Abstract
Ligand interactions are central to enzyme or receptor function, constituting a cornerstone in biochemistry and pharmacology. Here we discuss a ligand application that can be exploited to significantly increase the proportion of recombinant protein expressed in soluble form, by including ligands during the culture. Provided that a sufficiently soluble, cell-permeable and avid ligand is available, one can use it to stabilize nascently synthesized proteins, and in this manner promote solubility and prevent aggregation. To our knowledge, this concept has not been explored systematically and we provide here the first data on ligand supplementation in expression experiments across a whole human protein family: the short-chain dehydrogenases/reductases (SDR). We identified glycerrhitinic acid and its hemisuccinate ester, carbenoxolone (CBX), as ligands with variable affinities ranging from low nanomolar to micromolar binding constants against several SDRs. CBX was utilized as a culture additive in Escherichia coli expression systems against a total of approximately 500 constructs derived from 65 SDR targets, and significantly higher levels of soluble protein were obtained for more than four distinct targets. One of these, the glucocorticoid-activating enzyme type 1 11β-hydroxysteroid dehydrogenase (11β-HSD1), was solubly expressed only at a very low level (<10 µg/l culture) in the absence of ligand; however, soluble expression could be enhanced to mg/l levels by inclusion of CBX or other inhibitors. Other compounds with different chemical scaffolds were used against 11β-HSD1 in equivalent expression experiments yielding similar results. Taken together, if suitable ligands for a given protein are available, this approach could be tested quickly and might represent an easy and effective strategy to enhance soluble protein production, suitable for structural and functional characterization studies.
(A) Different Escherichia coli strains (BL21 with and without a co-expression plasmid harboring the chaperone GroEL/ES [GroE7]) were analyzed for soluble expression by SDS-PAGE (left lane for each construct is soluble homogenate and the right is the eluate from immobilized metal-affinity chromatography purification). Two different constructs (denoted G25 and G26, with different domain boundaries of human 11β-HSD1) were used to inoculate the cultures. After initial incubation, cultures were split and treated with CBX or solvent (DMSO). Arrow indicates the expected mass of 11β-HSD1. (B) Ligand supplementation of human 11β-HSD1 with distinct chemical scaffolds (cpd 1, cpd 2, 18α-GA and 18β-GA). Different 11β-HSD1 inhibitors were used in analogous expression experiments, as presented for CBX (A). Note the specificity for the GA isomers 18α-GA and 18β-GA.
CBX: Carbenoxolone; ctrl: Control; GA: Glycerrhitinic acid.
![Figure 1. Ligand supplementation (100 µM CBX during induction phase) leads to increased soluble expression of distinct human short-chain dehydrogenases/reductases, shown here by expression of type 11β-hydroxysteroid dehydrogenase (HSD).(A) Different Escherichia coli strains (BL21 with and without a co-expression plasmid harboring the chaperone GroEL/ES [GroE7]) were analyzed for soluble expression by SDS-PAGE (left lane for each construct is soluble homogenate and the right is the eluate from immobilized metal-affinity chromatography purification). Two different constructs (denoted G25 and G26, with different domain boundaries of human 11β-HSD1) were used to inoculate the cultures. After initial incubation, cultures were split and treated with CBX or solvent (DMSO). Arrow indicates the expected mass of 11β-HSD1. (B) Ligand supplementation of human 11β-HSD1 with distinct chemical scaffolds (cpd 1, cpd 2, 18α-GA and 18β-GA). Different 11β-HSD1 inhibitors were used in analogous expression experiments, as presented for CBX (A). Note the specificity for the GA isomers 18α-GA and 18β-GA.CBX: Carbenoxolone; ctrl: Control; GA: Glycerrhitinic acid.](/cms/asset/273e0079-da2a-4005-aba2-3d84e05584c3/ieru_a_11217000_f0001_b.jpg)
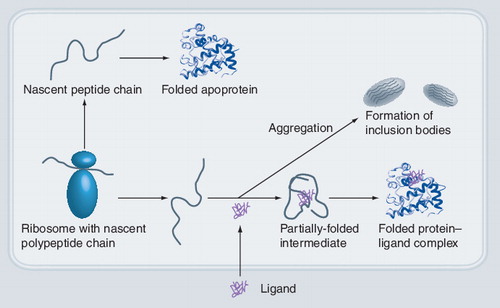
Nascent polypeptides can fold into apoproteins or aggregate and are then deposited in inclusion bodies. Inclusion of a ligand can promote and stabilize the folded protein, leading to a stable and soluble protein–ligand complex.
The advent of genome sequencing has resulted in the determination of an ever-increasing number of hundreds of thousands of coding sequences from numerous organisms of all forms of life. This vast amount of novel sequence data makes the structural and functional analysis of a comprehensive set of gene products a mandatory and highly challenging goal. To achieve this objective, heterologous protein production is necessary and therefore constitutes one of the cornerstones of functional and structural genomics initiatives. Cost-effective and simple systems are required to achieve the often large (i.e., milligram) amounts of protein that are necessary, for example, in activity and ligand screens or for structure determination purposes.
A multitude of choices for eukaryotic and prokaryotic expression systems of foreign genes exist, and thus make the production of a desired protein a straightforward procedure in many instances. Overexpression of target genes in Escherichia coli is most often the recommended first method of choice unless specific eukaryotic protein modifications are required, which cannot be carried out in prokaryotic systems Citation[1,2]. Prokaryotic expression in E. coli has received a first-option status because of the extensive knowledge of its genetics, the widespread availability of versatile vector systems and host strains, the ease of use, low costs and, importantly, the high-expression levels obtained, which in some cases exceeds more than 30% of total cellular protein.
However, despite the many advantages that heterologous protein production in E. coli offer, high-level protein expression is not necessarily routinely achieved. Surveys of structural genomics projects with several tens of thousands of attempted expression trials indicate that the current success rate to obtain soluble eukaryotic protein in E. coli is only approximately 10% Citation[3]. Among the many identified and reported causes preventing efficient heterologous protein production in E. coli are biased codon usage, gene product toxicity, target protein solubility, mRNA secondary structure and stability. Many of these obstacles can be circumvented by careful choices of expression vectors, by redesign and modification of target genes or by supplementation with rare codon genes Citation[1,2,4]. In many cases, protein expression levels are high, but the recombinant protein is found as insoluble material, most often in inclusion bodies. Despite many classical and straightforward attempts to increase the amount of soluble proteins (e.g., temperature shift or co-expression with chaperones Citation[5–7]), a large proportion of targets still appear to be refractory to successful soluble expression attempts. This is specifically observed in the case of troublesome targets such as integral or membrane-associated proteins, but also seen in other areas such as mammalian protein kinases or phosphodiesterases. Choices are often considerable and time-intensive fine-tuning efforts; for example, mutagenesis of surface residues or the obvious switch to an expression host such as baculovirus/insect cells or mammalian cells. The latter is probably the most employed and preferred method by the academic and pharmaceutical research community although, in our hands, further fine-tuning in construct design by exploring domain boundaries often enabled soluble protein production Citation[8]. Here, we would like to advocate another simple method that could be easily tested to eventually enhance or even achieve soluble protein production.
Protein–ligand interactions to enhance protein production
In many instances, analysis of protein–ligand interactions is the key to functional or structural investigations. Binding of a ligand to the active site of an enzyme or a receptor results, in most cases, in protein stabilization through polar or hydrophobic interactions. This principle has now found widespread applications in a general screening method for protein ligands; for example, in thermal shift assays that can detect ligand binding through changes in the midpoint transition temperature upon thermal denaturation of the protein Citation[9,10]. This generic method is mentioned here because it can rapidly deliver information on potential ligands of a protein in question, does not require specific activity assays and is performed reasonably fast in multiwell formats, provided a suitable ligand library is available. We propose that ligand identification, either through this or any other screening method or by literature knowledge could be utilized for a ligand application that has the potential to increase the soluble fraction in expression experiments.
Recently, we reported that inclusion of a small protein ligand during the protein-induction phase of a bacterial culture resulted in significantly (>100-fold) higher levels of soluble protein that are sufficient to allow functional analysis Citation[11]. We have utilized this principle to produce high levels of soluble human type 1 11β-hydroxysteroid dehydrogenase (HSD) by including a pharmacologically active arylsulfonamidothiazole Citation[11,12]. This currently pursued drug target in diabetes and obesity research is an enzyme, converting in an NADPH-dependent reaction cortisone to the glucocorticoid receptor ligand cortisol, is bound to the endoplasmic reticulum membrane and has been shown to be nearly completely insoluble when produced in E. coli under normal or attempted optimization conditions Citation[11,13]. We have now extended this initial observation by applying the ligand concept to expression trials towards a whole protein family. The objective was to test whether the concept was applicable to different ligand classes and, secondly, whether other targets were amenable to a possible pan inhibitor that would promote higher soluble protein levels. Within the target class of short-chain dehydrogenases/reductases (SDR), which includes the aforementioned 11β-HSD1, several expression constructs can be obtained that display higher levels of soluble protein by including carbenoxolone (CBX) as a culture additive. Furthermore, we tested several chemical compound classes, identified in classical high-throughput screens, for their ability to enhance soluble 11β-HSD1 protein levels and found them suitable as enhancing agents.
Carbenoxolone as a pan ligand of short-chain dehydrogenases/reductases
Human SDR enzymes constitute a heterogenous enzyme family with a multitude of different functions that comprise central roles in metabolic pathways of hormones, lipids, growth factors, carbohydrates and xenobiotics, or by acting as redox sensors Citation[14,15]. This protein family has received considerable attention as a potential drug class of enzymes, mainly of those targets that control central metabolic activation steps such as the 11β- or 17β-HSDs, involved in glucocorticoid or sex-steroid activation, respectively Citation[16,17]. Approximately 70 distinct genes have been identified in the human genome, of which approximately a third are orphan, that is, poorly or un-annotated genes, and well over 30% are predicted to be membrane bound through N- or C-terminal transmembrane helices Citation[14,15]. Whereas soluble expression has been observed for a substantial fraction of the human SDR family (∼45% in our laboratory [unpublished data]), in particular, the membrane-bound enzyme forms are consistently difficult to express and purify, even as transmembrane-deleted catalytic domains. In many cases, the catalytic core domain of the SDRs is straightforward to predict, due to the presence of a highly conserved Rossmann-fold motif, and hence leaves little opportunity in creating a multitude of differentially deleted expression constructs. We were therefore interested to apply the ligand supplementation concept to this protein family. We selected CBX as a possible pan ligand, for the following reasons: CBX has been reported as active against several SDR-type oxido-reductases (e.g., type 1 and type 2 11β-HSD) and we identified CBX in thermal shift compound screens as being active against several SDRs, prompting us to test the hypothesis that it might be a useful enhancing agent in expression trials. For this purpose, we generated a set of six to nine distinct expression constructs per target (a total of ∼500) and subjected all of them to expression testing in E. coli in the presence or absence of 100 µM CBX as culture supplement. The SDR genes were chemically synthesized, and were adapted to circumvent the codon bias often observed in E. coli as cause for low or missing expression Citation[2,4]. The constructs were tagged at the N-terminus with a hexahistidine sequence to enable rapid purification by immobilized metal affinity chromato-graphy (IMAC) using Ni-sepharose. The expression scale chosen was 50 ml in TB medium, to detect even low-expressing proteins, and experiments were carried out at 18°C culture and induction temperature. Subsequent to expression trials, lysates were obtained by high-pressure lysis and subsequent centrifugation, and were subjected to IMAC purification. The eluates were analyzed either by SDS-PAGE or on an Agilent 5100 Alp capillary electrophoresis system (Agilent Technologies) and quantified. These experiments were carried out three- to four-times; a representative SDS-PAGE result is displayed in . In total, we tested 65 distinct SDR genes with approximately 500 distinct constructs generated. Of these, 24 targets (corresponding to 70 distinct constructs) demonstrated some level of soluble expression. Inclusion of CBX during the culture significantly increased the amount of soluble targets to 28, corresponding to 86 soluble constructs. Enhancement of expression levels ranges from two- to threefold for most of the targets to a nonquantifiable increase; for example, for 11β-HSD1, no or very low levels were detected in expression experiments without ligand, which increased to mg/l levels when including CBX or other known inhibitor scaffolds . These results indicate that, first, CBX has the capacity to enhance soluble expression of a subset of human SDR members indicating (presumably) binding to their active sites and, second, that this effect appears to be more pronounced in the case of targets with initially low or absent solubility, such as 11β-HSD1 .
Different chemical scaffolds as culture additives
As outlined before, the most prominent effect of induction of soluble protein production was noted with 11β-HSD1, from very low levels of soluble protein to milligram amounts, which are clearly sufficient for structural and functional studies. Again, this points to the fact that ligand supplementation appears to be most effective and useful when initial solubility levels are low. In fact, in this case, the strategy of ligand supplementation was utilized to determine the high-resolution structure of human 11β-HSD1 in complex with CBX (PDB ID 2BEL) Citation[17]. The high-resolution crystal structure reveals that CBX binds through few polar contacts, namely through hydrogen bonds from CBX-O35 to O2n of the nicotinamide ribose and OH of Tyr183, and through contacts between CBX-O11 and OH of Ser170. Binding is otherwise driven by shape complementarities of large hydrophobic surfaces. This active site geometry allows binding of distinct scaffold such as steroids (glucocorticoids, oxysterols and bile acids), steroid-like ligands (CBX or glycerrhitinic acid) and nonsteroidal inhibitors (e.g., arylsulfonamidothiazoles) Citation[12].
We next investigated whether compound scaffolds other than CBX were effective to enhance soluble protein levels. For this purpose, we analyzed the influence of distinct 11β-HSD1 inhibitor scaffolds on expression levels, tested in an analogous manner to the experiments described earlier Citation[18–20]. As illustrated in , some of these compounds indeed display similar behavior as CBX, demonstrating the versatile utility of different inhibitors to provide solubility enhancement.
Discussion
We have discussed a heterologous protein expression strategy using small molecules as culture additives. The cause of increased solubility of certain targets by inclusion of ligands is unknown. Although mechanisms such as increased RNA stability or reduction of toxic effects on the expression host cannot be ruled out, the observed increased soluble expression of target proteins by small molecules is most likely due to induction of a correctly folded state of the target protein. This ligand complex formed in the host is more stable than the apo-structure, and thus likely promotes solubility and prevents aggregation Citation[11]. This is further supported by the notion that, upon ligand supplementation and subsequent purification, target proteins contain their ligand bound to the active site Citation[17]. Intuitively, a more stable and correctly folded protein appears to be less prone to aggregation (e.g., in inclusion bodies in E. coli) or to proteolytic degradation. This postulated mechanism for ligand-induced stabilization for heterologous protein production is schematically depicted in .
Previously, it was also found that even ligands with moderate affinity (i.e., with a low micromolar Kd constant) were suitable for enhancement of soluble expression Citation[11]. In our experiments we usually include compounds at 100 µM dissolved in dimethyl sulfoxide, which can be used as a general starting point and has led repeatedly to successful results. In the case of 11β-HSD1, we have now successfully utilized different chemical classes of inhibitors to promote solubility, comprising steroid-based, adamantyltriazole and arylsulfonamidothiazole scaffolds.
Although not investigated here in detail, it is conceivable that several factors are important and contribute to the outcome and design of this approach, such as compound toxicity in E. coli, cellular uptake, compound solubility, interference with the host metabolism and conversion of the compound itself by the host.
Previously, we utilized successfully ligand supplementation with one compound to obtain sufficient protein amounts of 11β-HSD1, and then exchanged by extensive dialysis the initial compound against another ligand of choice or produced ligand-free enzyme for kinetic analysis. This strategy proved to be useful in cases of limited solubility and insufficient uptake by E. coli, and indicates a way to circumvent some of the problems indicated above. Similar results were reported on ligand-induced expression of several kinases, where ligand exchange upon purification was possible Citation[21].
This ligand-supplementation approach is by no way limited to bacterial expression systems or to protein families such as oxidoreductases. Indeed, several groups have previously reported successful implementation of this concept to produce active protein; for example, in baculovirus expression systems Citation[21,22]. In an early report, Pyle et al. describe the use of protease inhibitors such as leupeptin and pepstatin, to produce intact human proapolipoprotein A1, which was otherwise degraded, in an insect-cell secretion system Citation[22]. In a recent paper, Strauss et al. present data on the expression of several tyrosine and serine/threonine kinases in the presence of inhibitors using a baculo-virus expression system Citation[21]. By using this strategy, the authors were able to dramatically increase the yield of soluble protein and produce sufficient and intact protein for structural and functional studies. In a similar manner, increased expression of difficult protein families, such as nuclear receptors, was also found by inclusion of ligands during culture using E. coli as the expression system Citation[23].
Furthermore, in a previous study we demonstrated that the combination of chaperone co-expression (using the chaperones GroEl/ES) and ligand supplementation resulted in an additive effect regards soluble protein levels by using an arylsulfonamidothiazole compound Citation[11]. However, this effect might be compound specific, because we did not observe statistically significant additive effects when using CBX in this study. Taken together, these and our present studies (although still few in number) indicate the suitability of the approach for obtaining soluble protein, which would otherwise have been difficult to achieve.
It is clear that some ligand information is essential for the supplementation approach. Identification of suitable ligands by either literature search, or directed experimental or in silico screening efforts could be used to generate ligand candidates that are subsequently tested in expression experiments. Of potential use here are pan inhibitors; compounds that display binding activity against several members of a given protein family (e.g., CBX used against human SDR enzymes). In many instances, pharmaceutical screening events identify compounds with broad off-target activity, which could be potentially useful in ligand supplementation approaches in case of expression of many related proteins.
Another conceivable possible strategy could be to utilize heterologous, engineered host systems that synthesize high-affinity pan ligands such as staurosporine for kinases. Successful transfer and engineering of a whole complex biosynthetic pathway has been demonstrated in yeast. In a recent study, a strain was stably modified with all enzyme components to produce steroids such as hydrocortisone Citation[24]. These organisms producing high-affinity ligands could, in turn, be used as expression hosts for complete protein families that are known to bind these pan ligands.
Expert commentary
Ligand supplementation adds to an increasing list of refined second-tier strategies for heterologous protein production, such as directed evolution approaches or chaperone co-expression Citation[25,26]. To our knowledge, this is a concept that has not been explored systematically. However, if suitable ligands for a given protein are available, this approach can be quickly implemented in test-expression experiments, and therefore represents an easy and effective approach to enhance soluble protein production.
Five-year view
Simple (that is, cost effective and robust) expression systems such as E. coli, will remain at the forefront of heterologous expression technologies. Likely a large fraction of, for example, membrane proteins will remain difficult to express in recombinant form in the foreseeable future, and only a fraction of achievable targets is routinely expressed in soluble form, suitable for further downstream characterization. Successful soluble expression of a given target is a function of several variables, including (among many others) construct design, choice of expression system and co-expression of auxiliary proteins such as chaperones, media composition or culture temperature. Screening for optimal expression conditions can easily be conducted utilizing suitable protein ligands, and implementation of the ligand supplement strategy is a possible route to enhance or achieve soluble expression of recombinant proteins. Successful use of this method will largely depend on knowledge of suitable ligands for distinct classes of protein targets. Large-scale chemical genomics efforts (e.g., the National Institutes of Health Roadmap Molecular Libraries Initiative) with the aim of identifying small-molecule ligands as tools for biological research have the potential to provide the necessary ligand data for distinct protein classes. Combined with generic ligand identification assays, these efforts will likely further the use of ligand supplementation as a method for protein production.
Key issues
| • | With an almost exponentially growing list of sequenced organisms, protein characterization will remain an integral component in functional genomics. | ||||
| • | Heterologous protein expression often constitutes a bottleneck in production of proteins of interest, at present a success rate well below 20% on large-scale expression trials of eukaryotic targets is achieved in Escherichia coli. | ||||
| • | Ligand supplementation as an easy way to test and possibly increase protein production in heterologous expression systems is discussed. | ||||
| • | Identification of suitable ligands or ligand classes for ligand supplementation and expression studies is mandatory. Whereas results from screening efforts conducted in the pharmaceutical industry are mostly not disclosed, public screening efforts embedded in the National Institutes of Health Roadmap Libraries Initiative, conducted by the various National Institutes of Health Chemical Genomics Centers, and deposited in PubChem Citation[27] will increasingly provide publicly available data on chemical probes that can be useful for ligand supplementation studies described in this paper. | ||||
Acknowledgements
We thank Stefan Svensson, Bjorn Elleby and Lars Abrahmsen (Biovitrum, Stockholm, Sweden) for collaboration on initial experiments, and Sujata Sharma (SGC Toronto, Canada) for cloning of SDR genes.
We are also indebted to Rolf Thieringer (Merck, Rahway, NJ, USA) and Nigel Vikker (Sterix, Bath, UK) for providing 11β-hydroxysteroid dehydrogenase inhibitors. Preliminary results were presented at the Peptalk meetings (Cambridge Healthtech, MA, USA) in San Diego in 2005 and 2007.
Financial & competing interests disclosure
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Hunt I. From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Protein Expr. Purif.40(1), 1–22 (2005).
- Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev.60(3), 512–538 (1996).
- Structural Genomics Consortium A, Berkeley Structural Genomics Center, China Structural Genomics Consortium et al. Guidelines for the production and purification of recombinant proteins in E. coli. Nat. Methods (2008) (In press).
- Wu X, Jornvall H, Berndt KD, Oppermann U. Codon optimization reveals critical factors for high level expression of two rare codon genes in Escherichia coli: RNA stability and secondary structure but not tRNA abundance. Biochem. Biophys. Res. Commun.313(1), 89–96 (2004).
- Wynn RM, Davie JR, Song JL, Chuang JL, Chuang DT. Expression of E1 component of human branched-chain a-keto acid dehydrogenase complex in Escherichia coli by cotransformation with chaperonins GroEL and GroES. Meth. Enzymol.324, 179–191 (2000).
- Ailor E, Betenbaugh MJ. Overexpression of a cytosolic chaperone to improve solubility and secretion of a recombinant IgG protein in insect cells. Biotechnol. Bioeng.58(2–3), 196–203 (1998).
- Widersten M. Heterologous expression in Escherichia coli of soluble active-site random mutants of haloalkane dehalogenase from Xanthobacter autotrophicus GJ10 by coexpression of molecular chaperonins GroEL/ES. Protein Expr. Purif.13(3), 389–395 (1998).
- Gileadi O, Knapp S, Lee WH et al. The scientific impact of the Structural Genomics Consortium: a protein family and ligand-centered approach to medically-relevant human proteins. J. Struct. Funct. Genomics8(2–3), 107–119 (2007).
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc.2(9), 2212–2221 (2007).
- Vedadi M, Niesen FH, Allali-Hassani A et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl Acad. Sci. USA103(43), 15835–15840 (2006).
- Elleby B, Svensson S, Wu X et al. High-level production and optimization of monodispersity of 11β-hydroxysteroid dehydrogenase type 1. Biochim. Biophys. Acta1700(2), 199–207 (2004).
- Hult M, Shafqat N, Elleby B et al. Active site variability of type 1 11β-hydroxysteroid dehydrogenase revealed by selective inhibitors and cross-species comparisons. Mol. Cell. Endocrinol.248(1–2), 26–33 (2006).
- Shafqat N, Elleby B, Svensson S et al. Comparative enzymology of 11β-hydroxysteroid dehydrogenase type 1 from glucocorticoid resistant (Guinea pig) versus sensitive (human) species. J. Biol. Chem.278(3), 2030–2035 (2003).
- Kallberg Y, Oppermann U, Jornvall H, Persson B. Short-chain dehydrogenase/reductase (SDR) relationships: a large family with eight clusters common to human, animal, and plant genomes. Protein Sci.11(3), 636–641 (2002).
- Oppermann U, Filling C, Hult M et al. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem. Biol. Interact.143–144, 247–253 (2003).
- Lukacik P, Kavanagh KL, Oppermann U. Structure and function of human 17b-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol.248(1–2), 61–71 (2006).
- Wu X, Lukacik P, Kavanagh KL, Oppermann U. SDR-type human hydroxysteroid dehydrogenases involved in steroid hormone activation. Mol. Cell. Endocrinol.265–266, 71–76 (2007).
- Su X, Vicker N, Ganeshapillai D et al. Benzothiazole derivatives as novel inhibitors of human 11β-hydroxysteroid dehydrogenase type 1. Mol. Cell. Endocrinol.248(1–2), 214–217 (2006).
- Vicker N, Su X, Ganeshapillai D et al. Novel non-steroidal inhibitors of human 11β-hydroxysteroid dehydrogenase type 1. J. Steroid Biochem. Mol. Biol.104(3–5), 123–129 (2007).
- Olson S, Aster SD, Brown K et al. Adamantyl triazoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. Lett.15(19), 4359–4362 (2005).
- Strauss A, Fendrich G, Horisberger MA et al. Improved expression of kinases in Baculovirus-infected insect cells upon addition of specific kinase inhibitors to the culture helpful for structural studies. Protein Expr. Purif.56(2), 167–176 (2007).
- Pyle LE, Barton P, Fujiwara Y, Mitchell A, Fidge N. Secretion of biologically active human proapolipoprotein A-I in a baculovirus-insect cell system: protection from degradation by protease inhibitors. J. Lipid Res.36(11), 2355–2361 (1995).
- Hassell AM, An G, Bledsoe RK et al. Crystallization of protein–ligand complexes. Acta Crystallogr. D Biol. Crystallogr.63(Pt 1), 72–79 (2007).
- Szczebara FM, Chandelier C, Villeret C et al. Total biosynthesis of hydrocortisone from a simple carbon source in yeast. Nat. Biotechnol.21(2), 143–149 (2003).
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol.17(7), 691–695 (1999).
- Yang JK, Park MS, Waldo GS, Suh SW. Directed evolution approach to a structural genomics project: Rv2002 from Mycobacterium tuberculosis.Proc. Natl Acad. Sci. USA100(2), 455–460 (2003).
- Inglese J, Auld DS, Jadhav A et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl Acad. Sci. USA103(31), 11473–11478 (2006).