
Abstract
Introduction: Inherited retinal degenerations (IRDs) have long been considered untreatable and incurable. Recently, one form of early-onset autosomal recessive IRD, Leber congenital amaurosis (LCA) caused by mutations in RPE65 (retinal pigment epithelium-specific protein 65 kDa) gene, has responded with some improvement of vision to gene augmentation therapy and oral retinoid administration. This early success now requires refinement of such therapeutics to fully realize the impact of these major scientific and clinical advances.
Areas covered: Progress toward human therapy for RPE65-LCA is detailed from the understanding of molecular mechanisms to preclinical proof-of-concept research to clinical trials. Unexpected positive and complicating results in the patients receiving treatment are explained. Logical next steps to advance the clinical value of the therapeutics are suggested.
Expert opinion: The first molecularly based early-phase therapies for an IRD are remarkably successful in that vision has improved and adverse events are mainly associated with surgical delivery to the subretinal space. Yet, there are features of the gene augmentation therapeutic response, such as slowed kinetics of night vision, lack of foveal cone function improvement and relentlessly progressive retinal degeneration despite therapy, that still require research attention.
1. Introduction
Inherited retinal degenerations (IRDs) are a group of blinding eye diseases now recognized to be caused by hundreds of different gene defects, mainly affecting photoreceptor cells and the retinal pigment epithelium Citation1, Citation2, Citation3. Prior to the era of gene discovery and known relationships of genes to specific diseases, clinicians specializing in IRDs tried to make sense out of the confusing array of symptoms and signs by subclassifying patients using Mendelian genetic type, age of disease onset, clinical features and retinal function. Clinical trials of treatment mainly sought to slow the natural history in groups of IRD patients using nutrient supplementation Citation4, Citation5.
The discovery of genetic causes of IRDs has continued at a steady pace over the last two decades with about 10 new genes discovered each year Citation[6]. Knowing the causative genes and mutations provides a basis for understanding human IRDs. Pathophysiological mechanisms can now be postulated and, in rare instances, science-based treatments have been devised for these historically incurable diseases. Rather than a limited number of animal models for IRDs based only on phenotype, now there are naturally occurring or de novo generated models with gene defects that may mimic features of the genetically corresponding human diseases. With a few exceptions, consortia of basic science, animal and human retinal specialists have not worked together to understand a newly molecularly defined group of patients and how an animal model relates to that human disease. Some of the models move forward to be used for proof-of-concept studies; and human therapies have even been proposed based on results in animals that may or may not be faithful mimics of the human IRD.
About 15 years ago, advances in science and medicine came together to pave the path toward therapies for an early-onset autosomal recessive IRD. The form of Leber congenital amaurosis (LCA) that has now been treated is caused by that is caused by an abnormality in the visual (retinoid) cycle resulting from deficiency of RPE65. Animal models of RPE65 deficiency were available and proof-of-concept studies for two forms of therapy in young animals showed efficacy. This review summarizes the stepwise progress to treat humans with this form of LCA and suggests further directions to take now that early clinical trials of treatment have been successful. The data presented herein are mainly those of the authors. There are many recent reviews of retinal gene therapy or, specifically, gene augmentation in RPE65-LCA and these cover a spectrum of viewpoints of this rapidly advancing field Citation7, Citation8, Citation9, Citation10, Citation11, Citation12, Citation13.
2. RPE65 and the retinoid cycle
Visual pigments in rod and cone photoreceptor cells detect light because they have the covalently linked light-sensitive chromophore, 11-cis-retinal. Light causes photoisomerization of this chromophore and its release as all-trans-retinal. Metabolic transformation of spent all-trans-retinal back to its light-sensitive form, 11-cis-retinal, is achieved by a series of transport and enzymatic processes termed the retinoid cycle. Combining recycled 11-cis-retinal with opsins to form visual pigments completes chromophore regeneration and permits phototransduction and visual perception to continue Citation14, Citation15, Citation16, Citation17, Citation18. One of the key enzymes of the retinoid cycle is retinoid isomerase, which is encoded by the RPE65 gene (). Deficiency of RPE65 leads to visual loss in human LCA. This visual disturbance is due not only to an inadequate supply of 11-cis-retinal but also to varying degrees of retinal degeneration Citation7, Citation19, Citation20, Citation21, Citation22.
Figure 1. RPE65 and the retinoid cycle. Schematic of a retinal pigment epithelial (RPE) cell and a photoreceptor cell outer segment with the flow of retinoids within and between the two different cells. There are intracellular and extracellular retinoid-binding proteins (such as CRALBP, CRBP1, IRBP). The retinoid isomerase, RPE65 (top center of the figure), the retinoid isomerase, is deficient in a form of LCA. See text for more details.
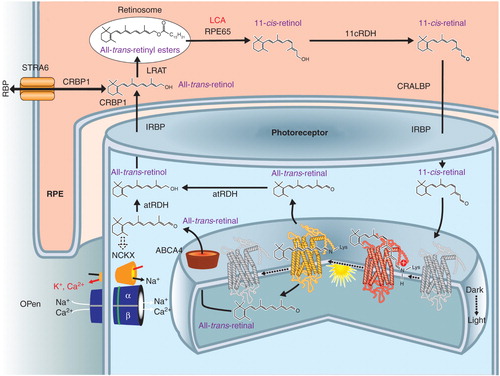
The topic of the rod versus the cone visual cycle is worthy of comment. During the last two decades, a significant amount of biochemical evidence has indicated that Müller cells are involved in regeneration of cone visual pigments through a specific visual cycle pathway Citation[23]. The key enzyme of this hypothetical pathway is thought to be the dihydroceramide desaturase-1 or Des-1 in conjunction with the key role of the cellular retinaldehyde-binding protein (CRALBP, encoded by RLBP1) in selection of the isomerization reaction products Citation[24]. The Müller cells are also a source of 11-cis-retinal bound to CRALBP that is expressed in both RPE and Muller cells Citation[25]. However, there is little genetic evidence from studies in humans and animals that provide proof of this pathway and its interplay with the ‘canonical’ RPE65-driven pathway.
3. Two preclinical experiments hold promise for treating human RPE65 deficiency
3.1 Remarkable improvement with oral 9-cis-retinoid in murine Rpe65 disease
Given knowledge of the retinoid cycle, a pharmacological replacement therapy was devised and preclinical studies were performed in Rpe65-deficient mice and dogs Citation26, Citation27, Citation28, Citation29, Citation30, Citation31, Citation32. Studies in young mice with Rpe65 deficiency but without retinal degeneration demonstrated the feasibility of increasing visual pigment and retinal function by oral administration of a 9-cis-retinoid, thus bypassing the retinoid cycle blockade in this genetic disease (). The pathway for oral 9-cis-retinoid action is proposed to involve absorption in the intestine, storage in the liver, secretion into the blood and transport via a binding protein to the RPE Citation[16]. 9-cis-retinal released from the RPE, possibly from storage sites composed of retinyl ester-containing lipid droplets, termed retinosomes Citation33, Citation34, combines with opsin to form isorhodopsin within photoreceptor cells. Isorhodopsin, investigated for more than half a century Citation[35], is a photosensitive visual pigment proposed to be the source of residual function originating from rod photoreceptors in Rpe65-deficient mice Citation[36] and the cause of improved vision after oral supplementation with 9-cis-retinoids Citation26, Citation27, Citation29, Citation30.
Figure 2. Preclinical studies showing improvement in retinal function in Rpe65-mutant mice treated with oral 9-cis-retinoids (A) or subretinal gene delivery of AAV2-Rpe65 (B). Left: Dark-adapted electroretinograms (ERGs) to increasing stimulus light intensity for a representative wild-type mouse. (A) ERG recordings in a representative 2-month-old Rpe65−/− mouse and in a 2.1-month-old Rpe65−/− mouse 48 h after oral 9-cis-retinal treatment. The treatment causes a lower stimulus threshold and larger amplitude ERGs. (B) ERG recordings comparing the untreated eye of a 2.9-month-old Rpe65rd12 mouse and the contralateral eye treated with subretinal gene therapy. The treatment effect was a lowering of threshold and larger amplitude ERGs.
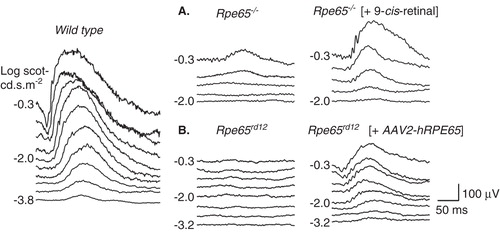
To explain further, opsin forms a stable pigment with 9-cis- 11-cis-retinal with two differences. First, the efficiency of photoisomerization for isorhodopsin is about one-third lower than that of rhodopsin. Second, the maximum absorption is shifted ∼ 7 – 10 nm to lower wavelengths (hypsochromic shift). Taking into consideration the range of human vision, these differences should be readily tolerated. Both isorhodopsin and rhodopsin when photoactivated form the same signaling intermediate termed metarhodopsin II Citation[37], thus providing a sound molecular basis of this approach.
3.2 Remarkable improvement after subretinal gene augmentation in canine and murine Rpe65 disease
Surgically delivered gene augmentation therapy was also used to correct the abnormal retinoid cycle in canine and murine models of human RPE65-LCA. Subretinal gene therapy improved visual function in three RPE65-mutant dogs using a recombinant adeno-associated virus (AAV) vector-carrying wild-type canine RPE65 cDNA Citation[38]. This proof-of-concept experiment was confirmed and extended in many additional studies. Visual function improvements were recorded at retinal, subcortical and cortical levels, and there were concordant biochemical, morphological and immunohistochemical observations Citation7, Citation39, Citation40.
Subretinal gene therapy using AAV, adenovirus (Ad) and lentiviral vectors was also performed in the Rpe65−/− (knockout) mouse model as well as the naturally occurring Rpe65rd12 mouse model. For the most part, there was improved retinal function () with supporting immunohistochemistry, rhodopsin biochemistry and cortical activity studies Citation[7].
4. Human RPE65 disease
Although there was promising preclinical evidence of efficacy with oral cis-retinoid and subretinal gene augmentation therapy in Rpe65-deficient animals, there were no studies specifically inquiring whether the disease expression in humans with RPE65 mutations resembled that in the animal models. It was assumed to be similar enough. Young canine and murine models of Rpe65 deficiency, however, exhibited near normal photoreceptor structure despite severe rod and cone dysfunction that was reversed by the therapies. Other than in certain rare congenital stationary night or day blinding disorders, normal photoreceptor structure would be unusual in man. High-resolution optical coherence tomography (OCT) was used to quantify photoreceptor layer thickness in RPE65-LCA patients (ages 11 – 53) to define the relationship of retinal structure to vision Citation[19]. Cone photoreceptor-rich central retina and rod-rich regions were specifically studied. Despite severely reduced cone vision, many RPE65-mutant retinas revealed a near-normal central microstructure. Absent rod vision was associated with a detectable but abnormally thinned photoreceptor cell layer. The human disease was thus not only a retinal dysfunction due to a biochemical blockade of the retinoid cycle but also a retinal degeneration. In contrast to other IRDs, however, RPE65-LCA patients showed greater photoreceptor nuclear layer integrity than predicted from their low level of vision Citation[19]. In the mouse model, it was asked whether abnormally thinned Rpe65-mutant retina with photoreceptor loss, such as found in the human disease, could respond favorably to treatment. Rpe65−/− mice at advanced disease stages show photoreceptor cell loss and this represented a more faithful mimic of the human disease. When gene therapy or oral retinoid treatment were administered to these late-stage degeneration mice, retinal function did improve but only in animals with better preserved photoreceptor structure. To accomplish early-phase clinical trial goals of testing safety and efficacy of subretinal gene therapy, retinal locations with retained photoreceptors would need to be identified (with OCT) and targeted. Otherwise, the goals of the clinical trials would either not be achieved or left to a trial-and-error approach. Oral cis-retinoid therapy clinical trials would also be best conducted if patients were evaluated pre-enrollment using noninvasive OCT imaging. In other words, RPE65-LCA patients should be ‘staged’ for severity of degeneration prior to entry into clinical trials (see Section 7). Results of the trials would then be more interpretable.
Further studies of retinal photoreceptor structure in young patients with RPE65-LCA (ages 6 – 17) revealed that there was considerable interindividual variation and a simple relationship of age to severity of degeneration could not be assumed Citation[21]. Summary maps of photoreceptor cell topography showed that superior-temporal and temporal pericentral retina was better preserved than other regions. Only more recently were natural history studies performed in the Rpe65 deficient dogs and mice and results used to decide when to administer treatment that would better approximate the human disease Citation41, Citation42.
5. Clinical trials of treatment
5.1 Subretinal gene augmentation therapy
Gene augmentation therapy for RPE65-LCA has been translated to the clinic and there are now multiple clinical trials worldwide with follow-up of at least 3 years showing safety and modest efficacy Citation7, Citation8, Citation9, Citation10, Citation11, Citation12, Citation13, Citation43, Citation44, Citation45, Citation46, Citation47. In the earliest versions of these trials, subretinal injection(s) of rAAV2-hRPE65 was administered to the worse-functioning eye at various dose levels. Primary outcomes were systemic and ocular safety. Secondary outcomes assayed visual function with a variety of methods, including visual acuity, dark-adapted full-field sensitivity testing, visual fields, pupillometry and mobility performance. Both cone- and rod-photoreceptor-based vision improved in treated areas Citation[46]. For extrafoveal cones, there were increases of up to 17 dB (50-fold); and for rods, there were gains of up to 48 dB (63,000-fold). A summary of the clinical trial (conducted by the authors) of 15 patients (age range, 11 – 30 years) showed there was no detectable systemic toxicity; any ocular adverse events were related to the retinal surgery Citation[48]. Visual function improved in all patients to different degrees; and improvements were localized to treated areas. Cone and rod sensitivities increased significantly in study eyes but not in control eyes (). Minor visual acuity improvements were recorded in many study as well as control eyes. Major acuity improvements occurred in study eyes with the lowest entry acuities and parafoveal fixation loci treated with subretinal injections. Other patients with better foveal structure lost retinal thickness and acuity after subfoveal injections. It was concluded that RPE65-LCA gene therapy was safe and efficacious to the extrafoveal retina. To treat the fovea was of no benefit and carried some risk. There was no evidence of age-dependent effects Citation[48].
Figure 3. Comparison of early efficacy post-treatment in subretinal gene therapy (A) and oral cis-retinoids (B) in RPE65-LCA. Pseudo-three-dimensional representation of fully dark-adapted sensitivities across the visual field of the (A) gene-treated eye of two patients at baseline and 30 days post-treatment. P9 had a single subretinal injection in the superior retina; P11 had two subretinal injections, one in the superior retina and another in the nasal retina. (B) Baseline and 5 – 6 days after beginning a week-long course of oral cis-retinoids in one eye of two patients. Part B is reproduced with permission from Citation[52].
![Figure 3. Comparison of early efficacy post-treatment in subretinal gene therapy (A) and oral cis-retinoids (B) in RPE65-LCA. Pseudo-three-dimensional representation of fully dark-adapted sensitivities across the visual field of the (A) gene-treated eye of two patients at baseline and 30 days post-treatment. P9 had a single subretinal injection in the superior retina; P11 had two subretinal injections, one in the superior retina and another in the nasal retina. (B) Baseline and 5 – 6 days after beginning a week-long course of oral cis-retinoids in one eye of two patients. Part B is reproduced with permission from Citation[52].](/cms/asset/17d8f19d-20e4-4f88-a275-c13a3fa64e03/ieod_a_1030393_f0003_c.jpg)
5.1.1 Unexpected results encountered to date
Detailed studies of the effects of gene therapy in patients with RPE65-LCA have produced several unexpected observations.
5.1.1.1 Slow rod kinetics in treated retina
To assess what fraction of full visual potential was restored by gene therapy (taking into account the degenerative component), the relationship of the degree of light sensitivity to the level of remaining photoreceptors within the treatment area was determined. It was found that treatment could overcome nearly all the loss of light sensitivity resulting from the biochemical blockade component of RPE65-LCA. The reconstituted retinoid cycle, however, was not completely normal. Newly treated rods were remarkably slow to resensitize after light exposure and required 8 h or more to attain full sensitivity as compared to < 1 h for normal eyes. Cone sensitivity recovery time was rapid. These results demonstrated dramatic but imperfect recovery of rod and cone photoreceptor-based vision after RPE65 gene therapy. In addition to theoretical interest in the basis of the slowed kinetics of rod adaptation, there is the practical implication that these patients need an extended time to dark adapt post-treatment or the visual gain in rod sensitivity would be underestimated. More concerning would be an apparent variability in magnitude of rod-mediated visual gain if insufficient (or different amounts of) dark adaptation was used to test for efficacy on different post-treatment assessments Citation[46].
5.1.1.2 Pseudo-fovea formation
A self-report by a patient about 1 year after gene therapy led to the observation that there could be late emergence of further visual gain in the treated eye. A pseudo-fovea developed and under certain conditions, the patient showed a preference to fixate at the treated retinal region (in the superior-temporal retina) rather than at the anatomical fovea. This suggested an experience-dependent plasticity of the adult visual system Citation[49]. Further examples of this behavior have been documented subsequently and recently reported Citation[50].
5.1.1.3 Continued progression of retinal degeneration, independent of therapy
A key question about the effects of gene therapy on photoreceptor longevity was addressed recently Citation[42]. As demonstrated previously, untreated RPE65-LCA patients show degeneration as well as dysfunction of photoreceptors even at the earliest ages. When examined serially over years, untreated RPE65-LCA outer photoreceptor nuclear layers (ONL) displayed progressive thinning. It has been expected, although unproven, that correction of the underlying cellular dysfunction by gene therapy could rescue the photoreceptors from degeneration. In treated RPE65-LCA, however, retinal degeneration also continued to progress despite the improved vision. This set of observations indicated that gene therapy in RPE65-LCA should not be considered as a permanent one-time treatment. An expanded view of the therapy is needed and ways to improve photoreceptor survival should be explored. Neuroprotective agents could be delivered independently to patients or in combination with gene augmentation and this could be tested experimentally in the canine model Citation[42].
5.2 Oral retinoid therapy
Oral synthetic retinoid treatment (ClinicalTrials.gov Identifier NCT01014052) advanced to clinical trials in 7 RPE65-LCA patients and in another molecular subtype of LCA (lecithin retinol acyltransferase, LRAT) that also involves the retinoid cycle. Results of light-adapted metrics, such as visual acuity and kinetic perimetry, were reported to be improved following a week-long course of treatment with most returning to baseline by 2 years post-treatment. A minority of patients had longer-term persistence of improved acuity or visual field responses Citation[51]. Considering the dramatic improvement in rod photopigment and rod physiology in the proof-of-concept studies of oral 9-cis retinoids in murine Rpe65 deficiency Citation19, Citation26, Citation27 it is of interest that results of dark-adapted perimetry were reported in two RPE65-LCA patients (ages 24 and 40) who received oral cis-retinoid Citation[52]. Increases in dark-adapted sensitivity averaged 12 dB at 10 – 50% of loci. With extended dark adaptation, there were more dramatic increases in sensitivity of, on average, 16 – 19 dB over baseline at 40 – 83% of loci; individual loci could be as much as 24 – 36 dB increased (). The fact that extended dark adaptation led to further increases in rod sensitivity leads to the speculation that, like the slowed kinetics of rod adaptation in gene treated RPE65-deficient retina Citation[46], the kinetics of rod adaptation after oral retinoid was also abnormally prolonged.
6. Comparison of the two treatments for RPE65-LCA
What are the differences between the results to date from surgically delivered gene augmentation therapy with AAV2-RPE65 and the oral cis-retinoid treatment? Ocular gene therapy involves subretinal injection(s) of a certain volume of vector gene that is expected to transfect a localized region of the retina. Within a few days after this surgical procedure in one eye, there was evidence of an increase in rod vision in that eye limited to the region of subretinal treatment Citation[48]. There is progressive retinal degeneration despite the improved vision Citation[42], suggesting that vision would also eventually be impacted by the underlying cellular losses.
Oral cis-retinoid therapy is expected to affect retinal areas in both eyes, presumably those that retain sufficient photoreceptors and RPE to support the improved vision. Within days of therapy, there was evidence of an increase in rod vision across both eyes. The length of time that improvement in rod vision is sustained after oral cis-retinoid has not been reported; if predicted from rodent proof-of-concept experiments Citation[29], it would be expected to diminish soon after dosing stops. It is also unknown whether the retinoid has any effect on the natural history of the progressive retinal degeneration in RPE65-LCA.
7. Next steps for treatment of RPE65-LCA
Ocular gene therapy of RPE65-LCA initially appeared deceptively simple with remarkable visual improvement occurring days to weeks after subretinal injection and apparent persistence of the positive effects for years. Treatment of the human disease (and later stages in animal models) has now been revealed to have limitations. Though augmenting wild-type RPE65 restored the retinoid cycle function (albeit not normally) and it improved rod and cone vision (but not foveal vision) in a measurable way, what was not measured, except in one of the clinical trials, was the degenerative component of this disease. If not quantified by serial optical imaging, the degenerative component would not have been noted to change until atrophy would become evident on ophthalmoscopic examination years to decades later. Clearly, the natural history of photoreceptor loss was independent of treatment Citation[42]. The time course of visual loss that would be expected to eventually accompany the photoreceptor losses in treated and untreated retinal regions has not been reported.
Based on what is known to date of the effects of gene therapy in RPE65-LCA, it is obvious that the existing therapy is not fully adequate. What can be done now to advance the application of gene therapy to RPE65-LCA and other potentially treatable retinopathies?
7.1 Staging the disease
There is no strategy yet to define in detail patient candidacy for receiving gene therapy. The stage of the RPE65-LCA disease at the time of clinical presentation should serve as a guide to whether a subretinal injection procedure is an appropriate recommendation for that patient. Age of a patient has been assumed to be a major determinant of candidacy and benefit. By extrapolating from data of animal models of various IRDs, it has been assumed that an invariant decline in retinal function and photoreceptor structure with age is probably also what occurs in the population of patients with RPE65-LCA, despite a spectrum of different causative mutant alleles and diversity of genetic backgrounds. Are there data in humans to support this reductionist assumption? Individual untreated patients with RPE65 mutations definitely show a decline in vision with age Citation[22]. The kinetics of retinal degeneration in untreated human RPE65-mutant retina has also been investigated Citation[42]. The conclusion from these studies is that there is no simple relationship between age and severity of disease when examining a population of patients with RPE65 mutations. The onset, degree and spatial distribution of retinal degeneration across patients differ such that some young patients can have relatively severe degenerative disease while some older patients can have far better retinal preservation determined by careful imaging and visual functional analyses Citation21, Citation22. There are in vitro results for certain mutant RPE65 alleles Citation53, Citation54, Citation55, but there are no data on the relationship of the many different mutations in the RPE65 gene (and any modifier genes) to severity of in vivo disease expression. Considerable variation of severity of retinal degeneration exists in the first two decades of life. For patients above the age of 30 years, however, evidence of limited function and structure should provoke serious consideration of the risks versus the benefits of administering gene therapy Citation20, Citation21, Citation22.
Staging for severity of retinal degeneration is akin to performing biopsies as is commonly used to stage progressive nonocular diseases in order to make decisions about treating or type of treatment. To stage RPE65-LCA patients is within modern clinical capabilities. In RPE65-LCA, it can be accomplished noninvasively by in vivo imaging of the retina with OCT. Segmentation of these images and calculation of the average photoreceptor layer thickness across a wide retinal area would allow a patient’s retinal disease to be classified into one of possibly three stages (namely mild, moderate or severe). Patients without measurable photoreceptors would not be candidates for this therapy. For example, the average (and standard deviation) of photoreceptor layer thickness was measured across the central retinal region (30° × 30° square, 870 loci) in 12 RPE65-LCA patients and the patients were grouped as mild, moderate or severe (4 patients per group). In this sample, the statistically different averages were: 31 (6.7), 22.5 (6.1) and 11 (3.5) µm, respectively. Retrospective or prospective staging of all patients in ongoing clinical trials could be related to treatment efficacy by using some visual function criterion such as dark-adapted full-field sensitivity testing Citation56, Citation57. From such data a decision could be made about the frequency of treatment efficacy for each stage. In the future, it would be ideal and fair to present data to a patient about the chance of a successful outcome, rather than just a list of possible adverse events associated with retinal surgery.
Methods for rapidly testing interventions, such as in cancer treatment or prevention, are of strong interest to develop in the future, but have yet to be realized Citation58, Citation59. Based on the preliminary report about improvement in dark-adapted measures of vision using oral 9-cis-retinoid in two RPE65-LCA patients Citation[52], it would seem worthwhile to consider this retinoid for use as a provocative test of the potential for efficacy in patients with RPE65-LCA before there is commitment to ocular surgery for gene therapy. This testing could be especially valuable for patients staged as ‘severe’ based on OCT average results. Some functional improvement from oral retinoid would suggest that a patient could be considered for gene therapy. A way to predetermine the value of gene therapy to a given patient would be a welcome addition to the therapeutic strategy for RPE65-LCA.
7.2 Disease management strategy
A long-term management strategy with gene therapy for RPE65-LCA also has not been proposed to date. All clinical trials began by treating one eye with a single subretinal injection. One clinical trial also treated two retinal locations in one eye in later cohorts to increase retinal coverage Citation[48] and there is a report of second eye treatment administered years later Citation[60]. Binocular strategies with single injections on separate days are also in progress (NCT00999609). Same eye retreatment has not been reported to date. Given a calculated stage of severity at first presentation, a photoreceptor life expectancy from time of initial OCT mapping can be determined using the delayed exponential model of the disease Citation[42]. This prognosis should help define the long-term therapeutic strategy.
A sequence from clinical diagnosis to staging to treatment is illustrated (). In the clinical staging column, there is a photoreceptor life expectancy graph. The horizontal axis is the time (years) since degeneration phase onset; ONL fraction remaining is plotted vertically in log units. The photoreceptor life expectancy for each stage (labeled below horizontal axis) is based on the patient’s average ONL fraction at time of imaging (marked by circle) until the ‘end-of-life’ criterion (−1 log unit ONL fraction, which is 10% of normal mean). A relatively simple gene transfer strategy with a series of single uniocular subretinal injections over years is shown. More complex strategies with two injections per eye and binocular treatments are possible. Initial administration targets the superior retina, as shown in the schematic of the fundus. Hypothetical outcomes are illustrated for each of three disease severities (see Staging above). Predicted visual improvement (at peak response) is shown as a square on a visual field map. After an interval determined by disease severity stage and the expected time to peak improvement, the nasal retina is treated. Treatment of the temporal retina could follow after a further interval, thereby providing a substantial total area of visual field that would otherwise be lost.
Figure 4. An algorithm for gene therapy in RPE65-LCA. Diagnostic workup is followed by clinical staging for severity of retinal degeneration. Photoreceptor life expectancy from time of initial mapping can then be calculated using the delayed exponential model of the disease. Simple or more complex strategies can then proceed with targeting of specific regions of retina proven to have photoreceptor integrity. Neuroprotection can be initiated as early as the diagnosis is made. See text for more details.
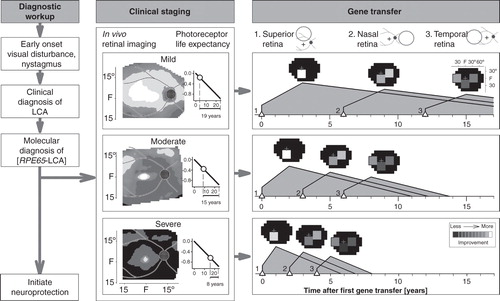
In any future management strategy, it must be remembered that RPE65-LCA disease and gene therapy treatment effects are dynamic processes. Given the addition of a neuroprotective agent Citation[42], the lifespan of photoreceptor cells could increase thus possibly lengthening the interval between follow-on treatments. Whether oral retinoid treatment becomes an alternative or a complement to gene therapy remains an important question.
7.3 Other treatment strategies
Nonviral gene therapy of Rpe65-deficient murine models has also been explored Citation61, Citation62, Citation63. Specifically, proof-of-concept experiments with subretinal DNA nanoparticles have been encouraging with long-term persistence of efficacy. Given further improvement of this method, concern over re-administration of AAV-mediated delivery could be eliminated if nonviral therapy was used. Problems with subretinal injection, however, would remain.
Staging of LCA patients with molecular evidence of disease-causing RPE65 mutations will lead to identification of those with such severe photoreceptor and RPE loss (or who have some other exclusion criterion) that the gene-specific methods described above may not be appropriate. Such patients deserve consideration as possible candidates for other current methods being developed to improve severe vision loss Citation[64]. For example, the development of a retinal prosthesis has a long history and there have been recent clinical trials; and further advances are expected. With evidence of extremely impaired vision, little or no measurable photoreceptors by OCT and presence of inner retinal laminae, this becomes one option Citation65, Citation66, Citation67, Citation68, Citation69. Another evolving method uses optogenetics to transform remaining inner retinal cells into light sensors Citation[70]. Light-gated ion channels via a gene therapy approach are introduced into the residual inner retinal neurons Citation71, Citation72. Such optochemical and optochemical-genetic approaches have also shown proof-of-concept efficacy in large and small animal models of retinal degeneration Citation[73].
Finally, the field of regenerative medicine is advancing such that stem cells of different origins can eventually become alternatives for improving vision in patients without sufficient photoreceptors to respond to current therapies Citation74, Citation75.
8. Expert opinion
The new challenge for clinicians will be to decide if or when novel therapies should be recommended and administered to specific IRD patients. As in all branches of medicine, treatment options create the need for decision-making. The most difficult decisions will require judgment based on knowledge of the topic(s). There will be pressure upon general ophthalmologists and retinal specialists who only rarely see IRD patients to be up-to-date (with the diseases, the natural history, the disease mechanisms) and be able to speak knowledgably about the latest therapies. Speaking from clinical experience will not be possible for many years. IRD patients tend to be well-informed about their diseases and will require more information from the physician than what they may have already gleaned from the popular media. There is now even more pressure than in past decades to administer therapy. Well-meaning patient organizations, for example, have fund-raised for decades to support basic and applied research, and have from the beginning promised clinical (not only basic scientific) progress. Their constituents have waited for these promises to be kept and there are accumulated feelings of obligation by organizations to announce and even promote treatments sometimes without the usual scientific scrutiny and complete knowledge of the outcomes and risks.
Will the workups and longer consultations needed to discuss novel therapies with the rare IRD patients fit into the active clinical schedules of ophthalmic practitioners? In the past, some of the busier practitioners found it expedient simply to provide a generally poor prognosis to their IRD patients. Ongoing needs of such patients including referrals for low vision and mobility training, career counseling, genetic counseling and even routine ophthalmic care to avoid complicating eye diseases were occasionally not pursued. Patients tended to learn from each other in support groups and at meetings of research-oriented foundations or associations to help the blind. The ophthalmic clinician will now have to practice an evolving form of personalized medicine for this group of orphan diseases.
For those who specialized in IRDs before the current novel therapeutics, personalized medicine has long been the only form of medicine practiced Citation[76]. Now, there are simply more branches to the decision tree. A modified version of the four approaches to the practice of personalized medicine by Meyer Citation[77] can be applied to RPE65-LCA. First is the assessment of an individual and the clinical diagnosis (from eye examination and electroretinography) and then molecular genetic testing. The latter demands a DNA sample and an infrastructure to submit it to a standardized approved laboratory and then the expertise in interpreting the result and explaining it to the patient/family Citation78, Citation79. Incidentally, there is no evidence to date that prenatal genetic diagnosis (e.g., in the case of a sibling with RPE65-LCA) is warranted, just as there is no evidence for administering gene therapy within the first years of life (see above). However, early diagnosis of RPE65-LCA remains a goal – mainly to be able to begin administration of a neuroprotective agent to alter the natural history Citation[42]. Improvement of central cone function (given foveal cone structural integrity) is another plausible reason for early diagnosis and possible intervention. The latter could be a pharmaceutical agent or a gene therapy targeting an alternative cone visual cycle Citation15, Citation24 with an intravitreal vector delivery (but not subretinal surgery with its attendant risk of foveal cell loss). Second is to ‘increase diagnostic precision’ beyond molecular diagnosis by defining the phenotype with ‘prognostic and therapeutic implications’ Citation[77]. Staging the retinal degenerative component of the disease using OCT is key to estimating the prognosis and deciding when to administer gene therapy (). Third is ‘tailoring of treatment to the individual characteristics of each patient’ Citation[77]. Even simple staging of the degree of degeneration would identify nonresponders. Improved tailoring of treatment to individuals could come from classifying patients by a predictable response to treatment derived from a consensus about how to measure efficacy in relationship to stage of degeneration in currently ongoing clinical trials (see above). The rapid move to higher phase trials has forced less rather than more information to be collected in the pursuit of ‘quality of life’ measurements. Focus should return to those details needed to understand a disease better and thereby improve the quality of its treatment. For example, gene therapy in RPE65-LCA has been popularized to an extent that it would seem to be in an advanced state, but some of the basics of the human disease, such as genotype–phenotype studies, have been bypassed in favor of a trial-and-error approach in the clinic and operating room. Clarifying whether patients with specific RPE65 mutant alleles have different natural histories than others and require a specifically timed treatment strategy is essential. A starting population to study could be the group of RPE65 patients with the same founder alleles in Israel to determine if there is a predictable natural history in this relatively homogeneous group Citation[47]. A fourth tenet of personalized medicine from Meyer Citation[77] is the ‘proper evaluation of objective and subjective clinical outcomes.’ OCT imaging, a feasible objective outcome for mechanism and safety, led to the observation that gene therapy did not alter the natural history of cell loss Citation[42] and that foveal cell loss can accompany subfoveal injection Citation44, Citation48. The full-field sensitivity test after extended dark adaptation has offered the best assessment of outcome from these trials Citation46, Citation48, Citation57. The clinical utility of the therapy will not be improved until the impaired kinetics of dark adaptation are explained and corrected. The gain in night vision, one of the major improvements as a result of this therapy, is now constantly in jeopardy of being of no clinical utility unless the patients somehow learn to attend to their light history, a difficult way to live life in our well-lit modern world. Most reports by patients in the popular media are essentially proclaiming gains of extrafoveal cone vision.
Gene correction of IRDs, perhaps by a CRISPR-Cas9 or related approach, will advance and extend to other retinopathies Citation8, Citation10. The dual pathomechanism of RPE65-LCA disease, dysfunction and degeneration, however, is unlikely to be replicated in many other IRDs. Most IRDs will require natural history studies performed with quantitative outcomes for structure and function to try to detect negative changes in relatively short periods of time. Yet, there could be conditions in which photoreceptor outer segment lengths in residual cells would increase as a result of therapy and this could lead to some visual restoration. There are also retinal disorders that may be permanently cured by gene augmentation such as forms of congenital stationary night blindness that do not have a degenerative component Citation[80].
In summary, treatment of IRDs seems to be at an inflection point, with early successes of gene therapy for RPE65-LCA deservedly infusing the field with optimism. However, careful clinical examination of these patients over time has revealed that simple, one-time gene therapy does not lead to a permanent therapeutic outcome and that there is a need to reexamine how clinical decisions are made relating to both initial treatment and follow-up options. This review has attempted to lay out a logical scaffold for dealing with these complex but ultimately approachable issues.
Inherited retinal degenerations (IRDs) are a group of blinding eye diseases that have long been considered untreatable and incurable.
An autosomal recessive IRD called Leber congenital amaurosis (LCA), caused by mutations in the key retinoid cycle gene encoding RPE65, has recently been in clinical trials.
Both gene augmentation therapy by subretinal injection and oral retinoid administration have led to some visual improvement in RPE65-LCA.
There remain unresolved issues related to both therapeutic approaches that warrant attention before commercialization of the products.
Most important is the issue of progressive photoreceptor cell loss (retinal degeneration) that continues in the treated regions even after vision-improving gene therapy.
Efforts are needed to improve the quality and longevity of this novel treatment and thereby advance retinal gene augmentation therapy for RPE65-LCA as well as for other IRDs that will become candidates for this form of intervention.
Declaration of interest
Supported by the National Eye Institute (National Institutes of Health) grants U10 EY017280, EY06855, EY17549; Foundation Fighting Blindness; Hope for Vision; Macula Vision Research Foundation; and The Chatlos Foundation, Inc. All procedures followed the Declaration of Helsinki and there was approval by the Institutional Review Board (IRB). Included were patient data from those taking part in a Phase I clinical trial (NCT00481546 registered at www.clinicaltrials.gov) evaluating the safety of rAAV2-CBSB-hRPE65. University of Pennsylvania, University of Florida and Cornell University hold a patent on the described gene therapy technology (United States Patent 20070077228, ‘Method for Treating or Retarding the Development of Blindness’). WWH and the University of Florida have a financial interest in the use of AAV therapies, and own equity in a company (AGTC, Inc.) that might, in the future, commercialize some aspects of this work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Notes
This box summarizes key points contained in the article.
Bibliography
- ANBramall, AFWright, SGJacobson, RRMcInnes. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci 2010;33:441-72
- AFWright, CFChakarova, MMAbd El-Aziz, SSBhattacharya. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet 2010;11:273-84
- CPHamel. Gene discovery and prevalence in inherited retinal dystrophies. C R Biol 2014;337:160-6
- ELBerson. Nutrition and retinal degenerations. Int Ophthalmol Clin 2000;40:93-111
- DRHoffman, KGLocke, DHWheaton, et al. A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for X-linked retinitis pigmentosa. Am J Ophthalmol 2004;137:704-18
- RetNet: retinal Information Network. Daiger SP and the University of Texas Health Science Center. RetNet, Houston, TX; 2014. Available from: https://sph.uth.edu/retnet/ [Last accessed 10 December 2014]
- AVCideciyan. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 2010;29:398-427
- PColella, AAuricchio. Gene therapy of inherited retinopathies: a long and successful road from viral vectors to patients. Hum Gen Ther 2012;23:796-807
- RBHufnagel, ZMAhmed, ZMCorrea, RASisk. Gene therapy for Leber congenital amaurosis: advances and future directions. Graefes Arch Clin Exp Ophthalmol 2012;250:1117-28
- SEBoye, SLBoye, ASLewin, WWHauswirth. A comprehensive review of retinal gene therapy. Mol Ther 2013;3:509-19
- LSCarvalho, LHVandenberghe. Promising and delivering gene therapies for vision loss. Vision Res 2014;10.1016/j.visres.2014.07.013
- EAPierce, JBennett. The status of RPE65 gene therapy trials: safety and efficacy. CSH Perspect Med 2015;10.1101/cshperspect.a017285
- DAThompson, RRAli, EBanin, et al. Advancing therapeutic strategies for inherited retinal degeneration: recommendations from the Monaciano symposium. Invest Ophthalmol Vis Sci 2015;56:918-31
- JCSaari. Biochemistry of visual pigment regeneration: the Friedenwald lecture. Invest Ophthalmol Vis Sci 2000;41:337-48
- GHTravis, MGolczak, RMoise, KPalczewski. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol 2007;47:469-512
- KPalczewski. Retinoids for treatment of retinal diseases. Trends Pharmacol Sci 2010;31:289-95
- PHTang, MKono, YKoutalos, et al. New insights into retinoid metabolism and cycling within the retina. Prog Retin Eye Res 2013;23:48-63
- KPalczewski. Chemistry and biology of the initial steps in vision: the Friedenwald Lecture. Invest Ophthalmol Vis Sci 2014;55:6651-72
- SGJacobson, TSAleman, AVCideciyan, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci USA 2005;102:6177-82
- SGJacobson, TSAleman, AVCideciyan, et al. Human cone photoreceptor dependence on RPE65 isomerase. Proc Natl Acad Sci USA 2007;104:15123-8
- SGJacobson, AVCideciyan, TSAleman, et al. Photoreceptor layer topography in children with Leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci 2008;49:4573-7
- SGJacobson, TSAleman, AVCideciyan, et al. Defining the residual vision in Leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci 2009;50:2368-75
- NLMata, RARadu, RCClemmons, GHTravis. Isomerization and oxidation of vitamin a in cone-dominant retinas: a novel pathway for visual-pigment regeneration in daylight. Neuron 2002;36:69-80
- JJKaylor, QYuan, JCook, et al. Identification of DES1 as a vitamin A isomerase in Muller glial cells of the retina. Nat Chem Biol 2013;9:30-6
- YXue, SQShen, JJui, et al. CRALBP supports the mammalian retinal visual cycle and cone vision. J Clin Invest 2015;125:727-38
- JPVan Hooser, TSAleman, YGHe, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA 2000;97:8623-8
- JPVan Hooser, YLiang, TMaeda, et al. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem 2002;277:19173-82
- ZAblonczy, RKCrouch, PWGoletz, et al. 11-cis-retinal reduces constitutive opsin phosphorylation and improves quantum catch in retinoid-deficient mouse rod photoreceptors. J Biol Chem 2002;277:40491-8
- TMaeda, AMaeda, GCasadesus, et al. Evaluation of 9-cis-retinyl acetate therapy in Rpe65-/- mice. Invest Ophthalmol Vis Sci 2009;50:4368-78
- TMaeda, AVCideciyan, AMaeda, et al. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum Mol Genet 2009;18:2277-87
- PMGearhart, CGearhart, DAThompson, SMPetersen-Jones. Improvement of visual performance with intravitreal administration of 9-cis-retinal in Rpe65-mutant dogs. Arch Ophthalmol 2010;128:1442-8
- AMaeda, KPalczewski. Retinal degeneration in animal models with a defective visual cycle. Drug Discov Tod Dis Models 2013;10:e163-72
- YImanishi, MLBatten, DWPiston, et al. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J Cell Biol 2004;164:373-83
- TOrban, GPalczewska, KPalczewski. Retinyl ester storage particles (retinosomes) from the retinal pigmented epithelium resemble lipid droplets in other tissues. J Biol Chem 2011;283:17248-58
- RHubbard, GWald. Cis-trans isomers of vitamin A and retinene in the rhodopsin system. J Gen Physiol 1952;36:269-315
- JFan, BRohrer, GMoiseyev, et al. Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc Natl Acad Sci USA 2003;100:13662-7
- KPalczewski. G protein-coupled receptor rhodopsin. Annu Rev Biochem 2006;75:743-67
- GMAcland, GDAguirre, JRay, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 2001;28:92-5
- GMAcland, GDAguirre, JBennett, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther 2005;12:1072-82
- GKAguirre, AMKomáromy, AVCideciyan, et al. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med 2007;4:e230
- RCCaruso, TSAleman, AVCideciyan, et al. Retinal disease in Rpe65-deficient mice: comparison to human Leber congenital amaurosis due to RPE65 mutations. Invest Ophthalmol Vis Sci 2010;51:5304-13
- AVCideciyan, SGJacobson, WABeltran, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA 2013;110:E517-25
- JWBainbridge, AJSmith, SSBarker, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008;358:2231-9
- AMMaguire, FSimonelli, EAPierce, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008;358:2240-8
- WWHauswirth, TSAleman, SKaushal, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 2008;19:979-90
- AVCideciyan, TSAleman, SLBoye, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA 2008;105:15112-17
- EBanin, DBandah-Rozenfeld, AObolensky, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther 2010;21:1749-57
- SGJacobson, AVCideciyan, RRatnakaram. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012;130:9-24
- AVCideciyan, WWHauswirth, TSAleman, et al. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med 2009;361:725-7
- AVCideciyan, GKAguirre, SGJacobson, et al. Pseudo-fovea formation after gene therapy for RPE65-LCA. Invest Ophthalmol Vis Sci 2014;56:526-37
- RKKoenekoop, SRufang, JSallum, et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet 2014;384:1513-20
- AVCideciyan, ATMoore, EZrenner, et al. Increased vision within days of oral cis-retinoid (QLT091001) treatment in blindness due to mutations in retinal pigment epithelium-specific protein 65kDa (RPE65) or lecithin retinol acyltransferase (LRAT). ARVO Meeting Abstract 2012;53:6965
- TMRedmond, EPoliakov, SYu, et al. Mutation of key residues of Rpe65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci USA 2005;102:13658-63
- ARPhilp, MJin, SLi, et al. Predicting the pathogenicity of RPE65 mutations. Hum Mutat 2009;30:1183-8
- SLi, TIzumi, JHu, et al. Rescue of enzymatic function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J Biol Chem 2014;289:18943-56
- AJRoman, SBSchwartz, TSAleman, et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp Eye Res 2005;80:259-72
- AJRoman, AVCideciyan, TSAleman, et al. Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol Meas 2007;25:N51-6
- BKEdwards, EWard, BAKohler, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010;116:544-73
- MVBlagosklonny. NCI’s provocative questions on cancer: some answers to ignite discussion. Oncotarget 2011;2:1352-67
- JBennett, MAshtari, JWellman, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med 2012;4:120ra15
- ZHan, SMConley, RMakkia, et al. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PLoS One 2012;7:e52189
- AKoirala, RSMakkia, SMConley, et al. S/MAR-containing DNA nanoparticles promote persistent RPE gene expression and improvement in RPE65-associated LCA. Hum Mol Genet 2013;22:1632-42
- AKoirala, SMConley, RMakkia, et al. Persistence of non-viral vector mediated RPE65 expression: case for viability as a gene transfer therapy for RPE-based diseases. J Control Release 2013;172:745-52
- SGJacobson, AVCideciyan. Treatment possibilities for retinitis pigmentosa. N Eng J Med 2010;363:1669-71
- GJChader, JWeiland, MSHumayun. Artificial vision: needs, functioning, and testing of a retinal electronic prosthesis. Prog Brain Res 2009;175:317-32
- JDWeiland, AKCho, MSHumayun. Retinal prostheses: current clinical results and future needs. Ophthalmology 2011;118:2227-37
- JFRizzoIII, DBShire, SKKelly, et al. Overview of the boston retinal prosthesis: challenges and opportunities to restore useful vision to the blind. Cong Proc IEEE Eng Med Biol Soc 2011;2011:7492-5
- SJGarg, JFederman. Optogenetics, visual prosthesis and electrostimulation for retinal dystrophies. Curr Opin Ophthalmol 2013;24:407-14
- SPicaud, J-ASahel. Retinal prostheses: clinical results and future challenges. C R Biol 2014;337:214-22
- LFenno, OYizhar, KDeisseroth. The development and application of optogenetics. Annu Rev Neurosci 2011;34:389-412
- SNirenberg, CPandarinath. Retinal prosthetic strategy with the capacity to restore normal vision. Proc Natl Acad Sci USA 2012;109:15012-17
- SGJacobson, ASumaroka, KLuo, AVCideciyan. Retinal optogenetic therapies: clinical criteria for candidacy. Clin Genet 2013;84:175-82
- BMGaub, MHBerry, AEHolt, et al. Restoration of visual function by expression of a light-gated mammalian ion channel in retinal ganglion cells or ON-bipolar cells. Proc Natl Acad Sci USA 2014;111(51):E5574-83
- SDSchwartz, CDRegillo, BLLam, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015;385(9967):509-16
- LAWiley, ERBurnight, AESongstad, et al. Patient-specific induced pluripotent stem cells (iPSCs) for the study and treatment of retinal degenerative diseases. Prog Retin Eye Res 2015;44:15-35
- LFPorter, GCMBlack. Personalized ophthalmology. Clin Genet 2014;86:1-11
- UAMeyer. Personalized medicine: a personal view. Clin Pharmacol Ther 2012;91:373-5
- EMStone. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc 2003;101:437-84
- BPBrooks, IMMacdonald, SJTumminia, et al. National Ophthalmic Disease Genotyping Network (eyeGENE). Genomics in the era of molecular ophthalmology: reflections on the National Ophthalmic Disease Genotyping Network (eyeGENE). Arch Ophthalmol 2008;126:424-5
- CZeitz, AGRobson, IAudo. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res 2015;45C:58-110