Abstract
Protein chemistry, such as crosslinking and photoaffinity labeling, in combination with modern mass spectrometric techniques, can provide information regarding protein–protein interactions beyond that normally obtained from protein identification and characterization studies. While protein crosslinking can make tertiary and quaternary protein structure information available, photoaffinity labeling can be used to obtain structural data about ligand–protein interaction sites, such as oligonucleotide–protein, drug–protein and protein–protein interaction. In this article, we describe mass spectrometry-based photoaffinity labeling methodologies currently used and discuss their current limitations. We also discuss their potential as a common approach to structural proteomics for providing 3D information regarding the binding region, which ultimately will be used for molecular modeling and structure-based drug design.
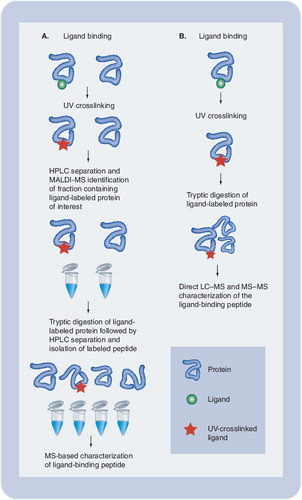
(A)Following interaction of the protein of interest with a photolabeled ligand, the complex is covalently linked by UV irradiation. Isolation of the covalent complex followed by MS allows for identification of binding protein, determination of the ligand:protein stoichiometry, and identification and sequencing of the ligand-binding region of the protein. We used this approach to localize the binding site for an inhibitor of the HIV-1 integrase protein. (B) Alternatively, if the receptor protein is known, the purified protein may be subjected to direct LC-MS following ligand binding, crosslinking and tryptic digestion. This approach may be advantageous when dealing with hydrophobic ligands that may be lost during HPLC separation.
HPLC: High-performance liquid chomatography; LC: Liquid chromatography; MALDI: Matrix-assisted laser desorption/ionization; MS: Mass spectrometry; UV: Ultraviolet.
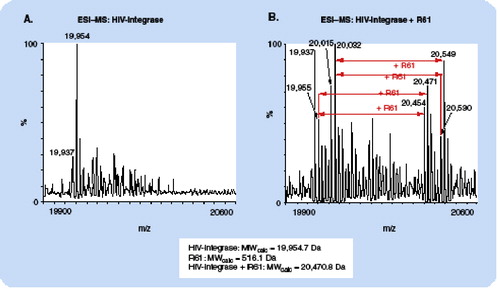
(A): the base peak at m/z 19,954 agrees precisely with the MW of HIV-integrase as calculated from the amino acid sequence. The additional peaks at higher m/z can be assigned to oxidation products and β-mercaptoethanol adducts. (B): due to the presence of the photoaffinity label, the peaks are shifted to higher m/z values by 516 Da, which agrees precisely with the expected mass increment for addition of one coumarin molecule.
ESI: Electrospray ionization; MS: Mass spectrometry; MW: Molecular weight.
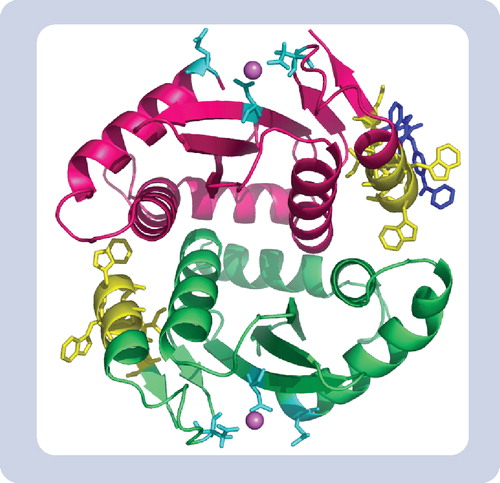
Yellow represents the inhibitor binding region peptide 128AACWWAGIK136. Blue represents one molecule of the coumarin inhibitor complexed within the binding site determined by PAL-MS. Cyan represents the integrase active site residues (DDE motif) and the sphere is Mg2+ chelating at this site.
MS: Mass spectrometry; PAL: Photoaffinity labeling.
In photoaffinity labeling (PAL), a ligand possessing a UV-photoactivating group is incubated with its receptor to form, first, a noncovalently bound ligand–receptor complex in which the ligand binds specifically to the binding site or pocket in the receptor due to its affinity. Some ligands, such as oligonucleotides, contain intrinsic photoactivating groups; alternatively, these groups can be introduced by synthesis into ligands for peptides and small molecules, such as drugs. It is important to establish that the incorporated photoactivating groups do not significantly alter the binding affinity of the ligand to its receptor and its functionality, compared with the nonderivatized ligand. Subsequent to the complex formation, UV irradiation triggers the photoactivating group in the ligand and leads to the conversion of the noncovalent binding into a covalent link between the ligand and its receptor at the binding site. A covalent ligand–receptor complex makes the study of the complex easier, since it permits the use of more rigorous but harsher analysis tools, such as SDS-PAGE, HPLC and mass spectrometry (MS), which would typically lead to the dissociation of the complex with information regarding the binding sites no longer being obtainable.
Photoaffinity labeling with radioactively labeled ligands is a widely used method to determine the competitive binding affinities of other molecules for the binding sites in the binding pocket occupied by the radioactively labeled ligand and, as such, is a valuable tool for drug screening. When photoaffinity techniques are combined with modern MS, this approach can provide additional structural and mechanistic information regarding the protein complex, such as the ligand–receptor stoichiometry, a map of the binding pocket, and even the exact identification of the amino acid residues, which are involved in the ligand–receptor interaction. The data obtained can be used to create a 3D model of the ligand-binding pocket that might provide better insight into the nature of the ligand–receptor interaction. This knowledge can be very valuable, not only for better determination of the mechanism of interaction, but also for structure-based drug design in order to generate more potent drugs.
General principles of photoaffinity labeling for structural analysis
Photoaffinity labeling is a valuable technique for studying the interaction of ligands with their target proteins, such as receptor proteins. Although the technique was initially developed and described more than four decades ago by Singh, Thornton, and Westheimer Citation[1], the basic principles of photoaffinity labeling remain essentially unchanged today. In this technique, ligands possessing photoactivating probes are irradiated with UV-light after the formation of the ligand–receptor complex. Photolysis results in the creation of highly reactive intermediates, mainly carbenes and nitrenes, which will form covalent bonds with receptor proteins and thereby covalently attach the ligand at their receptor-binding site. Three types of photoactivating probes are mainly used in photoaffinity labeling: azido groups, benzophenone groups and diazirenes. Furthermore, photoaffinity labeling containing additional groups, for example biotin, stable isotopes and fluorescence groups, facilitates the detection and identification of the photolabeled biomolecules. The ligand–receptor complex can be isolated via HPLC and analyzed by MS. The accurate measurement of the molecular weight of the complex provides information on the number of ligands covalently bound to each molecule of receptor, thus providing information on the stoichiometry of the complex and specificity of the photolabel. In order to identify the site of ligand–receptor interaction, the complex is typically digested and the peptide(s) containing the covalently linked ligand is analyzed by mass spectrometric or combined chromatographic and MS approaches. details general steps involved in photoaffinity labeling experiments combined with MS for structural analysis of protein–ligand complexes to determine receptor–ligand stoichiometry and identify ligand binding sites.
Photolabels
The choice of photoactivating group used to covalently link ligand and receptor is the critical design component of each photoaffinity labeling experiment. In most PAL experimental situations, the photolabeling reagent is typically a derivatized receptor substrate. An appropriate photoactivating group is usually synthetically inserted into the ligand of interest, although some ligands may contain moieties capable of intrinsic photo-activation (e.g., sulfides, enones, dienones and halogenated compounds) Citation[2]. A number of variables will affect the outcome of a PAL experiment and, therefore, selection of an appropriate photoactive group as a probe for any given ligand–receptor complex is not a trivial concern. The choice of probe should be well thought out and extensively evaluated with the ligand of interest to ensure that it satisfies certain critical criteria. Ideally, the photoreactive group chosen as a probe for a PAL experiment should demonstrate as many of the following characteristics as possible Citation[3,4]. First, the photophore molecule should be as small as possible so that when it is incorporated into the ligand it does not create any steric interference that would act to deleteriously impact ligand–receptor binding affinity. Once a probe is chosen, control experiments should be performed prior to PAL experiments to ensure that the binding affinity of the derivatized ligand for its receptor is not significantly altered in relation to that of the nonderivatized ligand. In addition to steric considerations, the photophore should demonstrate reasonable chemical stability under both ambient light conditions and in darkness.
Another critical consideration concerns the lifespan of the photo-activated state once activated by UV irradiation. The life span should be of short enough duration so that covalent linkage between the photophore and receptor occurs before disassociation of the ligand–receptor complex. Conversely, the excited state should last long enough for the photophore to remain in close contact with the binding site so that sufficient covalent bonding will be able to occur. The photophore should also be expected to generate only a single unambiguous covalent adduct in order to avoid uninterpretable results. Finally, an ideal photophore should be capable of attacking both C–H and nucleophilic X–H bonds at an activation wavelength that would not lead to photolytic damage to proteins. This last criterion is quite important, because a stable covalent complex permits the use of rigorous (but often harsh) analytical tools, such as electrophoresis and MS, in examining the structure of protein receptor–ligand complexes. Without a stable covalent linkage, the use of electrophoresis, chromatography and/or mass spectrometric techniques in downstream analyses would likely promote dissociation of the ligand–receptor complex and lead to ambiguous results.
The overwhelming majority of photophore reagents currently used in PAL studies of protein structure and function are based on the reactivity of nitrenes or carbenes. Nitrenes and carbenes demonstrate very similar chemistries and as they are strong electrophiles they are capable of reacting rapidly with double bonds or heteroatoms (e.g., N, O or S) with pairs of nonbonding electrons. However, the predominance of nitrene- or carbene-based reagents in photoaffinity labeling studies of protein interactions is largely due to the ability of these chemical groups to attack even ‘inert’ aliphatic C–H bonds Citation[3]. The broad reactivity of photoaffinity reagents based on these chemistries essentially means that almost all functional groups that are found in biomolecules such as proteins are available for forming covalent linkages.
The most commonly employed photoactivating probes currently utilized in photoaffinity labeling studies are aryl azides, aryl diazirines and benzophenone compounds Citation[5,4]. The aryl azide and diazirine photoactivating compounds are generally precursors of nitrene and carbene reactive groups, while the benzophenone based compounds rely on the photochemical reactivity of aryl ketones for the formation of covalent linkage between a ligand and receptor Citation[6]. While all three probe types are well represented in the photoaffinity labeling literature, benzophenone-based photoprobes have some distinct advantages that have led to them being particularly appealing for use in studies of protein structure and interaction. Benzophenone groups generally possess greater chemical stability than their azide and diazirine counterparts Citation[6], and they may be manipulated in ambient light without significant degradation and then activated at a wavelength sufficiently high enough to avoid protein denaturation that may result from the use of photophores that require activation at low UV wavelengths Citation[6]. In addition, the benzophenones retain their stability in many common protic solvents Citation[4] and another benefit of particular relevance to the study of biological molecules is that benzophenone photophores remain reactive with C–H bonds, even in the presence of water Citation[3,6]. Weber and Beck-Sickinger conducted a useful study comparing the crosslinking efficiency, influence of water on crosslinking, irraditation requirements and by-product formation for azide, diazirine and benzophenone photoprobes Citation[5]. The authors discuss some advantages and disadvantages that would be of concern to researchers in the planning stages of a PAL-based study.
To facilitate further study of the ligand–receptor complex, photoaffinity probes often contain a ‘tag’ in addition to the photoactivating moiety. This tag usually takes the form of a radioactive, fluorescent and stable isotope, or immunoreactive constituent, and is especially useful when attempting to identify the receptor for a ligand of interest in a complex matrix, such as a cell lysate. The tag enables the covalently linked ligand–receptor complex to be detected during subsequent isolation and characterization steps.
When combined with the power of MS, photoaffinity labeling techniques enable information on receptor–ligand interactions to be obtained on several levels. For instance, these techniques can be used to screen for binding proteins specific for a particular ligand or drug candidate. Once a binding protein is identified and isolated, the binding region itself can be localized by fragmentation of the protein into peptide fragments followed by MS-based sequencing. Finally, PAL facilitates identification of the amino acids involved in interaction with the ligand in the binding pocket. The data obtained at this level are useful in developing models of the 3D structure of the ligand–receptor complex, and this valuable information is a key component of effective structure based drug design Citation[4].
In this article, we will highlight some of the exciting ways in which photoaffinity labeling is being combined with proteomics techniques to answer questions at the forefront of structural proteomics and drug development. We will discuss the predominant techniques in light of both their promise and current limitations, as well as presenting a perspective on the future developments in photoaffinity labeling/MS-based proteomics in the coming years.
Protein–DNA interaction
Although the use of MS in conjunction with photolabeling is most often used to study protein–protein and protein–small molecule interactions, several reports illustrate the emerging utility of strategies involving photolabeling combined with MS for studying the molecular interactions between proteins and nucleic acids. Interactions between proteins and DNA play fundamental roles in cellular biology and disease processes. A variety of protein classes interact with DNA, including polymerases, histones, ribosomes and various transcription factors. Developing drugs to treat disease states involving these proteins necessitates a thorough understanding of how they interact with nucleic acids in the cell. NMR spectroscopy and X-ray diffraction are two of the most common traditional analytical methodologies for studying protein–DNA interactions Citation[7,8], but recently MS has emerged as a promising new technique in this field Citation[9]. Inducing covalent linkage between proteins and DNA through photo-activation of intrinsic photosensitive functional groups following complex formation has the advantage of stabilizing the often weak noncovalent interactions involved without greatly perturbing the binding interface. Covalent linkage paves the way for downstream purification and analysis of the interaction site sequence using electrophoresis, chromatography and MS. Another advantage of this approach is that it can provide information regarding the identity and location of binding sites with more detail, greater sensitivity, and less analysis time than the more traditional biophysical techniques.
Benzophenone Citation[10], azido-, thio-, iodo- and bromo-based Citation[11] photoactivating nucleotide analogs have been used to create covalent linkages between proteins and DNA, in addition to UV-induced crosslinking through intrinsic photoactive groups in native proteins and DNA. The advantage of using substituted photoactivating analogs is that activation can be achieved at wavelengths above 300 nm, thereby avoiding many problems associated with UV-induced protein degradation Citation[9]. Golden and colleagues demonstrated the utility of using a substituted DNA in conjunction with MS to characterize protein–DNA interfaces Citation[12]. By substituting photoactive 5-bromo-2´-deoxyuridine for thymidine and activating with a XeCl excimer laser at 308 nm they crosslinked a high-affinity single-stranded DNA aptamer to basic fibroblast growth factor (bFGF)155, an important protein regulator of cell proliferation and differentiation. The bFGF155 complex was then tryptically digested, purified and isolated by electrophoresis, digested again with phosphodiesterase/alkaline phosphatase, and then analyzed by HPLC/electrospray ionization (ESI)-MS. Comparison of mass spectra for the digested, uncrosslinked oligonucleotide and the digested complex revealed a single fragment unique to the complex. MS/MS sequencing of this fragment confirmed this as the site of covalent linkage, and provided the primary sequence of the binding pocket. However, a common drawback to this type of approach is the typical need for large amounts of starting protein due to the potential of low protein–DNA complex yields obtained with some photophores, and low complex recovery as a result of losses during the subsequent electrophoresis or liquid chromatography steps Citation[11] necessary to remove peptides and free DNA that may lead to MS ionization suppression Citation[13]. These drawbacks have limited the full exploitation of highly sensitive MS techniques as a complement to photolabeling for the study of protein–DNA interactions.
Geyer and colleagues recently described a novel approach to improve the sensitivity of MS analyses of protein–DNA complexes by eliminating the need for loss-inducing electrophoresis or reversed-phase HPLC separations prior to MSCitation[14]. They incorporated photoreactive 5-iododeoxyuridine (5-IdU) into a single site within the sequence of DNA known to interact with the bacterial restriction endonuclease MboI. Following UV-induced covalent linkage and proteolytic digestion, oligodeoxynucleotide-linked fragments of MboI were enriched by Fe3+-immobilized metal affinity chromatography (IMAC) Citation[13]. The size of the oligodeoxynucleotide was then reduced to a single dU residue attached to the peptide by hydrogen fluoride hydrolysis. MALDI-TOF-MS and MALDI-TOF MS/MS were then used to identify and characterize the DNA-binding region of MboI. This strategy not only reduces losses during covalent-complex purification, it greatly reduces the amount of unwanted analytes in the sample and thereby obviates the need for other purification steps, allowing protein–DNA interaction sites to be rapidly and efficiently characterized with less than 500 pmol of protein starting material. Such innovations, when coupled to continuing advances in MS capabilities, should only serve to solidify photolabeling coupled with MS as the method of choice for defining and characterizing sites of protein–DNA interaction.
Protein interactions with small proteins & peptides
Physical interactions between protein molecules are central to all biological processes. Understanding how these often complex networks of interacting proteins function in governing biochemical phenomena in both normal and diseased cells on a global level is one of the major aims of the emerging field of systems biology Citation[15] and in drug discovery Citation[16]. Consequently, research aimed at defining protein–protein interactions is of considerable importance, and photoaffinity labeling has been a valuable tool for elucidating the identity of interacting protein partners for some time Citation[17]. With the advent of the field of proteomics, however, the information that can be obtained regarding protein–protein interactions by combining photoaffinity labeling techniques with MS has grown tremendously Citation[16,18]. By integrating MS with PAL, researchers have not only identified binding partners for a particular ligand or receptor of interest, but also localized and characterized the sites of interaction at a molecular level Citation[19–25] with far greater efficiency and rapidity than could be achieved with more traditional biophysical techniques. At the protein level, comparing the mass of photolabeled protein–protein ligand–receptor complexes with that of unlabeled material can be used to generate information on the stoichiometry of interactions Citation[18]. By proteolytically cleaving and then purifying a protein–protein oligopeptide photoadduct by electrophoresis or chromatography, investigators can then use MS/MS-based peptide sequencing techniques to determine the amino acid sequence of the binding regions of both ligand and receptor protein. This capacity to define the primary structure of interaction sites makes combining PAL with MS a very powerful strategy for structure-based drug design Citation[4,16].
Recent advances aimed at employing new and novel MS techniques are expanding the potential of photoaffinity labeling as a protein–protein interaction discovery tool. Sachon and colleagues Citation[26] incorporated the use of biotin/streptavidin purification of photolabeled complexes Citation[20] with isotope-coded affinity tag (ICAT) methods Citation[27] to dramatically enhance the sensitivity of MS-based photoaffinity labeling techniques. By incorporating a deuterium-labeled p-benzoyl–phenylalanine photoreactive group into substance P and using this probe to label the human NK-1 receptor, the investigators used MALDI-MS to discriminate the photolabeled complex based on double peaks separated by the mass difference between the deuterated and nondeuterated species following digestion of the complex and biotin/streptavidin purification. Using this approach, very minute amounts of protein–protein complex in the subpicomolar range can be confidently characterized.
Jahn and colleagues recently reported the use of optimized liquid chromatography (LC) separation of photolabeled protein–protein complexes in conjunction with multiple ion chromatogram (MIC) on-line ESI-MS to characterize low abundance complexes Citation[18]. Special reversed-phase LC columns compatible with formic acid enabled them to eliminate the ion suppression effects commonly observed with the reversed-phase ion paring agent trifluoroacetic acid (TFA), resulting in a one order of magnitude increase in MS sensitivity. In addition, they demonstrated that the use of MIC in conjunction with optimized LC/MS can dramatically further increase sensitivity in experiments aimed at localizing protein–protein interaction sites in low abundance complexes Citation[28]. They extracted multiple ion chromatograms from LC total ion chromatograms in an extension of the selected ion extraction technique. This allowed for detection of corticotropin-releasing factor binding protein (CRFBP) and CRFBP–urocortin analog complexes from LC/ESI-MS chromatograms with an additional order of magnitude increase in sensitivity. As MS instrumentation and methods continue to evolve in conjunction with advances in techniques such as nanoscale LC, photoaffinity labeling techniques can be expected to become even more widely used tools for characterizing protein–protein interactions and for generating structural information for use in drug design.
Protein–small molecule interaction
Obtaining a thorough understanding of the molecular interactions between small bioactive ligands and their corresponding receptor proteins is a critical step in the drug discovery process. Protein interactions with a variety of chemical entities play a central role in the etiology of many disease processes, and proteins are often directly involved in metabolizing therapeutic compounds and mediating their transport into or out of cells. Consequently, understanding these phenomena on a molecular level is currently a high-priority area of research. Photoaffinity labeling-MS integrated techniques are exceptionally well suited to addressing many pertinent research questions regarding protein–small molecule interactions, including identifying binding partners of specific ligands or protein receptors of interest, elucidating or characterizing the binding or interaction site, determining the stoichiometry of interactions, and in assisting the development of structural models for use in design of new chemotherapeutic agents.
Identifying or characterizing the ligand-binding site within a protein receptor is perhaps the most common application of photoaffinity labeling-MS techniques at present. PAL in conjunction with MALDI-MS and/or LC/MS analysis of protein-small molecule covalent complexes enables investigators to rapidly and accurately identify sites of interaction based on detecting mass additions due to ligand attachment. Subsequent proteolytic digestion of the isolated protein–ligand complex and MS-based sequencing can, in turn, reveal the sequence of the amino acids in the ligand-binding region and provide valuable information regarding the potential binding chemistry. These techniques have been used to define a wide variety of binding sites on protein receptors including those for allosteric regulation of enzyme substrate specificity Citation[29], bile acid transport protein Citation[30], alkyl glucoside inhibitors of glucose cotransport protein Citation[31], allosteric inhibitors of protein–protein interactions Citation[32], inhibitors of enzyme activity Citation[33], visual cycle enzyme substrates Citation[34], substrate-binding sites of cytochrome P450 Citation[35], and substrate-binding domains of the drug-efflux mediator P-glycoprotein Citation[36–38]. In addition to identifying and characterizing binding sites in known protein receptors, several groups have demonstrated the promise of combining proteomics with PAL to identify protein targets for candidate drugs Citation[39,40].
Davidson and colleagues recently demonstrated the ability of PAL combined with micro-LC and Fourier-transform ion cyclotron resonance MS (FT-ICR/MS) to pinpoint the location of photolabel incorporation with atomic resolution Citation[41]. The unparalleled resolving power and mass accuracy achievable with FT-ICR/MS Citation[42], when combined with micro-LC techniques, can lead to highly accurate detection of peptides present as low as the attomolar range Citation[43]. Davidson and colleagues combined these techniques with benzophenone-based photolabeling to localize the binding site for inhibitors on the human papillomavirus (HPV) E2 protein that inhibits its interaction with HPV E1 helicase. Following proteolytic digestion of the complex, micro-reversed phase LC and on-line FT-ICR/MS characterization of the resulting peptides pinpointed the site of photolabel incorporation to the terminal carbon atom of a single Met residue within the ligand-binding pocket.
Another important application of MS in combination with photoaffinity labeling techniques is in the determination of the stoichiometry of protein–small molecule interactions. The accurate mass measurement of the molecular weight of the photoaffinity protein–ligand complex compared with the unlabeled protein provides information on the number of molecules of ligand covalently bound to each molecule of protein. ESI is a gentle ionization technique that is particularly amenable to accurate mass measurement of even large proteins, and Lafitte and colleagues demonstrated the potential of ESI-MS in determining the stoichiometry of various calmodulin–pyrazine derivative complexes Citation[44]. More recently, Wen and colleagues used ESI-MS to investigate both the site of substrate binding in cytochrome P450 and the stoichiometry of the interaction Citation[35]. In our own laboratory, we have employed ESI-MS in combination with photoaffinity labeling to characterize the binding site and stoichiometry for interaction between HIV integrase and an inhibitor of this protein, coumarin Citation[45]. shows the shift in molecular weight of HIV-integrase due to the incorporation of coumarin by photoaffinity labeling, and demonstrates how the accurate mass measurement obtainable with ESI-MS can easily discriminate the addition of just a single small molecule ligand to a protein receptor. A tryptic digest of the photolabeled integrase–coumarin complex produced peptides that were separated by HPLC. Peptide fractions demonstrating a difference in UV absorbance at 280 nm as compared with control digests were analyzed by LC/ESI-MS. Using this approach, we identified a single binding site for the inhibitor in the tryptic peptide 128AACWWAGIK136. Site-directed mutagenesis of the full-length integrase followed by in vitro inhibition assays implicate two specific residues, C130 and W132, as being involved in inhibitor binding. The approach we used here is particularly useful for characterizing the stoichiometry of protein–ligand complexes that are difficult to analyze by X-ray crystallography and NMR techniques.
With regard to drug development and design, once information regarding the location and stoichiometry of protein–ligand interactions is obtained, the next step is to develop 3D models of the complex Citation[32,38,46]. Structural models of protein-binding sites can then be used to conduct molecular modeling experiments to identify novel compounds that may have high affinity for the binding site, or screen the affinity of existing compounds computationally.
Based on PAL-MS experiments in our laboratory with HIV-integrase and the coumarin inhibitor, we identified and characterized a single novel site for coumarin attachment in the integrase and used the PAL data to conduct molecular modeling experiments in order to better define the inhibitor–enzyme complex and propose a mechanism of action. Molecular docking studies using the genetic optimization for ligand docking (GOLD) algorithm indicated the integrase inhibitor binds close to the dimeric interface of the integrase core domain (see ). Site-directed mutagenesis of selected amino acid residues in the PAL-MS-identified binding region conferred resistance to coumarin and confirmed this as the drug-binding site (). In addition, modeling enabled us to identify more than 300 molecules that are predicted to have high binding affinities for the same drug-binding site [unpublished results]. These results demonstrate the wealth of information that can be obtained from photoaffinity labeling-MS experiments when that data is combined with site-directed mutagenesis studies and molecular modeling techniques. The structural information derived from these types of studies can shed light on potential mechanisms of drug action and thereby assist in the design of additional therapeutic agents.
Expert commentary
3D structural data about binding sites and pockets in protein–ligand and protein–biomolecule complexes are of major interest for the biomedical research community since valuable information regarding mechanistic knowledge of protein–protein and protein–ligand interactions can be derived, which are crucial for structure-based drug design. Currently, the methods of choice for structural analysis of these interactions are X-ray crystallography and NMR as both techniques can achieve atomic resolution. Although tremendous improvements have been made in recent years in both techniques, they are still somewhat limited, especially for the analysis of large complexes under native conditions, as would be found with high-molecular-weight receptor proteins embedded in membrane vesicles. In general, the combined approach of photoaffinity labeling and MS is adequate to provide information regarding binding sites and pockets. We believe that this approach is particularly useful in cases where structural analysis of the complexes via X-ray and NMR is not possible. Especially constructive is the use of PAL-MS techniques in combination with molecular modeling to obtain 3D information regarding the interaction, as we have demonstrated for the drug binding sites of HIV-integrase.
Five-year view
The combined approach of photoaffinity labeling and MS has great potential as a complementary technique to X-ray crystallography and NMR. In general, the instrumentation and skills necessary for conducting PAL combined with MS experiments designed to identify ligand- or peptide-binding sites are accessible to most laboratories. The primary impediment to more widespread utilization of PAL-MS techniques is the need for expertise in several fields, (e.g., MS, biology, biochemistry, synthetic chemistry and molecular modeling). However, we feel that use of PAL combined with MS to address questions in structural biology will increase in the coming years as improvements in MS make it more practical for a larger number of laboratories.
One of the major technical challenges associated with PAL combined with MS is the detection and identification of the photolabeled products. In general, with increasing sample complexity detection becomes more difficult, due to mass spectrometric ionization suppression effects and instrumental limitations related to resolution, mass accuracy and dynamic range. However, recent instrumental and methodological improvements have demonnstrated that the limitations can be circumvented. For example, high-field FT-ICR and orbitrap mass spectrometers, instruments that are distinguished by ultra-high resolution and mass accuracy, and the highest dynamic range, respectively, offer promise in overcoming some of these limitations. In addition, novel mass spectrometric-based methods (such as top-down proteomics, where intact protein or protein complexes are directly analyzed using newly developed dissociation mechanisms such as electron capture dissociation [ECD] and electron transfer dissociation [ETD]) are showing great promise as useful tools for the combined approach of PAL and MS. ECD and ETD are mechanisms that lead mainly to fragmentation of the peptide backbone while keeping post-translation modifications intact, such that even very labile modifications such as carboxylated glutaric acid residues remain undamaged during MS-based peptide sequencing. Thus, these methods seem to be ideal to precisely identify the binding site of photolabels by sequencing photolabeled proteins and peptides.
In 5 years, we expect that with exploitation of these novel mass spectrometric techniques and methods, coupled with more sophisticated ‘MS friendly’ (isotopically labeled) photo-probes and better knowledge of the photochemical reaction, the combined approach of PAL and MS will be a significant alternative to NMR and X-ray crystallography. In particular, we believe that PAL combined with MS will have a great impact in the area of identifying drug-binding sites in membrane-located protein receptors (the main target proteins for drugs) and, therefore, this approach will be of great interest and use for the pharmaceutical industry.
Key issues
| • | Understanding protein–nucleic acid, protein–protein and protein–small molecule interactions on a molecular level is essential to drug discovery and development. | ||||
| • | Photoaffinity labeling is a very powerful biochemical tool for studying molecular interactions, especially when NMR and X-ray chrystallography is impractical. | ||||
| • | When combined with mass spectrometry (MS) techniques, photoaffinity labeling is a useful tool for identifying protein ligands, discovering what protein(s) in the cell a particular ligand or drug targets, determining the stoichiometry of protein–ligand interactions and elucidating the primary structure of protein ligand-binding regions. | ||||
| • | The data derived from photoaffinity-MS studies can be used to develop models for characterizing drug-target interactions as part of structure-based drug design efforts. | ||||
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, and Developmental Award #9P30 AI050410 from the University of North Carolina Center for AIDS Research (NC, USA).
References
- Sing A, Thornton ER, Westheimer FH. The photolysis of diazoacetylchymotrypsin. J. Biol. Chem.237(9), PC3006–PC3008 (1962).
- Fleming SA. Chemical reagents in photoaffinity labeling. Tetrahedron51(46), 12479–12520 (1995).
- Brunner J. New photolabeling and crosslinking methods. Ann. Rev. Biochem.62, 483–514 (1993).
- Dormán G, Prestwich GD. Using photolabile ligands in drug discovery and development. Trends Biotechnol.18, 64–77 (2000).
- Weber PJ, Beck-Sickinger AG. Comparison of the photochemical behavior of four different photo-activatable probes. J. Peptide Res.49(5), 375–383 (1997).
- Dormán G, Prestwich GD. Benzophenone photophores in biochemistry. Biochem.33(19), 5661–5673 (1994).
- Luger K, Mader AW, Richmond RK et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature389, 251–260 (1997).
- van Aalten DM, DiRusso CC, Knudsen J. The structural basis of acyl coenzyme A-dependent regulation of the transcription factor FadR. EMBO J.20, 2041–2050 (2001).
- Steen H, Jensen ON. Analysis of protein-nucleic acid interactions by photochemical cross-linking and mass spectrometry. Mass Spect. Rev.21, 163–182 (2002).
- Sastry S, Ross BM. RNA-binding site in T7 RNA polymerase. Proc. Natl Acad. Sci. USA95, 9111–9116 (1998).
- Meisenheimer K, Koch T. Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol.32, 101–140 (1997).
- Golden MC, Resing KA, Collins BD et al. Mass spectral characterization of a protein-nucleic acid photocrosslink. Protein Sci.8, 2806–2812 (1999).
- Steen H, Petersen J, Mann M et al. Mass spectrometric analysis of a UV-cross-linked protein–DNA complex: tryptophans 54 and 88 of E. coli SSB cross-link to DNA. Protein Sci.10, 1989–2001 (2001).
- Geyer H, Geyer R, Pingoud V. A novel strategy for the identification of protein–DNA contacts by photocrosslinking and mass spectrometry. Nucleic Acids Res.32(16), e132 (2004).
- Cusick ME, Klitgord N, Vidal M et al. Interactome: gateway into systems biology. Human Mol. Gen.14(2), R171–R181 (2005).
- Hatanaka Y, Sadakane Y. Photoaffinity labeling in drug discovery and developments: chemical gateway for entering proteomic frontier. Curr. Topics Med. Chem.2, 271–288 (2002).
- Price D, Park I, Avraham H. Methods for the study of protein–protein interactions in cancer cell biology. Methods Mol. Biol.218, 255–267 (2003).
- Jahn O, Eckart K, Tezval H et al. Characterization of peptide-protein interactions using photoaffinity labeling and LC/MS. Anal. BioAnal. Chem.378, 1031–1036 (2004).
- Mills JS, Miettinen HM, Barnidge D et al. Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J. Biol. Chem.273(17), 10428–10435 (1998).
- Girault S, Sagan S, Bolbach G et al. The use of photolabelled peptides to localize the substance-P-binding site in the human neurokinin-1 tachykinin receptor. Eur. J. Biochem.240, 215–222 (1996).
- Ploug M. Identification of specific sites involved in ligand binding by photoaffinity labeling of the receptor for the urokinase-type plasminogen activator. Residues located at equivalent positions in uPAR domains I and III participate in the assembly of a composite ligand-binding site. Biochemistry37, 16494–16505 (1998).
- Perrin MH, Fischer WH, Kunitake KS et al. Expression, purification, and characterization of a soluble form of the first extracellular domain of the human type 1 corticotropin realeasing factor receptor.J. Biol. Chem.276(34), 31528–31534 (2001).
- Jahn O, Eckart K, Brauns O et al. The binding protein of corticotrophin-releasing factor: ligand-binding site and subunit structure. Proc. Natl Acad. Sci. USA99(19), 12055–12060 (2002).
- Jahn O, Tezval H, Spiess J et al. Tandem mass spectrometric characterization of branched peptides derived from photoaffinity labeling. Int. J. Mass Spec.228, 527–540 (2003).
- Sachon E, Bolbach G, Lavielle S et al. Met174 side chain is the site of photoinsertion of a substance P competitive peptide antagonist photoreactive in position 8. FEBS Lett.544, 45–49 (2003).
- Sachon E, Tasseau O, Lavielle S et al. Isotope and affinity tags in photoreactive substance P analogues to identify the covalent linkage within the NK-1 receptor by MALDI-TOF analysis. Anal. Chem.75, 6536–6543 (2003).
- Goshe MB, Smith RD. Stable isotope-coded proteomic mass spectrometry. Curr. Opin. Biotech.14, 101–109 (2003).
- Jahn O, Hofmann B, Brauns O et al. The use of multiple ion chromatograms in on-line HPLC-MS for the characterization of post-translational and chemical modifications of proteins. Int. J. Mass Spec.214, 37–51 (2002).
- Olcott MC, Andersson J, Sjöberg B-M. Localization and characterization of two nucleotide-binding sites on the anaerobic ribonucleotide reductase from bacteriophage T4. J. Biol. Chem.273(38), 24853–24860 (1998).
- Kramer W, Sauber K, Baringhaus K-H et al. Identification of the bile acid-binding site of the ileal lipid-binding protein by photoaffinity labeling, matrix-assisted laser desorption ionization-mass spectrometry, and NMR structure. J. Biol. Chem.276(10), 7291–7301 (2001).
- Raja MM, Kipp H, Kinne RKH. C-terminus loop 13 of Na+ glucose cotransporter SGLT1 contains a binding site for alkyl glucosides. Biochem.43, 10944–10951 (2004).
- Last-Barney K, Davidson W, Cardozo M et al. Binding site elucidation of hydantoin-based antagonists of LFA-1 using multidisciplinary technologies: evidence for the allosteric inhibition of a protein–protein interaction. J. Am. Chem. Soc.123, 5643–5650 (2001).
- LeRiche T, Skorey K, Roy P et al. Using mass spectrometry to study the photo-affinity labeling of protein tyrosine phosphatase 1B. Int. J. Mass Spec.238, 99–106 (2004).
- Wu Z, Hasan A, Liu T, Teller D, Crabb J. Identification of CRALBP ligand interactions by photoaffinity labeling, hydrogen/deuterium exchange, and structural modeling. J. Biol. Chem.279 (26), 27357–27364 (2004).
- Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Fluorescent photoaffinity labeling of cytochrome P450 3A4 by lapechenole: identification of modification sites by mass spectrometry. Biochem.44, 1833–1845 (2005).
- Borchers C, Boer R, Klemm K et al. Characterization of the dexniguldipine binding site in the multidrug resistance-related transport protein P-glycoprotein by photoaffinity labeling and mass spectrometry. Mol. Pharm.61(6), 1366–1376 (2002).
- Ecker GF, Csaszar E, Kopp S et al. Identification of ligand-binding regions of P-glycoprotein by activated-pharmacophore photoaffinity labeling and matrix-assisted laser desorption/ionization mass spectrometry. Mol. Pharm.61(3), 637–648 (2002).
- Pleban K, Kopp S, Csaszar E et al. P-glycoprotein substrate binding domains are located at the transmembrane domain interfaces: a combined photoaffinity labeling-protein homology modeling approach. Mol. Pharm.67(2), 365–374 (2005).
- Webb Y, Zhou X, Ngo L et al. Photoaffinity labeling and mass spectrometry identify ribosomal protein S3 as a potential target for hybrid polar cytodifferentiation agents. J. Biol. Chem.274(20), 14280–14287 (1999).
- Shen D-W, Liang X-J, Gawinowicz M, Gottesman MM. Identification of cytoskeletal [14C]Carboplatin-binding proteins reveals reduced expression and disorganization of actin and filamin in cisplatin-resistant cell lines. Mol. Pharm.66(4), 789–793 (2004).
- Davidson W, McGibbon G, White P et al. Characterization of the binding site for inhibitors of the HPV11 E1-E2 protein interaction on the E2 transactivation domain by photoaffinity labeling and mass spectrometry. Anal. Chem.76(7), 2095–2102 (2004).
- Bogdanov B, Smith RD. Proteomics by FTICR mass spectrometry: top down and bottom up. Mass Spec. Rev.24, 168–200 (2005).
- Martin SE, Shabanowitz J, Hunt DF, Marto JA. Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI Fouier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem.72, 4266–4274 (2000).
- Lafitte D, Benezech V, Bompart J et al. Characterization of low affinity complexes between calmodulin and pyrazine derivatives by electrospray ionization mass spectrometry. J. Mass Spec.32, 87–93 (1997).
- Al-Mawsawi LQ, Fikkert, V, Dayam R et al. Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc. Natl Acad. Sci. USA103(26), 10080–10085 (2006).
- Ecker GF, Pleban K, Kopp S et al. A three-dimensional model for the substrate binding domain of the multidrug ATP binding cassette transporter LmrA. Mol. Pharm.66(5), 1169–1179 (2004).