
Abstract
Vaccines have been invaluable for global health, saving lives and reducing healthcare costs, while also raising the quality of human life. However, newly emerging infectious diseases (EID) and more well-established tropical disease pathogens present complex challenges to vaccine developers; in particular, neglected tropical diseases, which are most prevalent among the world’s poorest, include many pathogens with large sizes, multistage life cycles and a variety of nonhuman vectors. EID such as MERS-CoV and H7N9 are highly pathogenic for humans. For many of these pathogens, while their genomes are available, immune correlates of protection are currently unknown. These complexities make developing vaccines for EID and neglected tropical diseases all the more difficult. In this review, we describe the implementation of an immunoinformatics-driven approach to systematically search for key determinants of immunity in newly available genome sequence data and design vaccines. This approach holds promise for the development of 21st century vaccines, improving human health everywhere.
Neglected tropical & emerging infectious diseases: new challenges
Climate change and international travel have had a dramatic impact on the geographic distribution of pathogens infecting humans and animals. Old world pathogens such as Dengue and Chikungunya virus, previously restricted to the Middle East, Africa and Asia have now appeared in the Americas Citation[1]. Newer pathogens such as Middle East Respiratory Syndrome coronavirus (MERS-CoV), an entirely new coronavirus affecting humans, have been spreading beyond the region of the world from which they derive their names Citation[2]. Meanwhile, human populations in developing areas of the world continue to be threatened by neglected tropical diseases (NTD). More than two billion people – nearly 30% of the world’s population – suffer from one or more NTD Citation[3], which include leishmaniasis, lymphatic filariasis, onchocerciasis, schistosomiasis and soil-transmitted helminthiasis, among others . In addition to climate change and airline travel, economic conditions leading to transmigration contribute to the spread of NTD. Recent examples include the reemergence of Leishmania in Spain Citation[4] and Chagas disease in Texas Citation[5].
Table 1. The neglected tropical diseases.
Even while NTD expand their reach, vaccine development for these diseases lags behind. In contrast, vaccines for important emerging infectious diseases (EID, ) are being developed, to a certain extent, by large biotechnology companies, particularly when these companies receive guaranteed purchase agreements or other incentives to accelerate vaccine development. Examples include the development of a vaccine for H7N9 (an emerging avian influenza) by Novartis and Novavax in 2013 Citation[6,7] and work toward the development of a new MERS-CoV vaccine in 2014 Citation[8]. However, the standard approach to develop new vaccines for emerging (and reemerging) infectious disease threats, which is to implement previously existing vaccine design methodologies such as cloning and expressing the dominant surface antigen Citation[9], frequently results in the development of vaccines that are only effective when given with strong adjuvants Citation[10]. This approach is particularly unlikely to work for pathogens that have complex lifecycles (such as parasites) or are highly mutable (such as RNA viruses). This article will discuss new, computational approaches that may accelerate and improve the design of vaccines for NTD and EID.
Table 2. Emerging infectious diseases (as of 2014).
Truly effective vaccines do not exist for the majority of NTD. Although vaccines are in development for several NTD pathogens Citation[11], lack of financial incentive to invest in research and development programs for diseases concentrated in low-income countries has mired the progress of vaccine efforts among large pharmaceutical companies Citation[3]. Preventative chemotherapy mass drug administration (MDA) programs employing donated or extremely low-cost generic drugs are currently in progress to control lymphatic filariasis, onchocerciasis, leprosy, trachoma and helminthiases in many areas of the world Citation[12]. A complication inherent in this strategy is that because the regions affected by different NTD overlap, coinfections can be difficult to manage with antiparasitic agents Citation[13]. The success of these long-term efforts and off-target effects of mass-eradication campaigns at the individual and population levels remain to be determined Citation[14]. As has been observed for polio, geopolitical upheaval may hamper global efforts to eradicate NTD. Thus, effective NTD vaccines are still needed Citation[15]. New cost–effective design methodologies can help to bridge the gap between the great need for these vaccines and return on investment for pharmaceutical companies.
Fortunately, genomes for many newly emerging pathogens and neglected tropical disease-associated pathogens are becoming available due to research efforts worldwide Citation[16]. The availability of these genomes now makes it possible to apply computational vaccinology tools to these diseases of global health importance.
Basic principles of vaccinology
Host immune response to pathogens is mediated by the innate and adaptive arms of the immune system. Innate immune cells such as macrophages, neutrophils and natural killer cells are responsible for the first line of defense, while adaptive immunity provides a more targeted response to pathogens that establishes immune memory for more rapid responses upon repeated exposures. B cells produce antibodies which are capable of recognizing and neutralizing pathogenic antigens. T cells support antibody production, activation and memory development, and are capable of lysing infected cells. The potent response from T and B cells, however, comes at a cost. Whereas innate immune cells can respond within 24–72 h, the primary adaptive response normally takes 7–14 days to mature. Secondary adaptive responses driven by immune memory are much faster and much stronger. This is the principle behind vaccination: pre-exposure to pathogen-derived antigens can induce pathogen-specific immune memory. The discovery of critical antigens that drive protective memory is facilitated by new computational tools.
Indeed, the general principle that immune cells develop memory to specific pathogen components – has driven the development of genome-derived vaccines over the past two decades. Since T cells play a critical role in adaptive immunity and the development of immune memory required for an efficacious vaccine, computational tools have been used to search for small linear peptides (T-cell epitopes) derived from protein antigens that drive class I and class II T-cell responses. These peptides are displayed on the surface of APC by multiple alleles of the MHC. As human beings express multiple alleles of class I and class II MHC molecules, called human leukocyte antigens (HLA), computational vaccinologists now search for T-cell epitopes that can bind to the most common HLA alleles in the human population, reasoning that broad HLA coverage will contribute to the development of effective genome-derived vaccines. Computational tools can also be used to select epitope-rich surface proteins that are better immunogens to drive B-cell response. Fortunately, while B cells and antibodies generally recognize surface proteins, T cells recognize epitopes derived from a broader range of proteins, giving the computational vaccinologist many possible sources (internal and external proteins as well as secreted proteins) for the selection of T-cell epitopes for vaccines.
Exposure to a given pathogen generates memory T-cell clones capable of rapid and efficient response upon subsequent reinfection Citation[17]. This response may include T cell help for induction of higher antibody titers, T-cell-mediated lysis of infected cells and the expression of cytokines to coordinate other cell-mediated immune processes such as activation of APC. Breadth of T-cell response (responding to many different epitopes) appears to be correlated with protection from severe disease for many pathogens that affect humans. More specifically, for HIV, HBV, HCV, lymphocytic choriomeningitis virus and malaria, protection from disease has been correlated with broad T-cell epitope response to both ‘immunodominant’ and subdominant T-cell epitopes Citation[18–22]. These discoveries have contributed to the concept that vaccines can be made directly from genomes by selecting sets of epitopes that will stimulate immune responses and protect against diseases.
Based on the observation that broad T-cell response may be protective, computational vaccinologists have worked to define collections of T-cell epitopes that can recreate the requisite features of this response. T-cell-driven, epitope-based strategies for developing vaccines against EID and NTD are currently the focus of several NTD research laboratories. Proof of principle exists for a number of disease models: cellular immunity elicited by epitope immunization provided complete protection against respiratory syncytial virus challenge, partial protection of BALB/c mice against sporozoite challenge, elimination of malaria-infected hepatocytes in vitro, partial protection of BALB/c and CBA against encephalitis following intracerebral challenge with a lethal dose of measles virus, complete protection from intraperitoneal HSV challenge, protection against infection with malaria or influenza A virus and full protection of sheep against bovine leukemia virus (these examples are reviewed in Citation[23]). We have demonstrated complete protection against lethal vaccinia challenge Citation[24] and successful clearance of a chronic bacterial infection (Helicobacter pylori) following T-cell epitope-driven vaccination Citation[25]. In earlier studies, we achieved partial protection against an aerosolized bacterial pathogen (Tularemia Citation[26]) using a vaccine that contained only 14 epitopes. While mice are not humans, growing evidence that T-cell epitope-driven vaccines can be effective in humans has led to the establishment of a number of biotech startups and venture-backed companies focused entirely on T-cell epitope-based vaccines.
T-cell epitopes as ‘payload’
As described in the following sections, computational tools are being used to identify proteins or antigens of interest directly from the genomes of pathogens. In theory, a minimal set of antigens or epitopes that induce a competent immune response to a pathogen can be discovered using the new tools. Adjuvant triggers innate immunity, which is an essential component of the protective immune response, directing it toward inflammation rather than tolerance. When combined with the minimum antigenic components that comprise the ‘payload’ of a genome-derived vaccine, delivered in the right vehicle, may trigger protective immune response. The fundamental principle of the genome-derived epitope-driven vaccine approach is illustrated Tthus:
The importance of epitopes as key determinants of protective immune responses is reflected by the flurry of immunoinformatics activity over the past decades. A number of T-cell epitope mapping tools have been developed to accelerate the identification of these critical components of the immune response. Using methods such as frequency analysis, support vector machines, hidden Markov models and neural networks, researchers have developed highly accurate tools for modeling the MHC-peptide interface and predicting T-cell epitopes. Available tools have been comprehensively reviewed: see Brusic et al. Citation[27], De Groot and Martin Citation[28] and Sette et al. Citation[29]. The concept was at first called ‘vaccinomics’ by Brusic and Petrovsky in 2002 Citation[30], then ‘reverse vaccinology’ by Rappuoli in 2003 Citation[31] and, more recently, ‘immunome-derived’ or ‘genome-derived vaccine design’ by Pederson Citation[32]; De Groot and Martin Citation[33]; and Doytchinova, Taylor and Flower Citation[34].
B-cell epitopes: T-cell epitope content as a proxy for B-cell response?
Computational vaccinologists have been unable to successfully develop accurate tools for B-cell epitope prediction, even though one of the most commonly measured outcomes of vaccination and accepted determinant of protection is antibody generation Citation[35,36]. Thus, current computational vaccinology approaches to vaccine development must take B-cell response into consideration and develop approaches that include means of stimulating effective humoral immunity where it is required for protection against challenge. Given T-cell dependence for essential features of an effective antibody response, including B-cell affinity maturation, class switch recombination, plasma cell differentiation and memory B-cell differentiation Citation[37], T-cell epitope analysis and quantification have been used by our group as a proxy for identifying good B-cell immunogens, linking in silico sequence analysis to desired putative B-cell responses Citation[28].
The iVAX approach to design genome-derived epitope-driven vaccines
De Groot and colleagues have integrated epitope-mapping tools with a wider array of vaccine design algorithms into the web-based iVAX toolkit, which will be described in some detail in the following sections. The tools were initially used by EpiVax and collaborators Citation[23,38–42], and then expanded and refined for projects that have been in progress at the Institute of Immunology and Informatics (iCubed) Citation[43–45]. The iVAX toolkit is currently in use for NTD research at the iCubed and with academic collaborators under an agreement established between EpiVax and URI in 2009. iVAX tools are being used to evaluate the protective potential of existing NTD and EID vaccines Citation[46,47], to predict immune response to newly emerging pathogens Citation[9,10] and to design novel NTD vaccines composed of T-cell epitopes (for Chagas disease, Brugia malayi and several different species of Leishmania Citation[48]). In the following few sections, we describe the iVAX approach to design genome-derived epitope-driven vaccines for NTD and EID.
Selecting targets
One of the first questions facing computational vaccinologists is how to prioritize their search for antigenic proteins and epitope subunits. The entire set of proteins derived from a pathogen’s genome is an unlikely point of departure for epitope mapping, since many of these proteins may not be part of the ‘core genome’ for a set of bacterial or viral strains of the same pathogen. Others may be proteins that serve as ‘housekeeping’ genes that are also well conserved in harmless commensal organisms. On the other hand, proteins that are highly conserved across variant strains, that are pathogen-specific and those that are upregulated during interactions with the host, particularly those that are secreted by a pathogen (presumably in an attempt to alter the host environment), are excellent targets for vaccine development.
In addition to targeting upregulated, secreted and pathogen-specific antigens, other means of selecting antigens for epitope screening include identifying proteins that are more common in virulent as compared to avirulent strains, selection of genes differentially expressed in immunopathogenesis, prioritizing proteins exposed on the surface of the pathogen and focusing on proteins that are expressed early in the course of natural infection. The Expert Protein Analysis System (ExPASy) proteomics server of the Swiss Institute of Bioinformatics offers a wide variety of proteomics tools that can be used for this purpose, including tools related to protein identification and characterization. Our groups have adapted an approach first described by Gennaro et al. for Mycobacterium tuberculosis (Mtb) Citation[49], employing a series of ExPASy tools (SignalP, TMPred and Prosite Scan Citation[50]) to triage pathogen genomes, reducing the number of potential targets from thousands of proteins to several dozen candidate antigens. In our first test of this approach, we found that a subset of epitopes derived from the Mtb genome elicited IFN-γ response from Mtb-exposed human samples, and prototype epitope-based TB vaccines were shown to be robustly immunogenic in murine studies Citation[51].
Immunogenicity scale: ranking antigens
Once a set of antigens is selected using the methods described above, tools available through the iVAX toolkit can be used to rank proteins for relative immunogenicity, based on their putative T-cell epitope content. For this purpose, iVAX calculates an ‘immunogenicity score,’ which represents the deviation between the number of putative epitopes found in a given protein and the number we would expect to find in a randomly generated protein sequence, taking care to adjust for protein size. In this context, an immunogenicity score of zero reflects random or ‘average’ putative epitope content; positive scores reflect more epitope content than expected, while negative scores reflect less epitope content than expected Citation[52]. Large numbers of protein sequences derived directly from the genome of selected pathogens can be ordered by potential class I (CTL), class II (T helper) or both class I and class II epitope content and placed on an immunogenicity scale . This tool allows researchers to quickly rank a given set of proteins both in relative (i.e., relative to each other) and absolute (i.e., relative to a panel of known immunogens and nonimmunogenic proteins) terms Citation[53]. In our experience, epitope-rich proteins are good vaccine targets and elicit strong antibody responses – thus, as previously stated, T-cell epitope content is a useful proxy for overall immunogenic potential.
Figure 1. Lymphatic filariasis antigens on the EpiMatrix protein immunogenicity scale. All other factors being equal, the more HLA ligands (i.e., putative T-cell epitopes) contained in a given protein, the more likely that protein is to induce an immune response. To capture this concept, the EpiMatrix immunogenicity scale presents proteins by the EpiMatrix protein score, and compares them to other known immunogens. The EpiMatrix protein score is the difference between the number of predicted T-cell epitopes expected in a protein of a given size and the number of putative epitopes predicted by the EpiMatrix. The EpiMatrix protein scores are ‘normalized’ and can be plotted on a standardized scale. ‘Average’ proteins score near zero. Protein scores above zero indicate the presence of excess MHC ligands and denote a higher potential for immunogenicity, while scores below zero indicate the presence of fewer potential MHC ligands than expected and a lower potential for immunogenicity. The EpiMatrix protein score is correlated with observed immunogenicity in vitro and in vivo. As shown here, proteins scoring above +20, such as Brugia malayi antigen Juv-p120, are considered to have a significant immunogenic potential. Proteins scoring below –20, such as TPX-2 above, are less likely to be immunogenic in vivo.
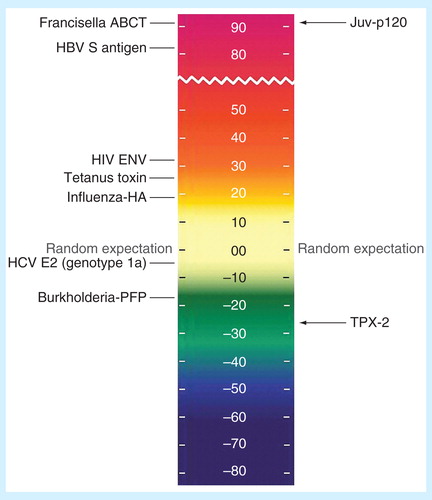
Antigen selection is particularly complicated when targeting parasitic organisms due to their comparatively massive genomes and multistaged life cycles, and ranking of these antigens may assist with the selection of better targets. For example, in , we show two candidate antigens derived from B. malayi, a causative agent of lymphatic filariasis, whose life cycle is divided into multiple larval stages including a microfilarial stage Citation[54]. In this case, TPX-2, a protein that has been identified as a potential vaccine target Citation[55], is shown to contain minimal T-cell epitope content, with an immunogenicity score of –27.61, and thus it may be less successful as a vaccine candidate. In contrast, Juv-p120, a B. malayi ortholog of a Litomosoides sigmodontis antigen implicated in conferring protection against microfilarial infection Citation[56] carries substantially more T-cell epitope content, scoring +94.14 on the immunogenicity scale, in the same range as other well-known immunogens. Furthermore, as is illustrated here in the case of B. malayi, we frequently find evidence that pathogens appear to reduce T-cell epitope content in key proteins to avoid human immune responses. Epitope deletion is an established means of immune evasion in HIV and HCV Citation[57,58]; thus, the mechanism may also be relevant in the context of infections that are associated with chronic infection caused by filaria, leishmania and other chronic NTD, particularly in stages associated with chronic parasitism and parasite persistence in the face of immune pressure. We will discuss additional means of immune evasion that can be uncovered by computational tools below.
EpiMatrix: T-cell epitope mapping of selected antigens
T-cell epitopes are short linear peptides that can bind to MHC molecules and engage T cells through their receptors (TCR), activating specific populations of CD8+ and/or CD4+ lymphocytes. These epitopes are key to forming the immunological synapse between antigen-presenting cells and T cells. Because TCRs are produced in a myriad of possible conformations (much like antibodies, to which they are related), MHC binding is the dominant event in immune recognition. In other words, most MHC ligands are also T-cell epitopes, and T-cell epitopes are by definition, MHC ligands. The MHC-peptide interaction is well characterized Citation[59,60]. Based on these characterizations, pattern-matching algorithms such as EpiMatrix have been developed to screen protein sequences for peptides that will bind MHC.
The human MHC molecules, or HLA, are among the most variable proteins in the human genome. This variation ensures that the surveillance capabilities of the human immune system are both broad and deeply redundant, making immune escape through mutation more difficult for pathogenic organisms. Fortunately, some alleles are much more common than others in the human population and the binding repertoire of many alleles significantly overlap.
By focusing on alleles that are both common (in the human population) and significantly different from each other (representative of human diversity), HLA alleles can be grouped into ‘supertypes,’ which can reduce the search space to a manageable number of evaluations. Six of these class I super-type alleles that ‘cover’ the genetic backgrounds of most humans worldwide have been used to define CTL epitopes: A*0101, A*0201, A*0301, A*2402, B*0702 and B*4403 Citation[61]. For class II T helper epitopes, mapping for a panel of eight common alleles: DRB1*0101, *0301, *0401, *0701, *0801, *1101, *1301 and *1501, gives broad T helper epitope coverage Citation[62]. The concept of supertype alleles is generally accepted and widely applied to vaccine design in the field of computational vaccinology Citation[61,62].
Using the set of selected protein antigens as a starting point, iVAX uses EpiMatrix to parse each into overlapping 9-mer frames where each 9-mer overlaps the last by eight amino acids. Each 9-mer is then scored for predicted binding affinity to a panel of class I or class II HLA alleles. The EpiMatrix algorithm compares the amino acid sequence of each given 9-mer peptide to the coefficients contained in stored probability matrices and produces a raw score. In order to compare potential epitopes across multiple HLA alleles, EpiMatrix raw scores are converted to a normalized ‘Z’ scale. Peptides scoring above 1.64 on the EpiMatrix ‘Z’ scale (typically the top 5% of any given sample) are likely to be MHC ligands Citation[63]. Evidence from animal studies suggests that the number of epitopes required for full protection is a small and definable subset (∼50) Citation[64,65]; thus, epitope-driven vaccines developed by our group generally contain a payload of 50–100 epitopes that provide broad coverage of human genetic backgrounds. With a combination of promiscuous class II epitopes and class I supertype epitopes, it is possible to attain >99% coverage of the HLA of most human populations Citation[61,62].
Eliminating regulatory or suppressor epitopes using JanusMatrix
A recent development in vaccine design includes the consideration of epitopes that induce regulatory or suppressive immune responses Citation[66]. Our group has been investigating epitope cross-conservation with the human genome and its association with diminished or regulatory immune responses. Using a recently developed tool called JanusMatrix we first determined that published effector T-cell epitopes can be distinguished from reported regulatory T-cell epitopes on the basis of TCR-specific cross-reactive potential with the human genome and human microbiome Citation[67]. JanusMatrix differs from whole-sequence alignment tools such as BLAST Citation[68] in its basis upon T-cell receptor homology. Pathogenic peptides whose TCR-facing residues are identical to the epitopes contained in multiple self may be recognized by T cells specific to those human proteins. Of course, even though the MHC-facing residues may differ, these peptides must still have the capacity to bind to the same MHC as the pathogen sequence, provided that binding is preserved. Taking this into account, JanusMatrix compares the TCR-facing contour of pathogen ligands to other genomes of interest, identifying matches therein that are predicted to bind the same MHC. TCR-homologous epitopes shared between pathogens and humans, or pathogens and other microbes, can be uncovered with remarkable speed using the JanusMatrix tool.
Exploring further, we have uncovered a high degree of host (human) homology in viruses that tend to establish chronic infections in humans such as EBV and CMV Citation[69]. Furthermore, ‘commensal’ viruses can be shown to contain significantly more human genome-homologous epitopes relative to those causing acute infection (e.g., Ebola, Marburg) Citation[69]. The limited clinical efficacy of some vaccines against selected microbial pathogens may, in fact, have been due to their extensive cross-conservation with the human genome Citation[10]. The JanusMatrix tool is currently being used by our team and collaborators to identify significant homology between candidate payload epitopes and proteins contained within the human genome and the human microbiome. Using the tool, we find that not only viruses but also bacteria that establish chronic infections in humans ‘deimmunize’ (remove T-cell epitopes) and ‘tolerize’ (modify epitopes to be more cross-reactive to human T-cell epitopes). Comprehensive studies of NTD genomes (and stage-by-stage analysis of parasite antigens) will be performed using the JanusMatrix tool in the near future.
It follows that careful selection of T-cell epitopes, and redesign of whole antigens, to avoid the inclusion of T-cell epitopes that may be highly cross-reactive with the human genome could improve the efficacy of whole-antigen and epitope-based vaccines. JanusMatrix complements recent research Citation[70] on the development of adaptive immunity and supports the hypothesis that adaptive T-cell responses are reinforced by cross-reactivity with the human microbiome Citation[71–73].
Cytoscape is an online tool that is usually used by bioinformaticians to illustrate the relatedness between proteins, for example, all of the intracellular proteins that might be involved in the stimulation of a cell through toll-like receptors. We have repurposed Cytoscape to describe the relationship between epitopes across proteins in groups of sequences (the human genome, the human microbiome, pathogen genomes Citation[67]). Using Cytoscape Citation[74], the results of JanusMatrix analysis (e.g., comparing a pathogen epitope to the human genome) can be visualized as networks where each epitope derived from a pathogen is linked to its TCR-matched counterparts in the search database, which themselves are linked to their source proteins. For example, an influenza T-cell epitope previously identified by Mark Davis and colleagues Citation[70] that stimulates T cells in subjects never exposed to influenza can be shown to have an extensive network of cross-reactive TCR-facing epitopes in the human microbiome. In contrast, an epitope from vaccinia virus synthesized and tested by Larry Stern’s group is shown to have extensive cross-reactivity with the human genome by JanusMatrix. This epitope was nonimmunogenic in vitro (by IFN-γ ELISpot) even though it was shown to bind to the correct class II MHC Citation[75]. This epitope fits the emerging in silico definition of a Treg epitope.
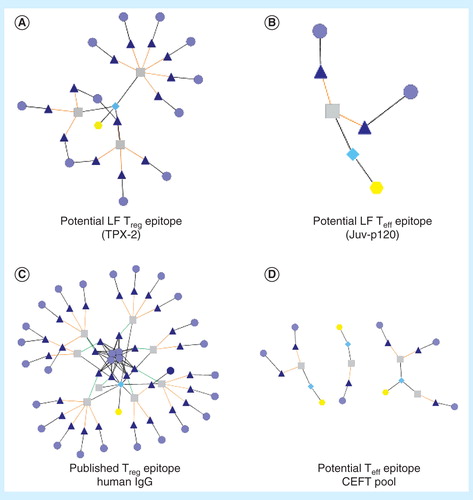
Due to their commensal nature and need to avoid human immune responses over many years of coexistence, it is even more likely for selected human parasites to share putative T-cell epitope content with their human hosts. In , we offer two example peptides from B. malayi antigens TPX-2 and Juv-p120, compared to published Treg epitopes from human immunoglobulin (Tregitopes) and effector epitopes, the CEFT pool (a set of peptides used as ‘control positive’ peptides in ELISpots Citation[67]). The potential cross-reactivity network differential is evident between the TPX-2 epitope, with many related epitopes derived from human sequences, and the Juv-p120 sequence, whose related human epitopes are few. This finding underscores the importance of validating the response phenotype of T cells stimulated by epitopes identified in silico prior to their inclusion in vaccine constructs, and also illustrates the importance of this type of analysis for the selection of candidate epitopes for NTD.
Figure 2. JanusMatrix analysis. This tool considers identity of TCR-facing residues to target proteins or genomes independently from residues that contribute to MHC binding. Peptides that have similar TCR-facing residues and are presented in the context of the same HLA can be identified. Extensive homology is easy to identify using the Cytoscape network visualization tool. The extent of the network can be used to distinguish potential regulatory T-cell epitopes (A) from potential effector T-cell epitopes (B). A published Treg epitope example is shown in (C), and several published Teff epitope examples are shown in (D). For these illustrations, yellow hexagons identify the source antigens, turquoise diamonds identify the source T-cell epitope clusters, gray squares indicate the source 9-mers, dark blue triangles indicate matched human 9-mers and light blue circles indicate human antigens in which matched 9-mers are found.
ClustiMer: finding promiscuous T-cell epitopes
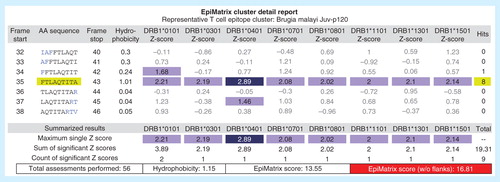
Promiscuous HLA binding potential is a feature of class II-restricted T-cell epitopes particularly exploitable for vaccine design purposes. It has been shown that putative epitopes for HLA class II are not often distributed evenly across protein sequences, but instead tend to cluster in specific regions, where it is not uncommon to observe several reactive 9-mer frames in close proximity Citation[76]. These ‘clusters’ of unusually high predicted epitope density can be identified in silico using the ClustiMer algorithm. In general, T-cell epitope clusters identified by the ClustiMer algorithm tend to be promiscuous MHC binders and are frequently T-cell epitopes Citation[52]. Due to overlapping peptide-binding preferences among HLA-DR alleles, it is also possible to identify single 9-mers capable of binding four or more HLA alleles Citation[76]. These sequences have been dubbed ‘EpiBars’ due to their horizontal, band-like signature in readout from EpiMatrix . T-cell epitope clusters can be very powerful, and EpiBars may be a characteristic feature of highly immunogenic, promiscuous class II epitopes. These compact, highly reactive peptides are relatively easy to deliver and show great promise as vaccine components when cross-reactivity with the human genome is limited (see above). We have used these clusters extensively in our own work Citation[24,43,51].
Figure 3. Example of an EpiBar: EpiMatrix analysis of candidate lymphatic filariasis epitope. In addition to providing an overall immunogenicity score, EpiMatrix can be used to analyze epitopes at the local level. A Brugia malayi Juv-p120 peptide is shown above, parsed into 9-mer frames and analyzed for predicted immunogenicity. EpiMatrix assessments above 1.64 constitute the top 5% of predicted HLA binders and are shaded medium blue, while scores above 2.32 fall in the top 1% and are shaded dark blue. This Juv-p120 peptide registers significant scores for all eight alleles in EpiMatrix in a single 9-mer frame, and based on the EpiMatrix method, has a cluster score of 16.81 (reflecting the number of predicted binders per amino acid length). Cluster scores higher than 10 are considered to be significant based on retrospective and prospective studies carried out by the EpiVax group. The band-like pattern illustrated in frame 35 is called an EpiBar and is characteristic of promiscuous epitopes.
Class I epitope selection
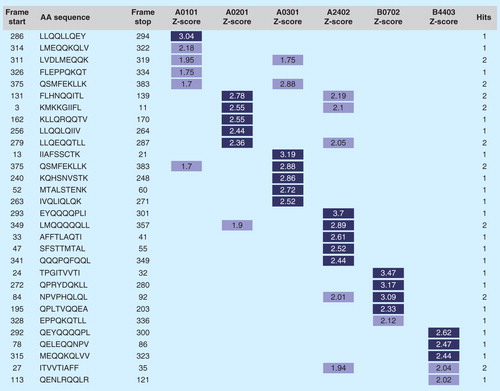
Promiscuous T-cell epitopes also exist, to a certain degree, for class I alleles; however, this is much less common than for class II. Some laboratories have demonstrated cross-presentation of peptides within HLA ‘superfamilies,’ such as the A3 superfamily: A3, A11, A31, A33 and A68 Citation[77]. Cross-MHC binding and presentation to T cells has been confirmed in HIV vaccine studies Citation[78]. However, we have found that weighting toward the selection of highly promiscuous class I epitopes may lead to identification of candidate epitopes that have lower binding affinities overall. Higher binding affinities appear to be a critical aspect of CTL epitope efficacy Citation[79], thus our group prefers to select a small set of the best-scoring putative epitopes for each of the six class I HLA superfamilies from a given protein or set of conserved peptides .
Figure 4. Class I epitope ‘staircase’ ranking. In the process of generating a selection of predicted high-affinity class I epitopes for inclusion in T-cell-driven vaccines, parsed 9-mers from any antigen are ranked by potential to bind supertype HLA alleles and collated in a ‘staircase’ report. In this example, the top five highest-scoring peptides from a given antigen are shown. In general, prioritizing class I epitopes by score for each of the individual alleles is preferred to define epitopes that bind across alleles.
EpiAssembler: generating immunogenic consensus sequences
Selecting epitopes that are broadly reactive across circulating strains can enhance broad applicability of new vaccines. The problem of pathogen variability significantly complicates the selection of epitopes for vaccine design. To address this problem, EpiVax has developed EpiAssembler Citation[80] to identify sets of overlapping, conserved and immunogenic epitopes and to assemble them into extended immunogenic consensus sequences (ICS, ).
Figure 5. EpiAssembler construction of immunogenic consensus sequences. This figure illustrates the process of assembling highly conserved T-cell epitopes into a single molecule. First, a highly conserved, promiscuous epitope is identified to form the 9-mer core of the ICS peptide (red bar). Overlapping conserved epitopes (pink, orange, green and blue bars) are then added to the N- and C-termini of the peptide until a suitable length is reached for binding in the class II HLA binding groove. This economical approach allows for targeting of multiple strains of a given pathogen using a single peptide, as illustrated by the blended bar at the bottom of the figure.
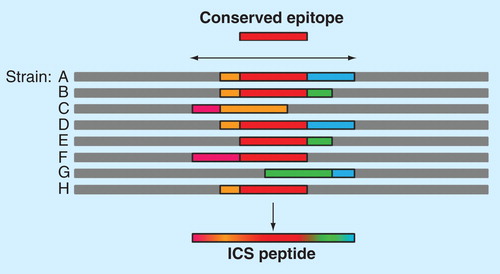
The theory behind developing ICS is that processing and presentation of these sequences would allow for presentation of the highly conserved class II-restricted epitopes contained in the ICS in the context of more than one MHC. The resulting peptide is not a ‘pseudo-sequence’ as such, since each constituent epitope occurs in its corresponding position in the native protein; adjacent epitopes may be similarly conserved but not in the same variant of the pathogen. The ICS approach has been useful for identifying highly immunogenic epitopes for HIV vaccine design Citation[38]. Using HIV as an example, while the full composite ICS peptides happen to be exactly conserved in a few individual strains of HIV, each peptide represents a significant percentage of circulating strains because every constituent overlapping epitope is conserved in a large number (range 893–2254) of individual HIV-1 strains Citation[38].
By extending the approach described above, it is possible to develop completely synthetic antigens whose sequences are optimized for T helper potential. With an eye to structural considerations, even recombinant protein-only vaccines could be optimized in this way, enabling primary cognate T help to be maximized and B-cell memory to be elicited. An ideal vaccine might include whole proteins in addition to some epitopes; some or all of these antigens could be optimized using the ICS approach. Linking ICS epitopes to a carrier protein (such as a surface protein target of B-cell response) would further maximize primary cognate T help, since B cells that capture the recombinant proteins would be able to process and present T helper epitopes derived from more variable proteins.
As compared with ICS, randomly selected counterparts, on average, contain half as many binding motifs and cover one-third fewer isolates Citation[40]. To develop vaccines of equivalent antigenic ‘payload’ using conventional methods would be prohibitively expensive, as it would require use of multiple variants of each antigen. We believe that this and similar approaches that harness conserved T help have tremendous potential and deserves careful consideration in vaccine design.
From genomes to vaccines: epitope annotation, validation & down-selection
After generating a preliminary list of candidate vaccine components, the next step in the iVAX approach is to review the putative epitopes produced by the EpiMatrix system, adding qualitative and quantitative annotations wherever possible, leading to an investigator-driven down-selection process. Putative epitopes derived from known antigens or from proteins overexpressed during early stages of infection or proteins known to be exposed to immune surveillance as reported in the literature may be prioritized. Furthermore, putative epitopes with the in silico profile of potential regulatory T-cell epitopes (based on JanusMatrix analysis) are removed from further consideration.
Individualized T-cell epitope measure
Algorithms can also be helpful to interpret vaccine component responses in preclinical and clinical studies. In studies of immune response to therapeutic proteins and vaccines, the authors have observed that subject-to-subject variation in T-cell response closely relates to subject HLA type and the number of motifs or peptides that match the subject’s HLA haplotype. To describe this relationship, EpiVax researchers have developed a metric that may be useful in clinical assessment of immune response to vaccines, called the ‘individualized T-cell epitope measure’ or iTEM. For a given T-cell epitope, an individual’s iTEM score can be calculated by weighting and summing the epitope’s EpiMatrix Z-scores for each HLA allele in a given subject’s haplotype. This calculated score allows for individualized immunogenic potential to be predicted based on the number of putative epitopes contained in a protein and a given individual’s HLA haplotype. Using this score, it is possible to analyze the contribution of haplotype to the corresponding T-cell response. In prospective and retrospective evaluations, significant correlations were found between the IFN-γ response to a given antigen and the iTEM scores for individual subjects Citation[42]. In addition, correlations between the iTEM score and patient HLA have been observed for antibody titers Citation[40,81,82], reflecting the importance of HLA-restricted T-cell responses to the genesis of a robust antidrug antibody response.
Iterative combinatorial analysis for vaccine design
A number of methods for enhancing epitope-based vaccines have been described and implemented Citation[83,84]. One approach is to align the individual epitopes in a protein or DNA vaccine construct as a ‘string of beads’ without any intervening sequences or spacers between the payload epitopes Citation[85]. However, the lack of spacers between the payload epitopes has raised concern that these sequences may contain junctional epitopes. VaccineCAD, an algorithm that iteratively analyzes epitope assemblies and minimizes the potential for junctional immunogenicity in any string-of-beads construct, has been developed to address this concern Citation[40]. Peptide sequences contained in the junctional regions between the target epitopes are evaluated for potential immunogenicity. The highest scoring junction is identified and the algorithm optimizes the order of epitopes by evaluating potential alternative sequences. The process is repeated until no additional reductions in junctional immunogenicity can be achieved or until all junctional immunogenic potential has been eliminated. When the potential for junctional immunogenicity cannot be sufficiently reduced, a cleavage promoting spacer sequence, typically ‘AAY’ for class I restricted constructs Citation[86] or a binding inhibiting ‘breaker’ sequence such as ‘GPGPG’ for class II restricted constructs Citation[87] is placed between the two offending epitopes. The ability to minimize junctional immunogenicity while simultaneously minimizing the presence of transmembrane domains or highly hydrophobic peptide segments which may be difficult to express would be a logical extension of this tool’s capabilities.
Successful vaccine design
The integration of computational tools for epitope discovery has enabled the development of genome-derived vaccines Citation[41,45,44]. Compared to conventional strategies, this approach has the potential to create more effective and safer next-generation vaccines, as carefully selected epitopes focus immune responses on the minimal, essential pathogen-specific antigenic elements; epitopes directed against conserved ‘self’ (host) antigens are eliminated. This approach is also well suited for highly variable pathogens, as selection of epitopes that are conserved across multiple strains or subtypes enables the development of a broadly applicable, multipathogen vaccine. The genome-derived vaccine strategy has been applied by our team to a wide range of pathogens, including F. tularensis, variola, HIV, Mtb, H. pylori and influenza. These studies demonstrate that immunoinformatic-predicted epitopes are immunoreactive in vaccinees and survivors of infection, and stimulate de novo, protective immune responses in vivo in HLA transgenic mice (e.g., Citation[24–26,51]).
Epitope-driven vaccines offer distinct advantages over traditional subunit vaccines. Multiple epitopes derived from several antigens can be packaged together. Thus, a broad-based immune response directed against several different antigenic proteins can be elicited without manufacturing and administering the entire protein, much of which will be immunologically irrelevant. This may reduce formulation challenges, cost and safety risk. The use of epitopes also mitigates safety concerns arising from the use of intact recombinant proteins that may have undesired biological activity.
This review of vaccine design tools developed by the EpiVax team is by no means comprehensive, and has mainly focused on antigen selection and design. Topics not covered in this review include formulation of epitope-driven vaccines, route of delivery (mucosal, intradermal, etc.), adjuvanting, selection of delivery vehicles and preclinical and clinical testing. A major caveat concerning the use of the iVAX toolkit is that none of the vaccines designed using these tools have advanced to the clinic. Given the cycle of vaccine development, this is not surprising (it may take up to 20 years to develop a vaccine with full industry support). Retrospective and prospective studies have provided extensive validation of the tools described here Citation[10,28,46,47]. Nonetheless, algorithms developed and applied by this group to a wide range of pathogens have met with significant preclinical success and are currently in use for the development of vaccines against NTD parasites, EID viruses and bioterror pathogens.
Access to nearly all of the tools described in this article is freely available to trained users through the iVAX toolkit Citation[88]. The website was developed with funding from the National Institutes of Health in 2010. Access to the iVAX toolkit and training on the tools is available for interested researchers under collaborative agreements with the University of Rhode Island (primarily for NTD, but other arrangements are possible). Commercial users are directed to EpiVax Citation[89], which provides a secure-access version of the iVAX website for commercial users.
Expert commentary
In general, the field of vaccine research has been slow to adopt new vaccine design tools, and even fewer NTD researchers are familiar with the use of the tools, despite proof of principle for the genome-derived vaccine approach and the fact that it significantly reduces time and effort to make vaccines. For EID, ‘tried and true’ approaches often win out over newer approaches, even though traditional approaches have no greater likelihood of success. The MERS-CoV vaccine development programs that have been implemented illustrate this principle; despite the fact that the virus belongs to a family of coronaviruses that have a history of rapid evolution, vaccination approaches are once again targeting the ‘spike’ protein. A similar approach was used during the emergence of SARS and completely failed to protect against rapidly evolving SARS viruses in animal challenge models Citation[9,90]. Application of advanced immunoinformatics tools to NTD vaccines has also lagged for a number of reasons. NTD researchers do not use the tools because they lack access to and familiarity with them, and there are no widely publicized examples focusing on diseases that impact the developing world.
A series of technical challenges for NTD vaccines have been described recently, including antigen discovery, process development, preclinical development, clinical trials in resource-poor settings and the immune response to NTD infection, including what is commonly referred to as the IgE trap, through which certain individuals, perhaps especially those in endemic regions, may have elevated preexisting IgE antibodies for potential NTD vaccine antigens, leading to increased risk with vaccination Citation[91]. Computational vaccinology cannot currently address all of these challenges; however, the approach described here offers a unique opportunity to address certain hurdles early in the developmental process. Early in the pipeline, antigen discovery using T-cell epitope prediction and ranking, along with candidate epitope triage using cluster analysis and cross-reactivity prediction provide valuable leads. Selected peptide candidates can be screened ex vivo in order to verify the phenotype of the immune response prior to inclusion in a final vaccine product.
Finally, T-cell epitope-based strategies are exceptionally platform-flexible, adaptable to synthetic peptide formulations deliverable in saline, emulsion or microparticle, or encoding into plasmid vectors for DNA vaccination or recombinant protein production, thus allowing for novel distribution strategies necessary to reach the world’s poorest. This flexibility extends to the antigen discovery approach as well, in that many kinds of targets may be explored using immunoinformatics tools. A pertinent example for NTD and EID applies to vector-based targets. Vaccine components based upon the salivary proteins of arthropod vectors are already under investigation Citation[92]. However, vector salivary antigens also have known immunomodulatory properties allowing for extended host tolerance Citation[93,94]. The same discovery and evaluation strategy described for pathogenic antigens could be applied to such proteins, potentially providing a mechanism through which to stimulate robust immune response in the absence of the immunomodulatory properties of the complete salivary antigens.
Five-year view
The amount of data generated through new technologies, such as next-generation sequencing, continues to expand exponentially. By applying these technologies to the study of EID and NTD causative agents, and expanding our genomic knowledge of these organisms, the feasibility of using high-throughput, informatics-based tools for the identification of putative protein and peptide targets increases. Vaccine efficacy may also improve, as the selection of targets can be refined by comparing the antigen to other genome sequences, such as the human genome and the human microbiome. The in silico-based approach to vaccine design may also alleviate many of the funding-associated challenges common to traditional vaccine design, by reducing the number of assays that need to be performed to select vaccine targets. Reduced cost should allow for the reallocation of critical funding to the testing of in silico-predicted targets and constructs. And finally, improved safety, by eliminating human genome cross-conserved epitopes, may reduce unwanted adverse effects. Looking further into the future, we are confident that the evolution of the tools described here will eventually contribute to the development of personalized, on-demand vaccines Citation[95].
Considering the importance of controlling infectious diseases to global economic stability, the integration of computational vaccinology tools and their application to the design of vaccines for NTD and EID is of paramount importance. Delay is no longer acceptable. Vaccine developers must implement computational vaccinology tools if they wish to contribute to improve world health in the 21st century.
New immunoinformatics tools have been developed that address critical problems in vaccine design.
These tools have been extensively validated in preclinical models.
The design of vaccines for neglected tropical diseases would benefit from expanded use of these tools.
Financial & competing interests disclosure
AS De Groot is CEO, CSO of EpiVax. FE Terry, L Moise and R Martin are employees of EpiVax. In addition, L Moise holds options in EpiVax and AS De Groot is a majority stockholder. This work was supported by National Institutes of Health Research Grant U19 AI082642. The American Recovery and Reinvestment Act funded the development of the iVAX website and the application of the suite of computational vaccinology tools to Neglected Tropical Diseases. The NIH/NIAID Filariasis Research Reagent Resource Center provided funding to M Torres, N Pilotte and SA Williams and provided all Brugia parasite materials. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
References
- Centers for Disease Control and Prevention (CDC). Chikungunya fever diagnosed among international travelers–United States, 2005-2006. MMWR Morb Mortal Wkly Rep 55(38):1040-2
- Milne-Price S, Miazgowicz KL, Munster VJ. The emergence of the Middle East Respiratory Syndrome coronavirus. Pathog Dis 2014. [Epub ahead of print]
- Zhang Y, MacArthur C, Mubila L, Baker S. Control of neglected tropical diseases needs a long-term commitment. BMC Med 2010;8:67
- Arce A, Estirado A, Ordobas M, et al. Re-emergence of leishmaniasis in Spain: community outbreak in Madrid, Spain, 2009 to 2012. Euro Surveill 2013;18(30):20546
- Montgomery SP, Starr MC, Cantey PT, et al. Neglected parasitic infections in the United States: chagas disease. Am J Trop Med Hyg 2014;90(5):814-18
- Settembre EC, Dormitzer PR, Rappuoli R. Bringing influenza vaccines into the 21st century. Hum Vaccin Immunother 2013;10:3
- Fries LF, Smith GE, Glenn GM. A recombinant viruslike particle influenza A (H7N9) vaccine. N Engl J Med 2013;369(26):2564-6
- Zhang N, Jiang S, Du L. Current advancements and potential strategies in the development of MERS-CoV vaccines. Expert Rev Vaccines 2014. [ Epub ahead of print]
- De Groot AS. How the SARS vaccine effort can learn from HIV-speeding towards the future, learning from the past. Vaccine 2003;21(27-30):4095-104
- De Groot AS, Moise L, Liu R, et al. Cross-conservation of T-cell epitopes: Now even more relevant to (H7N9) influenza vaccine design. Hum Vaccin Immunother 2014;10(2):256-62
- Hotez PJ, Dumonteil E, Heffernan MJ, Bottazzi ME. Innovation for the ‘bottom 100 million’: eliminating neglected tropical diseases in the Americas. Adv Exp Med Biol 2013;764:1-12
- Bockarie MJ, Kelly-Hope LA, Rebollo M, Molyneux DH. Preventive chemotherapy as a strategy for elimination of neglected tropical parasitic diseases: endgame challenges. Philos Trans R Soc Lond B Biol Sci 2013;368(1623):20120144
- Molyneux DH, Malecela MN. Neglected tropical diseases and the millennium development goals: why the “other diseases” matter: reality versus rhetoric. Parasit Vectors 2011;4:234
- Webster JP, Molyneux DH, Hotez PJ, Fenwick A. The contribution of mass drug administration to global health: past, present and future. Philos Trans R Soc Lond B Biol Sci 2014;369(1645):20130434
- Hotez PJ, Diemert D, Bacon KM, et al. The human hookworm vaccine. Vaccine 2013;31(Suppl 2):B227-32
- Eisen JA, MacCallum CJ. Genomics of emerging infectious disease: a PLoS collection. PLoS Biol 2009;7(10):e1000224
- Blattman JN, Sourdive DJ, Murali-Krishna K, et al. Evolution of the T-cell repertoire during primary, memory, and recall responses to viral infection. J Immunol 2000;165:6081-90
- Harrer T, Harrer E, Kalams SA, et al. Cytotoxic T-lymphocytes in asymptomatic long term nonprogressing HIV-1 infection. Breadth and specificity of the response and relation to in vivo viral quasispecies in a person with prolonged infection and low viral load. J Immunol 1996;156(7):2612-23
- Doolan DL, Hoffman SL, Southwood S, et al. Degenerate cytotoxic T-cell epitopes from P. falciparum restricted by multiple HLA-A and HLA-B supertype alleles. Immunity 1997;7(1):97-112
- Gianfrani C, Oseroff C, Sidney J, et al. Human memory CTL response specific for influenza A virus is broad and multispecific. Hum Immunol 2000;61(5):438-52
- Mok H, Lee S, Wright DW, Crowe JEJr. Enhancement of the CD8+ T-cell response to a subdominant epitope of respiratory syncytial virus by deletion of an immunodominant epitope. Vaccine 2008;26(37):4775-82
- Gillespie GM, Wills MR, Appay V, et al. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol 2000;74(17):8140-50
- De Groot AS, Berzofsky JA. From Genome to Vaccine - New Immunoinformatics tools for vaccine design. Methods 2004;34(4):425-8
- Moise L, Buller RM, Schriewer J, et al. VennVax, a DNA-prime, peptide-boost multi-T-cell epitope poxvirus vaccine, induces protective immunity against vaccinia infection by T cell response alone. Vaccine 2011;29(3):501-11
- Moss SF, Moise L, Lee DS, et al. HelicoVax: epitope-based therapeutic H. pylori vaccination in a mouse model of gastric cancer. Vaccine 2011;29(11):2085-91
- Gregory SH, Mott S, Phung J, et al. Epitope-based vaccination against pneumonic tularemia. Vaccine 2009;27(39):5299-306
- Petrovsky N, Brusic V. Bioinformatics for study of autoimmunity. Autoimmunity 2006;39:635-43
- De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol 2009;131(2):189-201
- Wang P, Sidney J, Dow C, et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol 2008;4(4):e1000048
- Elliott SL, Suhrbier A, Miles JJ, et al. Phase I trial of a CD8+ T-cell peptide epitope-based vaccine for infectious mononucleosis. J Virol 2008;82(3):1448-57
- Rappuoli R, Covacci A. Reverse vaccinology and genomics. Science 2003;302(5645):602
- Pederson T. The immunome. Mol Immunol 1999;36(15-16):1127-8
- De Groot AS, Martin W. From immunome to vaccine: epitope mapping and vaccine design tools. Novartis Found Symp 2003;254(57-72): discussion 72-6, 98-101:250-2
- Doytchinova IA, Taylor P, Flower DR. Proteomics in Vaccinology and Immunobiology: An Informatics Perspective of the Immunone. J Biomed Biotechnol 2003;2003(5):267-90
- Plotkin SA. Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis 2008;47(3):401-9
- Yao B, Zheng D, Liang S, Zhang C. Conformational B-cell epitope prediction on antigen protein structures: a review of current algorithms and comparison with common binding site prediction methods. PLoS One 2013;8(4):e62249
- Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011;29:621-63
- Koita OA, Dabitao D, Mahamadou I, et al. Confirmation of Immunogenic Consensus Sequence HIV-1 T-cell Epitopes in Bamako, Mali and Providence, Rhode Island. Hum Vaccin 2006;2(3):119-28
- McMurry JA, Gregory SH, Moise L, et al. Diversity of Francisella tularensis Schu4 antigens recognized by T lymphocytes after natural infections in humans: Identification of candidate epitopes for inclusion in a rationally designed tularemia vaccine. Vaccine 2007;25(16):3179-91
- De Groot AS, Marcon L, Bishop EA, et al. HIV vaccine development by computer assisted design: the GAIA vaccine. Vaccine 2005;23(17-18):2136-48
- De Groot AS, McMurry J, Marcon L, et al. Developing an epitope-driven tuberculosis (TB) vaccine. Vaccine 2005;23(17-18):2121-31
- Schanen BC, De Groot AS, Moise L, et al. Coupling sensitive in vitro and in silico techniques to assess cross-reactive CD4(+) T cells against the swine-origin H1N1 influenza virus. Vaccine 2011;29(17):3299-309
- Zhang S, Desrosiers J, Aponte-Pieras JR, et al. Human immune responses to H. pylori HLA class II epitopes identified by immunoinformatic methods. PLoS ONE 2014;9(4):e94974
- Mishra S, Losikoff PT, Self AA, et al. Peptide-pulsed dendritic cells induce the hepatitis C viral epitope-specific responses of naïve human T cells. Vaccine 2014;32(26):3285-92
- De Groot AS, Ardito M, Moise L, et al. Immunogenic consensus sequence t helper epitopes for a pan-burkholderia biodefense vaccine. Immunome Res 2011;7(2):7
- De Groot AS, Moise L, Martin W, , and et al. Analysis of ChimeriVax Japanese Encephalitis (JE) virus sequence for T cell epitopes and comparison to circulating wild type JE Virus strains. Vaccine 2007;25(47):8077-84
- De Groot AS, Ardito M, McClaine E, et al. Immunoinformatic comparison of T-cell epitopes contained in novel swine-origin influenza A (H1N1) virus with epitopes in 2008-09 conventional influenza vaccine. Vaccine 2009;27(42):5740-7
- Elfaki ME, Khalil EA, Degroot AS, et al. Immunogenicity and immune modulatory effects of in silico predicted L. donovani candidate peptide vaccines. Hum Vaccin Immunother 2012;8(12):1769-74
- Gomez M, Johnson S, Gennaro ML. Identification of secreted proteins of Mycobacterium tuberculosis by a bioinformatic approach. Infect Immun 2000;68(4):2323-7
- ExPASy bioinformatics resource portal. Available from: www.expasy.org
- McMurry JA, Kimball S, Lee JH, et al. Epitope-driven TB vaccine development: a streamlined approach using immuno-informatics, ELISpot assays, and HLA transgenic mice. Curr Mol Med 2007;7(4):351-68
- De Groot AS. Immunomics: discovering new targets for vaccines and therapeutics. Drug Discov Today 2006;11(5-6):203-9
- De Groot AS, Ardito M, Terry F, et al. Low immunogenicity predicted for emerging avian-origin H7N9: implication for influenza vaccine design. Hum Vaccin Immunother 2013;9(5):950-6
- Despommier DD, Gwadz RW, Hotez PJ, Knirsch CA. Parasitic diseases. 5th edition. Apple Trees Productions, LLC; New York, NY, USA: 2005
- Gnanasekar M, Rao KV, He YX, et al. Novel phage display-based subtractive screening to identify vaccine candidates of Brugia malayi. Infect Immun 2004;72(8):4707-15
- Hintz M, Schares G, Taubert A, et al. Juvenile female Litomosoides sigmodontis produce an excretory/secretory antigen (Juv-p120) highly modified with dimethylaminoethanol. Parasitology 1998;117(Pt 3):265-71
- Bowen DG, Walker CM. Mutational escape from CD8+ T cell immunity: HCV evolution, from chimpanzees to man. J Exp Med 2005;201(11):1709-14
- Bailey J, Blankson JN, Wind-Rotolo M, Siliciano RF. Mechanisms of HIV-1 escape from immune responses and antiretroviral drugs. Curr Opin Immunol 2004;16(4):470-6
- Falk K, Rotzschke O, Stevanovic S, et al. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 1991;351(6324):290-6
- Rotzschke O, Falk K, Stevanovic S, et al. Exact prediction of natural T cell epitope. Eur J Immunol 1991;21(11):2891-4
- Sette S, Sidney J. Nine major HLA Class I supertypes account for the vast preponderance of HLA-A and –B polymorphism. Immunogenetics 1999;50(3-4):210-12
- Southwood S, Sidney J, Kondo A, et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol 1998;160(7):3363-73
- Messitt TJ, Terry F, Moise L, et al. A comparison of two methods for T cell epitope mapping: “cell free” in vitro versus immunoinformatics. Immunome Res 2011;7(2):6
- Moutaftsi M, Peters B, Pasquetto V, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol 2006;24(7):817-19
- Moise L, De Groot AS. Putting immunoinformatics to the test. Nat Biotechnol 2006;24(7):791-2
- Cecil DL, Holt GE, Park KH, et al. Elimination of IL-10-inducing T-helper epitopes from an IGFBP-2 vaccine ensures potent antitumor activity. Cancer Res 2014;74(10):2710-18
- Moise L, Gutierrez AH, Bailey-Kellogg C, et al. The two-faced T cell epitope: examining the host-microbe interface with JanusMatrix. Hum Vaccin Immunother. 2013;9(7):1577-86
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215(3):403-10
- He L, De Groot AS, Gutierrez AH, et al. Integrated assessment of predicted MHC binding and cross-conservation with self reveals patterns of viral camouflage. BMC Bioinf 2014;15(Suppl 4):S1
- Su LF, Kidd BA, Han A, et al. Virus-specific CD4(+) memory-phenotype T Cells are abundant in unexposed adults. Immunity 2013;38(2):373-83
- Chung H, Pamp SJ, Hill JA, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012;149(7):1578-93
- Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010;330(6012):1768-73
- Lathrop SK, Bloom SM, Rao SM, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011;478(7368):250-4
- Saito R, Smoot ME, Ono K, et al. A travel guide to Cytoscape plugins. Nat Methods 2012;9(11):1069-76
- Calvo-Calle JM, Strug I, Nastke MD, et al. Human CD4+ T cell epitopes from vaccinia virus induced by vaccination or infection. PLoS Pathog 2007;3(10):1511-29
- Weber CA, Mehta PJ, Ardito M, et al. T cell epitope: Friend or Foe ? Immunogenicity of biologics in context. In Series “Optimizing the Future for Biotechnology Therapies, the Key Role of Protein Engineering”. Adv Drug Del Rev 2009;61(11):965-76
- Sette A, Sidney J. HLA supertypes and supermotifs: a functional perspective on HLA polymorphism. Curr Opin Immunol 1998;10(4):478-82
- De Groot AS, Bosma A, Chinai N, et al. From genome to vaccine: in silico predictions, ex vivo verification. Vaccine 2001;19(31):4385-95
- Jin X, Roberts CG, Nixon DF, et al. Identification of subdominant cytotoxic T lymphocyte epitopes encoded by autologous HIV type 1 sequences, using dendritic cell stimulation and computer-driven algorithm. AIDS Res Hum Retroviruses 2000;16(1):67-76
- De Groot AS, Bishop E, Khan B, et al. Engineering immunogenic consensus T helper epitopes for a cross-clade HIV vaccine. Methods 2004;34(4):476-87
- Koren E, De Groot AS, Jawa V, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol 2007;124(1):26-32
- Cohen T, Moise L, Ardito M, et al. A method for individualizing the prediction of immunogenicity of protein vaccines and biologic therapeutics: individualized T cell epitope measure (iTEM). J Biomed Biotechnol 2010;2010(pii):961752
- Thomson SA, Burrows SR, Misko IS, et al. Targeting a polyepitope protein incorporating multiple class II-restricted viral epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: a novel approach to vaccine design. J Virol 1998;72(3):2246-52
- Rodriguez F, Whitton JL. Enhancing DNA immunization. Virology 2000;268(2):233-8
- An LL, Rodriguez F, Harkins S, et al. Vaccine 2000;18(20):2132-41
- Velders MP, Weijzen S, Eiben GL, et al. Defined flanking spacers and enhanced proteolysis is essential for eradication of established tumors by an epitope string DNA vaccine. J Immunol 2001;166(9):5366-73
- Livingston BD, Newman M, Crimi C, et al. Optimization of epitope processing enhances immunogenicity of multiepitope DNA vaccines. Vaccine 2001;19(32):4652-60
- iVAX tool kit. Available from: www.immunome.org/ivax/ivax-tool-kit/
- EpiVax. Available from: www.EpiVax.com
- Yang ZY, Werner HC, Kong WP, et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci USA 2005;102(3):797-801
- Bethony JM, Cole RN, Guo X, et al. Petri W, Reed S, Valenzuela JG, Hotez PJ. Vaccines to combat the neglected tropical diseases. Immunol Rev 2011;239(1):237-70
- Abi Abdallah DS, Pavinski Bitar A, Oliveira F, et al. A listeria-based vaccine that secretes the sand fly salivary protein LJM11 confers long-term protection against vector-transmitted Leishmania major. Infect Immun 2014. [ Epub ahead of print]
- Oliveira F, de Carvalho AM, de Oliveira CI. Sand-Fly Saliva-Leishmania-Man: the trigger trio. Front Immunol 2013;4:375. eCollection
- Kazimírová M, Štibrániová I. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Microbiol 2013;3:43
- De Groot AS, Einck L, Moise L, et al. Making vaccines “on demand”: a potential solution for emerging pathogens and biodefense? Hum Vaccin Immunother 2013;9(9):1877-84
- Murray CJ, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2197-223