetic patients with thalassemia major. Open J Hematol 2012;3(4):1-7
- Farmaki K, Angelopoulos N, Anagnostopoulos G, et al. Effect of enhanced iron chelation therapy on glucose metabolism in patients with beta-thalassaemia major. Br J Haematol 2006;134(4):438-44
- Mangiagli A, Campisi S, De Sanctis V, et al. Effects of acarbose in beta-thalassaemia major patients with normal glucose tolerance and hyperinsulinism. Pediatr Endocrinol Rev 2004;2(Suppl 2):272-5
- Fernandes JL, Fabron A Jr, Verissimo M. Early cardiac iron overload in children with transfusion-dependent anemias. Haematologica 2009;94(12):1776-7
- Musallam KM, Rivella S, Vichinsky E, et al. Non-transfusion-dependent thalassemias. Haematologica 2013;98(6):833-44