Extensive validation of microarray-based gene signatures represents the significant challenge in the integration of such information into the standard of care of cancer patient management. Both retrospective and prospective validation, using multiple institutions or in cooperation with clinical trial groups, should be considered in the design of validation studies. In addition, biological pathway analysis and DNA copy number changes can provide a new dimension of data to verify the gene expression-based signatures. To obtain a comprehensive molecular and clinical picture of the patient, the information obtained from these signatures should then be used in concert with standard risk factors as well as data obtained from pathological and clinical evaluations.
Internal & external validations of gene expression-based signatures
Breast cancer is a heterogeneous disease that exhibits a wide variety of clinical presentations. Traditional prognostic factors are not always sufficient to predict patient outcomes accurately Citation[1,2]. Prognostic signatures generated from microarray studies have now emerged in the literature Citation[3–5]. Criticisms of microarray studies include the lack of sufficient sample size, lack of proper validation in independent patient cohorts, preferably in a multicenter setting, and lack of overlap of individual genes between various prognostic signatures.
A recent study validated two of these signatures Citation[6]. The sensitivities/specificities for 5-year time to distant metastasis (TDM) involving untreated lymph node-negative breast cancer patients were 90/42% for the 70-gene signature, and 90/50% and 97/34% for the 76-gene signature, respectively. In Cox univariate analysis for TDM, the 70-gene signature yielded a hazard ratio (HR) of 2.3, the 76-gene signature HRs of 7.4 and 5.8. Both gene signatures outperformed the clinicopathologic risk as defined by Adjuvant! Online software Citation[101] with adjusted HRs for 10-year TDM of 3.5 for the 70-gene signature and 5.1 for the 76-gene signature. Interestingly, a strong time dependency was found for both signatures in the analysis for TDM, peaking at 4 years with an adjusted HR of 9.2 for the 70-gene signature and at 5 years for the 76-gene signature with an adjusted HR of 13.6 Citation[6]. These independent multicenter validation studies demonstrate the reproducibility and robustness of these two-gene expression signatures.
Other validation studies are in progress. There are two recently started prospective randomized trials, Microarray In Node-negative Disease may Avoid Chemo-Therapy (MINDACT) in Europe and the Trial Assigning IndividuaLized Options for Treatment Rx (TAILORx) in the USA, both including lymph node-negative breast cancer patients and respectively based on the 70-gene signature derived from global gene-expression data using frozen samples and the 21-gene signature derived from a set of 250 previously published candidate genes Citation[7]. This latter assay was designed to analyze RNA isolated from formalin-fixed paraffin-embedded tissues by quantitative real time-PCR technology. It also has the advantage of potentially wider clinical applications due to the relative ease of logistics with respect to tumor handling, shipping and storage.
Biological pathways represented by the gene signatures
Most, if not all, published gene expression signatures are based on a combination of individual genes. It has been suggested that it might be more appropriate to interrogate the gene lists for biological themes, rather than just the individual genes. Moreover, identification of the distinct biological processes between subtypes of cancer patients is more relevant to help understand the mechanism of the disease development and for targeted drug development. Furthermore, gene expression profiles of a primary tumor are maintained throughout the metastatic process, suggesting that the genomic composition of the primary tumor might be relevant for the metastasis to be treated. Thus, identifying relevant pathways in the primary tumor might help to develop targeted drugs that may work in both the adjuvant and metastatic setting. Taking into account that many genes are correlated on gene expression arrays, especially genes involved in the same biological process, it is not surprising that many genes may be present in distinct signatures when different training sets of patients and statistical tools are used. Furthermore, genes are usually included in a classifier that applies stringent statistical criteria, and as a result of these strict significance levels there is only a small chance for any specific gene to be included.
We analyzed the biological processes associated with the tumor’s metastatic capability in a large set of 344 lymph node-negative breast cancer patients who had not received adjuvant systemic therapy. Since gene expression patterns of estrogen receptor (ER) subgroups of breast tumors are quite different, data analyses to derive gene signatures and subsequent pathways were conducted separately for ER-positive and -negative tumors. The datasets were re-sampled numerous times to construct a total of 1000 multiple gene lists whose expression correlated with patients’ distant metastasis-free survival Citation[8]. For ER-positive tumors, cell division-related processes were frequently found in the top 20 over-represented pathways. For ER-negative tumors, many of the top 20 pathways were related to RNA processing, transportation and signal transduction.
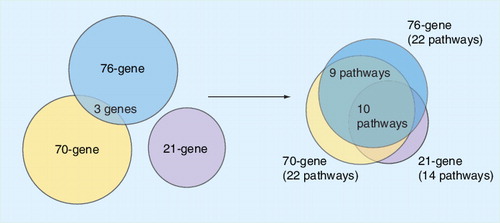
Since several prognostic gene signatures have a similar prognostic strength and are represented by genes playing a role in common biological pathways, the identification of the key biological processes, rather than the assessment of individual genes, provides potential targets for future drug development, as has been suggested. Furthermore, the identification of distinct biological pathways and their activation mechanisms that are targets for treatment may guide the use of the most promising combination of drugs targeting multiple pathways. Ultimately, knowledge of the tumor phenotype with respect to activated pathways may potentially lead to the implementation of personalized medicine in which each individual patient will be treated with the best combination of drugs. We and others have previously suggested that it might be more appropriate to interrogate the gene list for biological themes in the data, rather than for individual genes Citation[4,9,10]. To this end we analyzed the 76-gene signature Citation[4], the 70-gene signature Citation[3] and the 21-gene signature Citation[7]. Although only three genes overlap between the 76- and 70-gene signatures and there is no overlap of these two signatures with the 21-gene signature, pathway analysis reveals a significant overlap . Thus, although the genes in the signatures may differ, the biological pathways represented are nearly identical. We conclude that the combination of gene expression profiling and pathway data can be used to reduce the complexity of apparently dissimilar prognostic gene expression signatures.
Integration with gene expression & genetic information
The use of orthogonal datasets, employing changes in gene expression, copy number, DNA sequence and DNA methylation, combined with more traditional clinical parameters will generate an even more comprehensive and perhaps clearer view of the biological complexity. Several recent reports have illustrated this concept. For example, Sun and coworkers have demonstrated that breast cancer prognosis can be improved through a combination of clinical and genetic markers Citation[11]. A hybrid signature was developed that performed better than the original 70-gene signature Citation[3], clinical markers alone and the St Gallen criterion. For example, at 90% sensitivity, the hybrid signature demonstrated 67% specificity compared with 47% for the 70-gene signature and 48% for clinical markers. Two clinical markers used in the hybrid signature were tumor grade and angioinvasion.
Even when clinical information is not combined with molecular information, the use of different types of molecular information could have advantages. Unlike expression arrays, discrete data for genetic variation, using genotyping microarrays, provides a binary perspective on a patient’s relative risk for disease and/or correlation to therapeutic response Citation[12]. For example, combining information on single nucleotide polymorphisms and gene expression has shown utility. Liu and coworkers generated a 32-gene signature that distinguished p53-wild-type and mutant tumors and outperformed sequence-based assessments in predicting prognosis and response to therapy Citation[13]. The signature was also able to identify a set of aggressive tumors that did not display p53 sequence mutations but demonstrated expression patterns similar to those having p53 deficiency. Similarly, Troester and coworkers found a 52-gene expression signature associated with p53 loss that predicted relapse-free, disease-specific and overall survival Citation[14].
The combination of copy number changes and gene expression changes can also be used to paint either a more comprehensive or a simplified molecular portrait of a tumor. For example, Chin and coworkers identified associations between recurrent copy number aberrations, gene expression and outcome in breast tumors Citation[15]: they demonstrated that patient stratification was improved when both expression changes and copy number changes were measured. We have also combined the information obtained from copy number aberrations (using 100K single nucleotide polymorphism arrays from Affymetrix) and gene expression profiling. Our studies identified genes that had a consistent amplification or deletion and whose change in copy number correlated with a change in gene expression across 273 breast cancer patients. Analysis of the top 316 prognostic genes, as assessed by Cox regression modeling, revealed a striking similarity in copy number and gene expression changes [Zhang Y, Klijn J, Yu J et al. Multi-dimensional genomic analysis identifies a class of breast cancer patients with high metastatic outcome and differential response to chemotherapeutic drugs (2008), Submitted].
Lessons learned in the development of a colon prognostic assay
Despite the recent success of the oncogenomics field, there are significant technical challenges that we face in the development of gene signature-based tests in the clinic today. These challenges include:
| • | The type and quality of samples used for discovery and clinical use | ||||
| • | The transfer of technology from discovery to development of a robust and reproducible assay | ||||
| • | The inclusion of multiple samples from test sites that introduce site-to-site variability | ||||
| • | The sensitivity of the platform to detect moderately expressed but significant changes in expression | ||||
For patients diagnosed with stage II colon cancer, approximately 20–25% will progress to later stages of the disease and have a poor prognosis for survival Citation[16]. We previously published the performance of a 23-gene expression signature for determining the recurrence of distant metastases for colon cancer patients Citation[4]. Others have also validated our 23-gene signature with independent samples while identifying additional signatures for the same prognostic purpose or derived altogether different signatures Citation[17–19]. We further refined our signature from 23 genes to seven genes and, because the seven-gene signature was relatively small, we were able to consider the performance of these genes by using quantitative PCR that confirmed our microarray results [Zhang Y, Klijn J, Yu J et al. Development of a clinically feasible molecular assay to predict recurrence of stage II colon cancer (2008), Submitted].
New challenges may arise during validation of markers since clinical samples will potentially reflect differences in previous practices for sample storage or changes in the detection of disease. For example, access to primary tumors from pathology collections over the past 10 years may represent larger tumor sizes versus the samples collected today because screening improvements enable the detection of smaller tumors. Assays run on samples that are freshly collected, fixed and embedded may also demonstrate different technical performance than with retrospective samples, hence the need for prospective studies.
Site-to-site variability may also be caused by different clinical characterization of samples. For example, there is a wide range in the number of lymph nodes that are tested across different geographical regions for suspicion of colon cancer Citation[20]. This difference in practice demonstrates that some of the colon cancers are mis-staged when too few lymph nodes are sampled and should be an important component of sample characterization across different sites.
In summary, our studies and those of other investigators have demonstrated the importance of validation of gene signatures for cancer diagnosis and prognosis in multicenter trials with well-annotated patient cohorts. Combining molecular information with traditional pathological and clinical assessment will create a new era in systems biology and medicine, paving the way for the development of molecular interaction maps Citation[21], nomograms Citation[22], and personalized therapies Citation[23].
Financial & competing interests disclosure
The authors are employees of Veridex, LLC, a Johnson and Johnson company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Goldhirsch A, Wood C, Gelber RD et al. Meeting highlights: updated international expert consensus on the primary therapy of early breast cancer. J. Clin. Oncol.21, 3357–3365 (2003).
- Eifel P, Axelson JA, Costa J et al. National Institutes of Health Consensus Development Conference Statement: adjuvant therapy for breast cancer. J. Natl Cancer Inst.93, 979–989 (2001).
- Van’t Veer LJ, Dai H, van de Vijver MJ et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature415, 530–536 (2002).
- Wang Y, Klijn JG, Zhang Y et al. Gene expression profiles to predict distant metastasis of lymph node negative primary breast cancer. Lancet365, 671–679 (2005).
- Foekens JA, Atkins D, Zhang Y et al. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J. Clin. Oncol.24, 1665–1671 (2006).
- Desmedt C, Piette F, Loi S et al. TRANSBIG Consortium. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin. Cancer Res.13, 3207–3214 (2007).
- Paik S, Shak S, Tang G et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med.351, 2817–2826 (2004).
- Yu JX, Sieuwerts AM, Zhang Y et al. Pathway analysis of gene signatures predicting metastasis of node-negative primary breast cancer. BMC Cancer7, 182 (2007).
- Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics23(8), 980–987 (2007).
- Adler AS, Lin M, Horlings H et al. Genetic regulators of large-scale transcriptional signatures in cancer. Nat. Genet.38, 421–430 (2006).
- Sun Y, Goodison S, Li J et al. Improved breast cancer prognosis through the combination of clinical and genetic markers. Bioinformatics23(1), 30–37 (2007).
- Lanfear DE, McLeod HL. Pharmacogenetics: using DNA to optimize drug therapy. Am. Fam. Physician76, 1179–1182 (2007).
- Miller LD, Smeds J, George J et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl Acad. Sci. USA102, 13550–13555 (2005).
- Troester MA, Herschkowitz JI, Oh DS et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer6, 276 (2006).
- Chin K, DeVries S, Fridlyand J et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell10, 529–541 (2005).
- Allegra CJ, Paik S, Colangelo LH et al. Prognostic value of thymidylate synthase, Ki-67, and p53 in patients with Dukes’ B and C colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project collaborative study. J. Clin. Oncol.21, 241–250 (2003).
- Barrier A, Boelle PY, Roser F et al. Stage II colon cancer prognosis prediction by tumor gene expression profiling. J. Clin. Oncol.24, 4685–4691 (2006).
- Arango D, Laiho P, Kokko A et al. Gene-expression profiling predicts recurrence in Dukes’ C colorectal cancer. Gastroenterology129(3), 874–884 (2005).
- Grade M, Hörmann P, Becker S et al. Gene expression profiling reveals a massive, aneuploidy-dependent transcriptional deregulation and distinct differences between lymph node-negative and lymph node-positive colon carcinomas. Cancer Res.67, 41–56 (2007).
- Baxter NN, Virnig DJ, Rothenberger DA, Morris AM, Jessurun J, Virnig BA. Lymph node evaluation in colorectal cancer patients: a population-based study. J. Natl Cancer Inst.97, 219–225 (2005).
- Kohn KW, Aladjem MI, Kim S et al. Depicting combinatorial complexity with the molecular interaction map notation. Mol. Syst. Biol.2, 51 (2006).
- Stephenson AJ, Scardino PT, Eastham JA et al. Preoperative nomogram predicting the 10-year probability of prostate cancer recurrence after radical prostatectomy. J. Natl Cancer Inst.98, 715–717 (2006).
- West M, Ginsburg GS, Huang AT et al. Embracing the complexity of genomic data for personalized medicine. Genome Res.16, 559–566 (2006).
Websites
- Adjuvant! Online: decision making tools for health care professionals www.adjuvantonline.com