Abstract
Stroke is a significant cause of morbidity and mortality in children and adults with sickle cell disease. Great advances in the past couple of decades have enabled identification and treatment of children at risk of stroke, with a resultant dramatic reduction in stroke incidence in children. However, prevention and treatment of silent cerebral infarcts remain a challenge. This article reviews our current understanding of the epidemiology, risk factors and pathophysiology of small and large vessel disease with a focus on pediatric patients. The presentation and acute management of stroke in patients with sickle cell disease are discussed. Current recommendations for primary and secondary stroke prevention, as well as ongoing research studies and potential new therapies are reviewed.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/experthematology; (4) view/print certificate.
Release date: 14 June 2013; Expiration date: 14 June 2014
Learning objectives
Upon completion of this activity, participants will be able to:
• Understand the epidemiology of stroke among patients with SCD
• Assess risk factors for stroke among patients with SCD
• Distinguish best practices in the acute management of stroke among patients with SCD
• Evaluate secondary prevention strategies against stroke among patients with SCD
Financial & competing interests disclosure
EDITOR
Elisa Manzotti
Publisher, Future Science Group, London, UK
Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Charles P Vega, MD
Department of Family Medicine, University of California, Irvine, CA, USA
Disclosure: Charles P Vega, MD, has disclosed no relevant financial relationships.
AUTHORS AND CREDENTIALS
Jennifer Webb
Division of Hematology, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA and Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Disclosure: Jennifer Webb has disclosed no relevant financial relationships.
Janet L Kwiatkowski
Division of Hematology, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA and Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
Disclosure: Janet L Kwiatkowski has disclosed no relevant financial relationships.
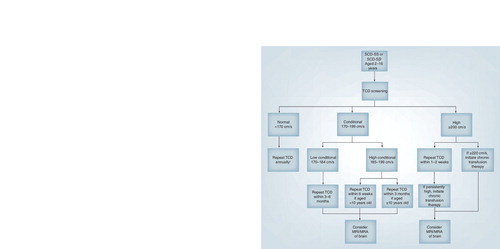
†Consider repeating TCD more frequently in those with additional risk factors such as silent infarcts, family history of stroke or abnormal TCD or abnormal MRA.
MRA: Magnetic resonance angiography; SCD-SS: Homozygous sickle-cell disease; TCD: Transcranial Doppler ultrasound.
Stroke is a significant cause of morbidity and mortality in children and adults with sickle cell disease (SCD). Strokes or cerebrovascular accidents (CVAs) are felt to represent the culmination of large and small vessel disease and altered cerebral autoregulation, as well as the sequelae of chronic inflammation, hemolysis and anemia. Although the precise pathophysiology of stroke in SCD is still an area of active research, clinical research studies have made significant advances in understanding and identifying those patients who are at highest risk of stroke and in implementing strategies for reducing that risk.
Epidemiology
CSSCD, the largest US multicenter longitudinal observational study of complications of SCD, reported an overall prevalence of stroke of 3.75% in all patients with SCD Citation[1]. The prevalence of CVA was 11% in patients under 20 years of age with homozygous sickle-cell disease (SCD-SS). In childhood, the highest incidence (1.02 per 100 person-years) was found in children between 2 and 5 years of age with SCD-SS Citation[1]. The rates of CVA vary by sickle cell genotype. The age-adjusted incidence of CVA is highest for those with SCD-SS (0.61 per 100 person-years) compared with SCD-SC (0.15 per 100 person-years) or hemoglobin Sβ+ or Sβ0 thalassemia (0.09 per 100 person-years and 0.08 per 100 person-years, respectively) Citation[1].
In CSSCD, strokes were classified as ischemic, hemorrhagic and transient ischemic attacks (TIAs). Ischemic strokes disproportionately affect the youngest and oldest patients with SCD, while hemorrhagic strokes have the highest prevalence in patients between the ages of 20 and 29 years with SCD Citation[1].
Silent infarcts, defined in CSSCD as an increased T2 signal abnormality on multiple views on MRI without corresponding neurologic deficit, were identified in 21.8% of children between 6 and 19 years of age with SCD-SS Citation[2]. Further studies have shown an estimated cumulative incidence of silent infarct as high as 37% in patients with SCD by 14 years of age Citation[3]. The increased prevalence of silent infarcts may be due to changes in the definition of silent infarct, as well as improvements in MRI techniques. Currently, the most widely accepted definition of silent infarct in pediatric patients requires an abnormality of at least 3 mm in greatest linear dimension visible on at least two planes of T2-weighted MRI sequences Citation[4].
The pathogenesis of silent infarcts appears to begin very early in life. Screening MRIs performed at early ages have demonstrated a prevalence of silent infarcts of 13% in infants and toddlers with SCD-SS at an average age of 13.7 months Citation[5]. Other studies have demonstrated silent infarcts in 27.7% of children with SCD-SS less than 6 years of age Citation[6]. In a longitudinal follow-up of the CSSCD cohort, silent infarcts progressed in number and in size over time Citation[2]. In girls with SCD-SS, most lesions appeared by 6 years of age. In boys with SCD-SS, new lesions continued to appear through to 10 years of age. Of those children in this cohort with normal MRIs at baseline, only 2.5% developed new silent infarcts during the follow-up period, compared with 24.5% of those with silent infarcts already present on their baseline MRI Citation[2]. Although initially felt to be clinically ‘silent’, research has demonstrated an association between silent infarcts and neurocognitive deficits and academic difficulties in pediatric patients Citation[7–13]. It is unclear whether the age at which a silent infarct occurs impacts neurocognitive outcomes.
In adults with SCD, silent infarcts are defined as 5-mm signal hyperintensity in T2-weighted images with corresponding hypointensity in the T1-weighted images due to the propensity for adults to naturally accumulate T2 hyperintensities as a function of the aging process Citation[14]. In a large study comparing neuroimaging abnormalities and neuropsychiatric outcomes in adults with SCD-SS without a history of overt stroke to healthy controls, individuals with SCD-SS were found to perform worse on tests of global cognitive functioning, working memory, processing speed and executive function Citation[14]. Although the proportion of SCD-SS patients with evidence of silent infarcts was higher than healthy controls, it was not statistically significant, nor was it independently correlated with poorer performance on cognitive tests Citation[14]. Other studies have demonstrated a prevalence of silent infarct in up to 50% of the adults with SCD-SS, although there is substantial variability in the literature Citation[15,16].
Pathophysiology
Ischemic strokes
Strokes in patients with SCD were traditionally hypothesized to be due to increased viscosity of sickled red blood cells causing stasis and downstream ischemia. However, this hypothesis does not fully explain the etiology of the large vessel CVAs that mark this disease. Although vasculopathy and stenosis of the large arterial branches off of the circle of Willis are frequently seen in patients with SCD-SS or SCD-Sβ0 Citation[17], multiple other factors, such as impaired vascular autoregulation, contribute to the ultimate event.
Current research suggests that early events in the pathogenesis of stroke are the attachment of sickled cells to vascular endothelium where endothelial activation and damage occur Citation[18]. Patients with SCD have been shown to have high expression of endothelial and erythrocyte adhesion molecules Citation[19], which have been implicated in the pathogenesis of vaso-occlusion including integrins Citation[20,21], endothelial selectins Citation[22,23], soluble adhesion molecules Citation[24] and immunoglobulin family members Citation[25]. Procoagulants, such as von Willebrand factor Citation[26], fibrinogen Citation[27] and thrombin Citation[28], have also been implicated. Chemoattractants, cytokines and adhesion molecules recruit leukocytes that can cause microvascular obstruction and ischemia Citation[29,30]. This proinflammatory state leads to intimal hyperplasia, fibrosis and, ultimately, thrombosis Citation[18].
Additional research into the role of nitric oxide (NO), a locally acting vasodilator, has implicated NO and NO-related pathways as part of the pathophysiology of stroke. Much of the research has focused on pulmonary hypertension (PH), a vascular complication of SCD similar to stoke that is associated with increased risk of mortality in adults Citation[31]. In patients with PH, hemolysis of sickled cells leads to increased plasma-free hemoglobin, which scavenges local NO Citation[32,33]. Furthermore, patients with SCD have a relative depletion of l-arginine compared with healthy controls, due to increased release of erythrocyte arginase from sickled red blood cells. The lack of l-arginine, a substrate for NO production, prevents reconstitution of NO in the acute setting. Among patients with SCD, arginase activity has been demonstrated to be highest in those with concomitant PH and is correlated with severity of PH Citation[31]. A recent review noted that further disruption of the arginine–NO pathway due to the downstream effects of uncoupling of NO synthase, formation of polyamines and l-proline, competition for intracellular transport and overproduction of reactive oxygen species may have roles in the development of sickle cell-related pulmonary vasculopathy Citation[32], although its role in stroke is less clear. Supporting this mechanism is the evidence that chronic transfusion therapy (CTT), which has long been the treatment for primary and secondary stroke prevention, has been shown to decrease levels of free plasma hemoglobin Citation[34] and decrease the rate of hemolysis Citation[35,36]. Additionally, lactate dehydrogenase level, a marker of intravascular hemolysis, has been showed to correlate significantly with transcranial Doppler (TCD) ultrasound velocities, a marker of stroke risk in children with SCD-SS Citation[37].
Hemorrhagic strokes
Imaging studies have demonstrated a high prevalence of cerebral vasculopathy in patients with SCD. Patients with SCD have been shown to develop cerebral aneurysms, although their exact prevalence is unknown. Aneurysms are the most common identified cause of hemorrhagic stroke in adult patients with SCD Citation[38]. Compared with patients without SCD, aneurysms in SCD patients are often multiple, have an increased propensity for the posterior cerebral circulation and may be prone to rupture at smaller sizes Citation[39]. Moyamoya disease, a progressive stenosis of the internal carotid arteries resulting in the formation of fragile arterial collaterals, has been found to be present in 20–35% of the patients with SCD who undergo cerebral angiography Citation[40]. If present, moyamoya syndrome is an indication of severe cerebrovascular disease. Patients with this finding are at high risk of recurrent stroke in spite of the initiation of CTT for secondary stroke prevention Citation[41].
Silent infarcts
Silent infarcts may represent a different pathogenesis from either ischemic or hemorrhagic strokes as they involve small vessels in watershed distributions instead of the larger cerebral vessels Citation[42]. Their watershed distribution suggests defective regulation of cerebral perfusion pressure or diminished reserve as potential physiologic mechanisms Citation[43,44]. However, these theories are under active research. Further supporting a difference in pathogenesis, transcranial Doppler ultrasound velocities are not predictive of risk of silent infarct, unlike with overt ischemic stroke Citation[45]. Additionally, silent infarcts may continue to progress in spite of initiation of CTT Citation[46].
Risk factors
Ischemic strokes
The CSSCD identified several risk factors for ischemic stroke in patients with SCD including prior TIA, frequency of acute chest syndrome (ACS), ACS in the 2 weeks preceding the event, and increased systolic blood pressure Citation[1]. Prior silent infarct has also been found to increase the risk of ischemic stroke 14-fold in pediatric patients with SCD Citation[15,47]. Nocturnal hypoxemia (<90%) has been associated with an increased risk of CVAs and other CNS events in patients with SCD-SS Citation[48]. Studies have further shown that there is a high incidence of sleep disordered breathing in patients with SCD Citation[49], and one large cohort study using Medicaid data on 768 children with SCD showed a decreased rate of outpatient visits, emergency room visits and inpatient hospital days for stroke and TIAs in children who had undergone adenotonsillectomy Citation[50].
Various imaging techniques have been used to prospectively identify that pediatric patients with SCD who are at risk for ischemic stroke. TCD ultrasound, which measures the time-averaged mean of the maximal (TAMM) velocity in large intracranial arteries, is a powerful, noninvasive tool that can be utilized to classify an individual patient’s risk of stroke Citation[51]. Children with SCD-SS with abnormal velocity (TAMM: ≥200 cm/s) in the middle cerebral artery or distal internal carotid artery or its bifurcation have a 40% risk of stroke within 40 months. Children with TAMM velocities of 170–199 cm/s (conditional TCD) have a modestly elevated risk of stroke of approximately 7% in the subsequent 40 months, while those with normal studies (TAMM: <170 cm/s) have the lowest risk of stroke (<1% per year) Citation[52,53]. Children with abnormal TAMM who also have evidence of silent infarcts or brain magnetic resonance angiography (MRA) abnormalities are at the highest risk of stroke Citation[42,54].
Extracranial internal carotid artery vasculopathy has also been associated with ischemic strokes and silent infarcts Citation[55,56]. However, many of these children have concomitant intracranial vasculopathy. Further research is necessary to determine its prognostic significance and implications for intervention.
In children with SCD-SC, TAMM velocities are significantly lower than in those with SCD-SS and have not been shown to predict stroke risk Citation[57]. In adult patients with SCD, TCD velocities have been shown to be higher than adult controls without SCD Citation[58,59]; however, they are lower than in children with SCD and have not been found to be predictive of vascular stenosis or CVAs Citation[60].
Hemorrhagic strokes
The CSSCD also identified risk factors for hemorrhagic stroke including older age, low steady state hemoglobin and high steady state leukocyte count Citation[1]. A multicenter, retrospective case–control study of pediatric patients with primary hemorrhagic stroke compared with those with ischemic stroke demonstrated that hemorrhagic stroke was associated with older age, a reported history of hypertension, transfusion within the 2 weeks prior to the events, higher frequency of admission to the hospital for acute pain event in the preceding year, and treatment with corticosteroids or nonsteroidal anti-inflammatory medication within 2 weeks of the event Citation[61].
Silent infarcts
Associations have been found between silent infarcts and leukocytosis, low pain event rate, SEN haplotype and prior history of seizures Citation[62]. Additional studies have shown an association between silent infarcts and lower baseline hemoglobin Citation[3,63], as well as identified higher systolic blood pressure and male gender as additional risk factors Citation[3,63]. In patients under 6 years of age, silent infarcts were associated with lower rates of vaso-occlusive pain and ACS Citation[6]. No association has been found between silent infarcts and abnormal TCD velocity Citation[45] or hemoglobin F percentage Citation[62]. Two studies have found associations between vasculopathy identified using MRA and silent infarcts Citation[6,64], but this requires further study. Glucose-6-phosphate dehydrogenase deficiency has also been associated with silent infarcts Citation[64]; however, its role remains unclear due to conflicting data on its association with other CNS risk factors and ischemic strokes Citation[3,65–67].
Concomitant α-thalassemia in patients with SCD is associated with reduced rates of silent infarct and stroke Citation[1,35,68,69], as well as with decreased frequency of abnormal TAMM velocities Citation[65]. Risk factors for the various types of CVAs are reviewed in .
The phenotypic variation among patients with SCD with regard to CVAs indicates that there are additional disease modifiers. Sibling studies of patients with SCD have shown significant correlation in TCD values Citation[70], as well as in the frequency of CVA Citation[71]. Genetic studies have found that polymorphisms in the promoter for TNF-α were associated with a greater than threefold increased risk of large vessel stroke in children Citation[72,73] and various HLA alleles have been shown to convey differential risk for large and small vessel strokes, further suggesting different underlying pathophysiologic mechanisms Citation[74,75]. Multiple other candidate genes suspected of modifying stroke risk are currently under study, including VCAM-1 Citation[72,76] and IL4R Citation[72]. However, as the effect of individual polymorphisms on overall stroke risk may be moderate, there is a need for simultaneous evaluation of multiple candidate genes in a combinatorial fashion in order to predict presymptomatic vasculopathy Citation[77,78].
Clinical presentation
Acute ischemic stroke often presents as it would in others without SCD. Frequent symptoms include motor deficits including hemiparesis and monoparesis, aphasia or seizure Citation[79,80]; however, symptoms may vary by the size and location of the stroke. Posterior circulation strokes may present with ataxia, headache, vertigo or vomiting. In young children, symptoms may be subtle and mistaken for other illnesses. Hemorrhagic stroke may present with acute, severe headache. TIAs may present similarly to acute ischemic strokes; however, they spontaneously resolve.
Fortunately, of those pediatric patients with SCD with an overt stroke, many are able to become physically independent with few difficulties with self-care activities Citation[79]. However, most demonstrate persistent cognitive deficits that range from language problems and reduced intelligence quotient to moderate mental retardation Citation[81]. Evidence of poor educational attainment and neurocognitive disabilities in this population has been an impetus to implement ongoing evaluation and standardized cognitive rehabilitation programs Citation[82,83].
Acute treatment of stroke
Few studies have investigated the optimal medical management for acute ischemic stroke in pediatric patients with SCD and it is unclear if initial management plays a role in later outcomes. The initial assessment of a pediatric patient with SCD where there is a clinical concern for stroke should focus on stabilization, vital sign support and oxygenation. A history and physical exam, including a complete neurological exam, should be performed. Initial laboratory evaluation should include a complete blood count with differential and reticulocyte count, coagulation studies, hemoglobin quantification to determine the hemoglobin S percent, blood chemistries and evaluation for meningitis if clinically indicated. The patient’s blood should be typed and screened for any red blood cell antibodies. A noncontrast CT scan of the brain should be performed as soon as possible if there is a concern of acute hemorrhage. However, CT scans are limited in their ability to detect acute infarct. If there is enough clinical concern, MRI/MRA/magnetic resonance venography with diffusion-weighted imaging is the preferred modality for evaluating ischemic stroke, vasculopathy and eliminating dural venous sinus thrombosis as the etiology of the event Citation[84]. Intravenous fluids should be carefully monitored to maintain euvolemia. Additional supportive care measures to control glucose, maintain cerebral perfusion pressure and reduce fever should be performed. Currently, there is no role for anticoagulation or antifibrinolytics as they have not been studied in patients with SCD and may increase the risk of hemorrhagic conversion.
A manual or automated exchange transfusion should be performed with a goal of decreasing the hemoglobin S to below 30% and increasing the hemoglobin to 10–12 g/dl to improve oxygen carrying capacity. However, if exchange transfusion leads to significant delays in treatment initiation, straight transfusions may be used to increase the hemoglobin to approximately 10 g/dl. Increasing the hemoglobin higher than 12 g/dl in the acute setting may lead to increased viscosity and should be avoided. In a retrospective, multicenter cohort study of pediatric patients with SCD presenting with stroke, those who received a straight transfusion rather than exchange transfusion as part of their initial management had a fivefold greater risk of secondary stroke over the subsequent 5 years in spite of initiation of CTT Citation[80].
Patients with acute hemorrhagic stroke should receive similar supportive care measures as clinically indicated. Additionally, acute neurosurgical evaluation is required as there may be a role for placement of coils or embolization if an aneurysm is identified Citation[85]. Depending upon the size of the hemorrhage, a patient may require surgical decompression and control of vasospasm as well Citation[86].
Secondary stroke prevention
CTT has long been used to prevent secondary ischemic strokes. Transfusions are generally administered every 3–4 weeks with the goal to maintain the hemoglobin S under 30% of the total hemoglobin. The post-transfusion hemoglobin is increased to between 10 and 12 g/dl. Without treatment, the risk of recurrent stroke is approximately 70% Citation[38], but with CTT, that risk is reduced to 10–20% Citation[87–89]. In a retrospective review of 137 pediatric patients treated at 14 centers with CTT due to a history of ischemic stroke, 22% of the patients had a recurrent stroke (2.2 per 100 person-years) Citation[88]. However, vasculopathy and silent infarcts, when present in patients who also have a history of acute stroke, can progress despite optimal transfusion Citation[46,90–93]. Children with more severe vasculopathy at initiation of CTT appear to have a greater risk of progression. This supports the need for routine TCD surveillance and the use of primary stroke prevention strategies (see below).
Attempts to discontinue CTT after periods of up to 10–12 years resulted in a high rate of recurrent stroke, so lifelong treatment is recommended Citation[94]. CTT is complicated by potential risks of alloimmunization Citation[95], infectious disease exposure Citation[96] and iron overload Citation[97], which has prompted a search for alternative therapies. Erythrocytapheresis, automated red blood cell exchange, can limit iron loading and obviate the need for chelation therapy, but is associated with a two- to three-fold greater donor exposure and requires adequate venous access Citation[98–101]. Strategies that relax transfusion goals after several years to maintain a hemoglobin S under 50% have reduced the amount of blood products required and the resulting iron load without an increased risk of infarctive stroke; however, the long-term effect on progression of vasculopathy and silent infarcts remains to be studied Citation[102,103].
Single institutions have explored hydroxyurea as an alternative therapy for secondary stroke prevention in pediatric patients with SCD in countries where blood products are scarce. One institution in Jamaica followed 43 children with SCD with evidence of acute ischemic stroke between 2000 and 2009 Citation[104]. Of these, ten initiated therapy with hydroxyurea alone. In those patients, the incidence of recurrent stroke was two per 100 person-years, compared with 29 per 100 person-years in the untreated group Citation[104].
In a single-center study, 35 children with SCD and a history of stroke were transitioned to hydroxyurea after a mean of 56 ± 33 months of CTT Citation[105]. The rate of stroke recurrence was 5.7 events per 100 person-years. When an overlap period of transfusions and hydroxyurea was employed, the recurrence rate was further reduced to 3.6 events per 100 person-years. In addition, 26 subjects were able to tolerate phlebotomy to reduce iron load Citation[105]. This set the stage for the multicenter stroke with transfusions changing to hydroxyurea (SWiTCH) trial. In that study, subjects who had initiated CTT after an overt stroke and had evidence of iron overload were randomized to either continue CTT or transition to hydroxyurea and scheduled phlebotomy Citation[106]. While increasing to the maximally tolerated dose of hydroxyurea, subjects continued to receive transfusion therapy. The primary end point was a composite end point allowing for a slight increased stroke risk, but with superior removal of iron as measured by liver iron concentration. In total, 10% of subjects (n = 7) randomized to the hydroxyurea arm developed strokes; however, none of the subjects who continued with CTT had strokes. In addition, no significant difference in liver iron concentration was noted at the planned interim analysis, although the long overlap period of transfusions in the hydroxyurea arm may have contributed to the lack of significant improvement in iron reduction. These findings led to the study’s premature closure by the Data and Safety Monitoring Board Citation[106]. Therefore, chronic red cell transfusion therapy remains the mainstay of secondary stroke prevention. See for a summary of stroke recurrence rates with hydroxyurea Citation[104–109].
Moyamoya disease, if identified, predicts a high risk of recurrent stroke in patients with SCD in spite of optimal transfusion therapy Citation[41]. Several case series have shown benefit of neurosurgical revascularization procedures in this setting; however, those series have had small numbers of patients with limited follow-up Citation[110,111].
For patients who have suffered a hemorrhagic stroke, there is no clear role for CTT in the prevention of a secondary hemorrhagic stroke Citation[61]. Placement of clips or coils may prevent ongoing bleeding if an aneurysm is identified Citation[86].
Currently, limited data on the treatment or prevention of silent infarcts is available. Among children with both abnormal TCD and silent infarcts, those who received transfusion therapy were significantly less likely to develop new silent infarcts Citation[42]. A subset of those patients later had transfusions discontinued and demonstrated an increased frequency of progression of silent infarcts compared with those who continued transfusion therapy Citation[112]. However, a recent study looking at transfusion in patients with overt stroke found progression of silent infarcts in 25% of subjects despite being optimally transfused Citation[46]. The role of transfusion therapy for children with silent infarcts without abnormal TCD is currently being explored in the SIT trial (ClinicalTrials.gov identifier: NCT00072761) Citation[201]. Subjects are randomized to observation or monthly transfusion therapy. The primary outcome is progression of silent infarcts or overt stroke. This study has completed recruitment, but is not expected to complete data analysis until December 2013 Citation[4]. Studies investigating the effect of hydroxyurea on silent infarcts are lacking.
Hematopoietic stem cell transplant (HSCT) is currently the only curative therapy for SCD. In 2001, a multicenter investigation of HLA-identical sibling bone marrow transplantation (BMT) for pediatric patients with symptomatic SCD demonstrated the safety and efficacy of myeloablative BMT in this population Citation[113]. Of the 59 subjects who underwent BMT, 50 had stable allografts and were free of SCD complications at a median follow-up of 42.2 months (range: 11.8–115 months) following transplantation Citation[113]. Further follow-up of this cohort of subjects demonstrated a 5-year event-free survival of 85% and overall survival of 97% Citation[114]. However, because of an increase in neurologic events during transplant, especially in those subjects with prior history of ischemic stroke, this study established the need for specific supportive care guidelines for this population including seizure prophylaxis, strict hematologic goals (maintenance of hemoglobin between 9 and 11 g/dl and platelets at ≥50,000/µl), rapid correction of hypomagnesemia and strict control of blood pressure Citation[115]. Of the 29 subjects with stroke or CNS disease as their indication for BMT, 25 experienced stable engraftment with no further overt CNS events or progression of disease on MRI at a median of 3.2 years after transplant. Among the ten subjects with silent infarcts on baseline MRI prior to transplant, seven out of eight with follow-up MRIs performed a median of 1.7 years after transplant had smaller or stable lesions. In addition, no CNS disease developed following HSCT in ten subjects with prior normal MRI Citation[116]. A study in France of 87 pediatric and young adult subjects with SCD who received HLA-matched sibling BMTs using a variety of conditioning regimens between 1988 and 2004 showed variable effects on cerebrovascular stenosis (five resolved, 16 were unchanged and two progressed), persistence of vascular occlusions and worsening cortical atrophy in two subjects, but a significant reduction in arterial velocities in all 49 patients who were assessed Citation[117]. Of those with a history of stroke (n = 36), one subject experienced a TIA 10 days after transplantation and one died from intracranial hemorrhage 32 days after transplant Citation[117]. Overall, the effect on pre-existing CNS disease was favorable.
Historically, the frequency of HSCT in this population is limited by the number of HLA-matched sibling donors, availability of matched unrelated donors, high frequency of prior alloimmunization, resistance from families, lack of psychosocial support, high risk of acute toxicity during transplant and graft-versus-host disease Citation[118,119]. Advances in conditioning regimens Citation[117] and improved understanding of the therapeutic value of mixed chimerism in nonmalignant conditions Citation[120–122] may reduce the transplant-related toxicity and increase the frequency with which HSCTs are performed in this population. Currently, there is limited experience with cord blood transplants (CBT) in patients with SCD; however, this would increase the potential donor pool and allow for a greater degree of HLA mismatching. Several case reports and case series have shown that outcomes with regard to engraftment and graft-versus-host disease are better in those receiving related CBT compared with those receiving unrelated CBT Citation[123–129]. However, the CBT arm of SCURT was suspended after five out of eight subjects who received unrelated CBT in the setting of reduced-intensity conditioning experienced graft rejection by day 42 post-transplant Citation[130]. Experience with haploidentical or mismatched stem cell transplantation is even more limited than CBT; however, this also would increase the potential donor pool available Citation[131–133].
As the mechanism of stroke has been studied more closely, potential novel therapeutic targets have been identified. NO and sildenafil are being studied as therapeutic options for patients with PH Citation[134]. Although initial studies of sildenafil as potential treatment for PH in patients with SCD were promising, a randomized, double-blind, placebo-controlled study of sildenafil in adult SCD patients with PH was closed early due to increased frequency of admission due to vaso-occlusive events with no evidence of treatment effect Citation[134,135]. In mouse models, supplementation with arginine has been shown to decrease hemolytic rate and NO scavenging Citation[136]. Unfortunately, results of human studies remain inconclusive Citation[137,138]. Statins, which increase NO and decrease leukocyte adhesion, are also being studied Citation[139]. Senicapoc, a Gardos channel blocker that prevents dehydration of red blood cells, has been shown to increase hemoglobin and hematocrit in adult patients with SCD due to decreased hemolysis. However, a recent Phase III trial was closed early due to failure to achieve their primary end point of decreased vaso-occlusive events Citation[140]. Currently, the mainstay for acute treatment of stroke and prevention of secondary stroke in pediatric patients with SCD remains CTT.
Primary stroke prevention
As the utility of TCD as a reproducible, noninvasive and highly predictive method of prospectively identifying risk of stroke in patients with SCD became known, studies focused on the ability to prevent first stroke in pediatric patients with SCD. In 1998, Adams et al. published the results of STOP, which identified children with SCD-SS or SCD-Sβ0 as being at high risk for stroke based on blood flow velocity in the internal carotid artery or middle cerebral artery Citation[141]. Subjects with a TAMM velocity of greater than or equal to 200 cm/s (abnormal) were randomly assigned to CTT to maintain the hemoglobin S percentage at under 30% versus observation. Overall, 9.7% of the screened subjects had abnormal TCD results. Among subjects randomized to standard treatment, ten cerebral infarctions and one intracerebral hematoma occurred compared with only one infarction in the group receiving CTT representing a 90% reduction in the risk of stroke with prophylactic transfusion therapy Citation[141]. This led to premature closure by the Data and Safety Monitoring Board with the recommendation that all children with abnormal TCD be treated with CTT.
Current guidelines for the care of patients with sickle cell disease recommend annual screening with TCD in patients with SCD-SS or SCD-Sβ0 between the ages of 2 and 16 years. Velocity measurements ≥200 cm/s are considered abnormal and the TCD should be repeated within 1–2 weeks to confirm the finding. If the velocity remains abnormal, CTT is recommended. Velocities of ≥220 cm/s do not need to be confirmed. It is imperative to initiate CTT as early as possible in children with abnormal TCD readings to protect against stroke. In STOP, one subject developed a stroke while awaiting randomization and was withdrawn from the study Citation[141].
The recommendations for the timing of rescreening with TCD are based on the need to identify children who convert from a lower risk TCD to a higher risk TCD in a timely fashion so as to implement a proven prevention strategy as soon as possible in those who convert to abnormal velocities. In a follow-up study of children who were screened to participate in STOP whose initial TCD velocities were not abnormal at the time of screening, 9% converted to abnormal velocities during the 22-month follow-up period. Conversion to an abnormal TCD was highest in younger children and in those with a ‘high conditional’ TAMM velocity (185–199 cm/s) at first exam Citation[142]. shows the authors’ current screening protocol for children with SCD. For high conditional TCD, the study is repeated within 6 weeks for children under 10 years old and within 3 months for children 10 years and older. Children with low conditional TCD (170–184 cm/s) have their TCD repeated within 3–6 months, while those with a normal TCD (<170 cm/s) without other risk factors have their TCD repeated annually Citation[143].
TCD has been shown to be feasible in infants with SCD as young as 7 months; however, infants have a lower mean velocity than children ≥2 years of age. In 192 infants enrolled in the BABY HUG study, none had a TAMM velocity ≥200 cm/s Citation[144]. It is unclear if a new cutoff can be established to identify those infants at highest risk to develop stroke or abnormal TCD in the future, potentially allowing for even earlier intervention. Interestingly, in the BABY HUG study, which randomized young infants from 9 to 18 months of age with SCD-SS or SCD-Sβ0 to hydroxyurea or placebo for a 2-year period, the average increase in TAMM velocity was significantly lower in those subjects who had received hydroxyurea. Data regarding the impact this had on future strokes and silent infarcts is still pending Citation[145,146].
The impact of TCD screening and treatment programs on the prevalence of stroke in children with SCD-SS appears to be profound. Studies using hospital-based prevalence data from California (USA), as well as several large single institutions, have demonstrated the effectiveness of the implementation of routine TCD screening on reduction of incidence of stroke in pediatric patients with SCD on a population level Citation[3,147–149]. A recent retrospective analysis of national inpatient admissions data demonstrated a 45% decrease in incidence rates for stroke in pediatric patients with SCD from 1999 to 2009 (the period following STOP results), as compared with 1993 to 1998 (the period prior to STOP results) Citation[150].
Conventional TCD does not rely on direct visualization of the vessels and is considered a nonimaging technique. In contrast, imaging TCD sonography (TCDi), relies on direct visualization of the vessels, which can improve confidence in anatomic location. TCDi can be performed with and without angle correction. With angle correction, the operator can correct for the difference in angle between the course of the artery and the ultrasound beam to provide angle-corrected measurements of velocity Citation[151,152]. To adhere as closely as possible to the original STOP criteria, which used nonimaging TCD, angle correction may not be recommended as there are no large studies establishing appropriate thresholds for intervention Citation[153]. Additionally, studies have shown that with rigorous protocols, TCDi without angle correction may be equivalent to standard TCD Citation[154,155] However, some studies suggest that TCDi velocities without angle correction can be 10–15% lower than standard nonimaging TCD Citation[51,156]; therefore, a lower cutoff of 185 cm/s is often used to define an abnormal study Citation[157]. However, as no trials have compared TCD with TCDi screening techniques for primary stroke prevention, conventional TCD remains the standard of care.
MRI techniques that include T1, T2, FLAIR, diffusion imaging and susceptibility imaging and MRA protocols that include 3D time-of-flight techniques and reconstruction provide accurate, noninvasive, high-quality images of the cerebral vasculature and evidence of silent infarcts Citation[158,159]. As increasing evidence demonstrates the importance of identifying silent infarcts due to their impact upon neurocognitive abilities, some programs have recommended obtaining a screening brain MRI and MRA at least once for school-aged children with SCD-SS Citation[16]. Brain MRI/MRA is also indicated for children with abnormal or conditional TCD velocities, evidence of neurocognitive difficulties or other high-risk features. Follow-up studies are recommended for children with evidence of prior silent infarct or vasculopathy on prior MRI/MRA due to the risk of progressive disease. Functional imaging techniques, such as PET or single-photon emission computed tomography, are being studied in pediatric patients with SCD as they may able to detect perfusion deficits prior to the development of silent infarcts Citation[160]. Additionally, blood oxygen level-dependent MRI sequences, which can differentiate between deoxyhemoglobin and oxyhemoglobin, are being studied for their ability to detect even minor imbalances in oxygen consumption and delivery Citation[161]. These new techniques may allow providers to identify prospectively those patients at the highest risk of developing silent infarcts prior to any impact on neurocognitive outcomes.
Discontinuation of primary stroke prevention strategies
In the STOP 2 trial, children with abnormal TCD velocities without significant vasculopathy on MRA who had received at least 30 months of transfusion therapy with reversion to normal TCD were randomized to either continue with monthly transfusions or discontinue transfusion therapy. Of the 41 subjects removed from transfusion therapy, 14 reverted to abnormal TAMM velocities and two developed overt strokes. Neither of these end points was observed in the group who continued transfusion therapy Citation[162]. As such, the current recommendation is to continue lifelong transfusion therapy. However, given the downsides to transfusion, alternatives are being actively sought.
Several case series and retrospective reviews have found a benefit of hydroxyurea in lowering TCD velocities in patients with abnormal TCD velocities, but no history of stroke Citation[163,164]. As mentioned previously, in the BABY HUG study, TCD velocities increased at a slower rate in infants who received hydroxyurea compared with those who received standard care Citation[145]. A prospective Phase II study of hydroxyurea in 37 children with SCD with at least one TCD velocity ≥140 cm/s demonstrated a significant reduction in TCD velocity after a mean of 10 ± 5 months Citation[165]. Children with the highest velocities at baseline saw the greatest improvement in their TCD velocities. Five out of six subjects with TCD velocities in the abnormal range whose families declined transfusion therapy experienced a reduction in their TCD velocity into the conditional range on hydroxyurea, and 14 out of 15 subjects with conditional baseline TCD velocities showed improvement to the normal range by the end of follow-up (median: 24 months) Citation[165]. This preliminary data set the stage for the TWiTCH study. TWiTCH is an ongoing, Phase III noninferiority trial of daily hydroxyurea versus continued monthly transfusion therapy to prevent progression of TCD abnormalities in patients with SCD who have had at least 1 year of transfusion therapy for primary stroke prevention. Secondary outcomes include evaluation of stroke events in both arms of the study. This study is currently ongoing and it is not expected to complete until December 2016 (ClinicalTrials.gov identifier: NCT01425307) Citation[201].
Sickle cell trait
Although most patients with sickle cell trait are asymptomatic, individuals with sickle cell trait are at increased risk of renal papillary necrosis, hematuria, renal medullary carcinoma, hyposthenuria, splenic infarction, exertional rhabdomyolysis and exercise-related sudden death Citation[166]. Interestingly, in a study of 21 pediatric patients with sickle cell trait, two were classified as having mild abnormalities on MRI and four were identified as having vascular tortuosity. When compared with a population of patients without sickle cell trait, the subjects with sickle cell trait were significantly more likely to have arterial tortuosity and it was correlated with hemoglobin S percent at the time of the evaluation Citation[167]. This was considered mild vasculopathy and has not been replicated. Thus far, no definitive evidence links sickle cell trait with stroke risk, but larger studies are warranted to examine the association.
Expert commentary
Strokes cause significant morbidity and mortality in pediatric patients with SCD. Overt stroke occurs in approximately 10% of unscreened pediatric patients. Fortunately, TCD can identify children at high risk of developing ischemic stroke and CTT is very effective at preventing stroke in the high-risk group. This primary prevention strategy represents a significant advance in the care of patients with SCD. The rate of overt stroke in children with SCD has declined greatly since TCD screening and treatment programs have been more widely adopted. However, CTT must be continued indefinitely as the risk of stroke rises with discontinuation. Ongoing studies are assessing if alternative treatments, such as hydroxyurea, can be utilized.
Patients with SCD who have had an ischemic stroke have a very high risk of recurrence without prophylactic therapy. CTT as secondary stroke prevention following an initial stroke has been shown to be effective, but must also be continued indefinitely. CTT in pediatric patients requires adequate venous access and patient and parent compliance. Chronic transfusions may also result in alloimmunization, infection and iron overload, so alternative therapies are needed. Unfortunately, a trial of hydroxyurea for secondary stroke prevention was stopped early due to an increased risk of stroke without a benefit of superior reduction in iron burden. BMT with a matched sibling donor or matched unrelated donor is an alternative therapy for lasting protection from further CNS events.
Silent cerebral infarcts occur in approximately a third of children with SCD-SS. These infarcts are not benign, as the nomenclature suggests, but rather are associated with neurocognitive abnormalities and an increased risk of developing overt stroke. The optimal treatment of silent infarcts remains unknown, but ongoing research is addressing this problem.
Five-year view
The results of several large, ongoing studies in SCD will likely transform the treatment of this disease in pediatric patients over the next 5 years. The BABY HUG study has already demonstrated that TCD velocities are lower in subjects who received hydroxyurea compared with placebo. As these subjects age, it should become more clear whether these lower TCD values translate into a lower risk of stroke. Information on the role of hydroxyurea in primary stroke prevention following a period of CTT will be available with the TWiTCH trial analysis.
In the next 5 years, therapies to prevent silent infarcts or halt progression of CNS injury in patients who already have silent infarcts will be tested. The ongoing SIT trial will determine whether CTT is a suitable therapy for the prevention of progression of silent infarcts and neurological injury. Hydroxyurea and other treatments will probably also be tested. Once a therapeutic option is available, recommendations for screening with brain MRI/MRA will be made. Hopefully, as more patients are identified, additional educational and therapeutic services will become available to mitigate the sequelae of silent infarcts. Newer MRI techniques such as blood oxygen-level-dependent imaging and other CNS imaging techniques, such as PET, will potentially be able to identify patients at risk of CNS injury perhaps allowing for earlier intervention.
Novel targeted therapies for prevention and treatment of stroke should become available as the pathways and mechanisms of damage that contribute to cerebral infarcts are elucidated. As BMT in this population becomes safer and more donor sources become available, increased numbers of patients will receive this treatment, perhaps earlier in the course of the disease prior to the development of significant organ toxicity. Gene therapy trials will also offer insight into this curative treatment option.
Table 1. Risk factors for cerebrovascular events.
Table 2. Rates of stroke recurrence with hydroxyurea.
Key issues
• Ischemic, hemorrhagic and ‘silent’ infarcts cause significant morbidity and mortality in pediatric patients with sickle cell disease (SCD)-SS.
• Clinical risk factors associated with increased risk of ischemic stroke include prior transient ischemic attack, prior silent infarct, frequent or recent acute chest syndrome, increased systolic blood pressure, nocturnal hypoxemia and abnormal transcranial Doppler (TCD).
• Screening children with SCD-SS with TCD ultrasound is an effective method to identify patients at highest risk of developing ischemic strokes and then initiate a primary prevention strategy. Since the implementation of TCD screening, the rate of first stroke in patients with SCD has decreased.
• Brain MRI/magnetic resonance angiography evaluations for evidence of ‘silent’ infarcts and/or vasculopathy should be considered for SCD-SS patients with conditional or abnormal TCD, family history of stroke in SCD or evidence of neurocognitive abnormalities.
• The mainstay of primary and secondary stroke prevention is chronic transfusion therapy.
• Currently, research supports indefinitely continuing chronic transfusion therapy as no studies have demonstrated a safe alternative.
• Bone marrow transplant is an alternative for those patients with SCD and a high risk of stroke who have a matched sibling bone marrow donor or significant sequelae from SCD.
References
- Ohene-Frempong K, Weiner SJ, Sleeper LA et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 91(1), 288–294 (1998).
- Pegelow CH, Macklin EA, Moser FG et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood 99(8), 3014–3018 (2002).
- Bernaudin F, Verlhac S, Arnaud C et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 117(4), 1130–1140; quiz 1436 (2011).
- Casella JF, King AA, Barton B et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr. Hematol. Oncol. 27(2), 69–89 (2010).
- Wang WC, Pavlakis SG, Helton KJ et al.; BABY HUG Investigators. MRI abnormalities of the brain in one-year-old children with sickle cell anemia. Pediatr. Blood Cancer 51(5), 643–646 (2008).
- Kwiatkowski JL, Zimmerman RA, Pollock AN et al. Silent infarcts in young children with sickle cell disease. Br. J. Haematol. 146(3), 300–305 (2009).
- Bernaudin F, Verlhac S, Fréard F et al. Multicenter prospective study of children with sickle cell disease: radiographic and psychometric correlation. J. Child Neurol. 15(5), 333–343 (2000).
- Armstrong FD, Thompson RJ Jr, Wang W et al.; Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease. Pediatrics 97(6 Pt 1), 864–870 (1996).
- Schatz J, Brown RT, Pascual JM, Hsu L, DeBaun MR. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology 56(8), 1109–1111 (2001).
- Schatz J, Buzan R. Decreased corpus callosum size in sickle cell disease: relationship with cerebral infarcts and cognitive functioning. J. Int. Neuropsychol. Soc. 12(1), 24–33 (2006).
- Schatz J, Finke RL, Kellett JM, Kramer JH. Cognitive functioning in children with sickle cell disease: a meta-analysis. J. Pediatr. Psychol. 27(8), 739–748 (2002).
- Schatz J, White DA, Moinuddin A, Armstrong M, DeBaun MR. Lesion burden and cognitive morbidity in children with sickle cell disease. J. Child Neurol. 17(12), 891–895 (2002).
- Brown RT, Davis PC, Lambert R, Hsu L, Hopkins K, Eckman J. Neurocognitive functioning and magnetic resonance imaging in children with sickle cell disease. J. Pediatr. Psychol. 25(7), 503–513 (2000).
- Vichinsky EP, Neumayr LD, Gold JI et al.; Neuropsychological Dysfunction and Neuroimaging Adult Sickle Cell Anemia Study Group. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA 303(18), 1823–1831 (2010).
- Kugler S, Anderson B, Cross D et al. Abnormal cranial magnetic resonance imaging scans in sickle-cell disease. Neurological correlates and clinical implications. Arch. Neurol. 50(6), 629–635 (1993).
- DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 119(20), 4587–4596 (2012).
- Adams RJ, Nichols FT 3rd, Aaslid R et al. Cerebral vessel stenosis in sickle cell disease: criteria for detection by transcranial Doppler. Am. J. Pediatr. Hematol. Oncol. 12(3), 277–282 (1990).
- Platt OS. Preventing stroke in sickle cell anemia. N. Engl. J. Med. 353(26), 2743–2745 (2005).
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr. Opin. Hematol. 9(2), 101–106 (2002).
- Joneckis CC, Ackley RL, Orringer EP, Wayner EA, Parise LV. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood 82(12), 3548–3555 (1993).
- Swerlick RA, Eckman JR, Kumar A, Jeitler M, Wick TM. Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood 82(6), 1891–1899 (1993).
- Natarajan M, Udden MM, McIntire LV. Adhesion of sickle red blood cells and damage to interleukin-1 beta stimulated endothelial cells under flow in vitro. Blood 87(11), 4845–4852 (1996).
- Embury SH, Matsui NM, Ramanujam S et al. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood 104(10), 3378–3385 (2004).
- Hillery CA, Scott JP, Du MC. The carboxy-terminal cell-binding domain of thrombospondin is essential for sickle red blood cell adhesion. Blood 94(1), 302–309 (1999).
- Gee BE, Platt OS. Sickle reticulocytes adhere to VCAM-1. Blood 85(1), 268–274 (1995).
- Kaul DK, Nagel RL, Chen D, Tsai HM. Sickle erythrocyte-endothelial interactions in microcirculation: the role of von Willebrand factor and implications for vasoocclusion. Blood 81(9), 2429–2438 (1993).
- Wautier JL, Pintigny D, Wautier MP et al. Fibrinogen, a modulator of erythrocyte adhesion to vascular endothelium. J. Lab. Clin. Med. 101(6), 911–920 (1983).
- Ataga KI, Moore CG, Hillery CA et al. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica 93(1), 20–26 (2008).
- Okpala I. Leukocyte adhesion and the pathophysiology of sickle cell disease. Curr. Opin. Hematol. 13(1), 40–44 (2006).
- Frenette PS. Sickle cell vasoocclusion: heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation 11(2), 167–177 (2004).
- Morris CR, Kato GJ, Poljakovic M et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 294(1), 81–90 (2005).
- Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology Am. Soc. Hematol. Educ. Program 177–185 (2008).
- Morris CR, Suh JH, Hagar W et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 111(1), 402–410 (2008).
- Lezcano NE, Odo N, Kutlar A, Brambilla D, Adams RJ. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke. 37(6), 1424–1426 (2006).
- Adams RJ, Kutlar A, McKie V et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am. J. Hematol. 45(4), 279–282 (1994).
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 21(1), 37–47 (2007).
- O’Driscoll S, Height SE, Dick MC, Rees DC. Serum lactate dehydrogenase activity as a biomarker in children with sickle cell disease. Br. J. Haematol. 140(2), 206–209 (2008).
- Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am. J. Med. 65(3), 461–471 (1978).
- Brandão RA, de Carvalho GT, Reis BL, Bahia E, de Souza AA. Intracranial aneurysms in sickle cell patients: report of 2 cases and review of the literature. Surg. Neurol. 72(3), 296–299; discussion 299 (2009).
- Moritani T, Numaguchi Y, Lemer NB et al. Sickle cell cerebrovascular disease: usual and unusual findings on MR imaging and MR angiography. Clin. Imaging 28(3), 173–186 (2004).
- Dobson SR, Holden KR, Nietert PJ et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood 99(9), 3144–3150 (2002).
- Pegelow CH, Wang W, Granger S et al.; STOP Trial. Silent infarcts in children with sickle cell anemia and abnormal cerebral artery velocity. Arch. Neurol. 58(12), 2017–2021 (2001).
- Waterston JA, Brown MM, Butler P, Swash M. Small deep cerebral infarcts associated with occlusive internal carotid artery disease. A hemodynamic phenomenon? Arch. Neurol. 47(9), 953–957 (1990).
- Dowling MM, Quinn CT, Rogers ZR, Buchanan GR. Acute silent cerebral infarction in children with sickle cell anemia. Pediatr. Blood Cancer 54(3), 461–464 (2010).
- Wang WC, Gallagher DM, Pegelow CH et al. Multicenter comparison of magnetic resonance imaging and transcranial Doppler ultrasonography in the evaluation of the central nervous system in children with sickle cell disease. J. Pediatr. Hematol. Oncol. 22(4), 335–339 (2000).
- Hulbert ML, McKinstry RC, Lacey JL et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood 117(3), 772–779 (2011).
- Miller ST, Macklin EA, Pegelow CH et al.; Cooperative Study of Sickle Cell Disease. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J. Pediatr. 139(3), 385–390 (2001).
- Kirkham FJ, Hewes DK, Prengler M, Wade A, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet 357(9269), 1656–1659 (2001).
- Goldstein NA, Keller R, Rey K et al. Sleep-disordered breathing and transcranial Dopplers in sickle cell disease. Arch. Otolaryngol. Head Neck Surg. 137(12), 1263–1268 (2011).
- Tripathi A, Jerrell JM, Stallworth JR. Cost–effectiveness of adenotonsillectomy in reducing obstructive sleep apnea, cerebrovascular ischemia, vaso-occlusive pain, and ACS episodes in pediatric sickle cell disease. Ann. Hematol. 90(2), 145–150 (2011).
- Bulas DI, Jones A, Seibert JJ, Driscoll C, O’Donnell R, Adams RJ. Transcranial Doppler (TCD) screening for stroke prevention in sickle cell anemia: pitfalls in technique variation. Pediatr. Radiol. 30(11), 733–738 (2000).
- Adams RJ, Nichols FT, Figueroa R, McKie V, Lott T. Transcranial Doppler correlation with cerebral angiography in sickle cell disease. Stroke. 23(8), 1073–1077 (1992).
- Adams RJ, McKie VC, Carl EM et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann. Neurol. 42(5), 699–704 (1997).
- Abboud MR, Cure J, Granger S et al.; STOP study. Magnetic resonance angiography in children with sickle cell disease and abnormal transcranial Doppler ultrasonography findings enrolled in the STOP study. Blood 103(7), 2822–2826 (2004).
- Gorman MJ, Nyström K, Carbonella J, Pearson H. Submandibular TCD approach detects post-bulb ICA stenosis in children with sickle cell anemia. Neurology 73(5), 362–365 (2009).
- Deane CR, Goss D, Bartram J et al. Extracranial internal carotid arterial disease in children with sickle cell anemia. Haematologica 95(8), 1287–1292 (2010).
- Deane CR, Goss D, O’Driscoll S et al. Transcranial Doppler scanning and the assessment of stroke risk in children with HbSC [corrected] disease. Arch. Dis. Child. 93(2), 138–141 (2008).
- Valadi N, Silva GS, Bowman LS et al. Transcranial Doppler ultrasonography in adults with sickle cell disease. Neurology 67(4), 572–574 (2006).
- Sampaio Silva G, Vicari P, Figueiredo MS, Filho AC, Valadi N, Massaro AR. Transcranial Doppler in adult patients with sickle cell disease. Cerebrovasc. Dis. 21(1–2), 38–41 (2006).
- Silva GS, Vicari P, Figueiredo MS, Carrete H Jr, Idagawa MH, Massaro AR. Brain magnetic resonance imaging abnormalities in adult patients with sickle cell disease: correlation with transcranial Doppler findings. Stroke 40(7), 2408–2412 (2009).
- Strouse JJ, Hulbert ML, DeBaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics 118(5), 1916–1924 (2006).
- Kinney TR, Sleeper LA, Wang WC et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics 103(3), 640–645 (1999).
- DeBaun MR, Sarnaik SA, Rodeghier MJ et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 119(16), 3684–3690 (2012).
- Thangarajh M, Yang G, Fuchs D et al. Magnetic resonance angiography-defined intracranial vasculopathy is associated with silent cerebral infarcts and glucose-6-phosphate dehydrogenase mutation in children with sickle cell anaemia. Br. J. Haematol. 159(3), 352–359 (2012).
- Bernaudin F, Verlhac S, Chevret S et al. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood 112(10), 4314–4317 (2008).
- Rees DC, Lambert C, Cooper E et al. Glucose 6 phosphate dehydrogenase deficiency is not associated with cerebrovascular disease in children with sickle cell anemia. Blood 114(3), 742–743; author reply 743 (2009).
- Miller ST, Milton J, Steinberg MH. G6PD deficiency and stroke in the CSSCD. Am. J. Hematol. 86(3), 331 (2011).
- Hsu LL, Miller ST, Wright E et al.; Stroke Prevention Trial (STOP) and the Cooperative Study of Sickle Cell Disease (CSSCD). Alpha thalassemia is associated with decreased risk of abnormal transcranial Doppler ultrasonography in children with sickle cell anemia. J. Pediatr. Hematol. Oncol. 25(8), 622–628 (2003).
- Gill FM, Sleeper LA, Weiner SJ et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 86(2), 776–783 (1995).
- Kwiatkowski JL, Hunter JV, Smith-Whitley K, Katz ML, Shults J, Ohene-Frempong K. Transcranial Doppler ultrasonography in siblings with sickle cell disease. Br. J. Haematol. 121(6), 932–937 (2003).
- Driscoll MC, Hurlet A, Styles L et al. Stroke risk in siblings with sickle cell anemia. Blood 101(6), 2401–2404 (2003).
- Hoppe C, Klitz W, Cheng S et al.; CSSCD Investigators. Gene interactions and stroke risk in children with sickle cell anemia. Blood 103(6), 2391–2396 (2004).
- Hoppe C, Klitz W, D’Harlingue K et al.; Stroke Prevention Trial in Sickle Cell Anemia (STOP) Investigators. Confirmation of an association between the TNF(-308) promoter polymorphism and stroke risk in children with sickle cell anemia. Stroke 38(8), 2241–2246 (2007).
- Styles LA, Hoppe C, Klitz W, Vichinsky E, Lubin B, Trachtenberg E. Evidence for HLA-related susceptibility for stroke in children with sickle cell disease. Blood 95(11), 3562–3567 (2000).
- Hoppe C, Klitz W, Noble J, Vigil L, Vichinsky E, Styles L. Distinct HLA associations by stroke subtype in children with sickle cell anemia. Blood 101(7), 2865–2869 (2003).
- Taylor JG 6th, Tang DC, Savage SA et al. Variants in the VCAM1 gene and risk for symptomatic stroke in sickle cell disease. Blood 100(13), 4303–4309 (2002).
- Flanagan JM, Frohlich DM, Howard TA et al. Genetic predictors for stroke in children with sickle cell anemia. Blood 117(24), 6681–6684 (2011).
- Adams GT, Snieder H, McKie VC et al. Genetic risk factors for cerebrovascular disease in children with sickle cell disease: design of a case–control association study and genomewide screen. BMC Med. Genet. 4, 6 (2003).
- Ohene-Frempong K. Stroke in sickle cell disease: demographic, clinical, and therapeutic considerations. Semin. Hematol. 28(3), 213–219 (1991).
- Hulbert ML, Scothorn DJ, Panepinto JA et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J. Pediatr. 149(5), 710–712 (2006).
- Hariman LM, Griffith ER, Hurtig AL, Keehn MT. Functional outcomes of children with sickle-cell disease affected by stroke. Arch. Phys. Med. Rehabil. 72(7), 498–502 (1991).
- King AA, DeBaun MR, White DA. Need for cognitive rehabilitation for children with sickle cell disease and strokes. Expert Rev. Neurother. 8(2), 291–296 (2008).
- King AA, White DA, McKinstry RC, Noetzel M, Debaun MR. A pilot randomized education rehabilitation trial is feasible in sickle cell and strokes. Neurology 68(23), 2008–2011 (2007).
- Steen RG, Reddick WE, Mulhern RK et al. Quantitative MRI of the brain in children with sickle cell disease reveals abnormalities unseen by conventional MRI. J. Magn. Reson. Imaging 8(3), 535–543 (1998).
- McQuaker IG, Jaspan T, McConachie NS, Dolan G. Coil embolization of cerebral aneurysms in patients with sickling disorders. Br. J. Haematol. 106(2), 388–390 (1999).
- Kirkham FJ, DeBaun MR. Stroke in children with sickle cell disease. Curr. Treat. Options Neurol. 6(5), 357–375 (2004).
- Sarnaik S, Soorya D, Kim J, Ravindranath Y, Lusher J. Periodic transfusions for sickle cell anemia and CNS infarction. Am. J. Dis. Child. 133(12), 1254–1257 (1979).
- Scothorn DJ, Price C, Schwartz D et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J. Pediatr. 140(3), 348–354 (2002).
- Pegelow CH, Adams RJ, McKie V et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J. Pediatr. 126(6), 896–899 (1995).
- Kwiatkowski JL, Yim E, Miller S, Adams RJ; STOP 2 Study Investigators. Effect of transfusion therapy on transcranial Doppler ultrasonography velocities in children with sickle cell disease. Pediatr. Blood Cancer 56(5), 777–782 (2011).
- Bishop S, Matheus MG, Abboud MR et al. Effect of chronic transfusion therapy on progression of neurovascular pathology in pediatric patients with sickle cell anemia. Blood Cells Mol. Dis. 47(2), 125–128 (2011).
- Brousse V, Hertz-Pannier L, Consigny Y et al. Does regular blood transfusion prevent progression of cerebrovascular lesions in children with sickle cell disease? Ann. Hematol. 88(8), 785–788 (2009).
- Bader-Meunier B, Verlhac S, Elmaleh-Bergès M et al. Effect of transfusion therapy on cerebral vasculopathy in children with sickle-cell anemia. Haematologica 94(1), 123–126 (2009).
- Wang WC, Kovnar EH, Tonkin IL et al. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J. Pediatr. 118(3), 377–382 (1991).
- Venkateswaran L, Teruya J, Bustillos C, Mahoney D Jr, Mueller BU. Red cell exchange does not appear to increase the rate of allo- and auto-immunization in chronically transfused children with sickle cell disease. Pediatr. Blood Cancer 57(2), 294–296 (2011).
- Vichinsky EP. Current issues with blood transfusions in sickle cell disease. Semin. Hematol. 38(1 Suppl. 1), 14–22 (2001).
- Fung EB, Harmatz P, Milet M et al.; Multi-Center Study of Iron Overload Research Group. Morbidity and mortality in chronically transfused subjects with thalassemia and sickle cell disease: a report from the multi-center study of iron overload. Am. J. Hematol. 82(4), 255–265 (2007).
- Kim HC, Dugan NP, Silber JH et al. Erythrocytapheresis therapy to reduce iron overload in chronically transfused patients with sickle cell disease. Blood 83(4), 1136–1142 (1994).
- Adams DM, Schultz WH, Ware RE, Kinney TR. Erythrocytapheresis can reduce iron overload and prevent the need for chelation therapy in chronically transfused pediatric patients. J. Pediatr. Hematol. Oncol. 18(1), 46–50 (1996).
- Hilliard LM, Williams BF, Lounsbury AE, Howard TH. Erythrocytapheresis limits iron accumulation in chronically transfused sickle cell patients. Am. J. Hematol. 59(1), 28–35 (1998).
- Singer ST, Quirolo K, Nishi K, Hackney-Stephens E, Evans C, Vichinsky EP. Erythrocytapheresis for chronically transfused children with sickle cell disease: an effective method for maintaining a low hemoglobin S level and reducing iron overload. J. Clin. Apher. 14(3), 122–125 (1999).
- Cohen AR, Martin MB, Silber JH, Kim HC, Ohene-Frempong K, Schwartz E. A modified transfusion program for prevention of stroke in sickle cell disease. Blood 79(7), 1657–1661 (1992).
- Miller ST, Jensen D, Rao SP. Less intensive long-term transfusion therapy for sickle cell anemia and cerebrovascular accident. J. Pediatr. 120(1), 54–57 (1992).
- Ali SB, Moosang M, King L, Knight-Madden J, Reid M. Stroke recurrence in children with sickle cell disease treated with hydroxyurea following first clinical stroke. Am. J. Hematol. 86(10), 846–850 (2011).
- Ware RE, Zimmerman SA, Sylvestre PB et al. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. J. Pediatr. 145(3), 346–352 (2004).
- Ware RE, Helms RW; SWiTCH Investigators. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood 119(17), 3925–3932 (2012).
- Ware RE, Zimmerman SA, Schultz WH. Hydroxyurea as an alternative to blood transfusions for the prevention of recurrent stroke in children with sickle cell disease. Blood 94(9), 3022–3026 (1999).
- Sumoza A, de Bisotti R, Sumoza D, Fairbanks V. Hydroxyurea (HU) for prevention of recurrent stroke in sickle cell anemia (SCA). Am. J. Hematol. 71(3), 161–165 (2002).
- Lefèvre N, Dufour D, Gulbis B, Lê PQ, Heijmans C, Ferster A. Use of hydroxyurea in prevention of stroke in children with sickle cell disease. Blood 111(2), 963–964; author reply 964 (2008).
- Fryer RH, Anderson RC, Chiriboga CA, Feldstein NA. Sickle cell anemia with moyamoya disease: outcomes after EDAS procedure. Pediatr. Neurol. 29(2), 124–130 (2003).
- Vernet O, Montes JL, O’Gorman AM, Baruchel S, Farmer JP. Encephaloduroarterio-synangiosis in a child with sickle cell anemia and moyamoya disease. Pediatr. Neurol. 14(3), 226–230 (1996).
- Abboud MR, Yim E, Musallam KM, Adams RJ; STOP II Study Investigators. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 118(4), 894–898 (2011).
- Walters MC, Patience M, Leisenring W et al.; Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell Disease. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 7(12), 665–673 (2001).
- Panepinto JA, Walters MC, Carreras J et al.; Non-Malignant Marrow Disorders Working Committee, Center for International Blood and Marrow Transplant Research. Matched-related donor transplantation for sickle cell disease: report from the Center for International Blood and Transplant Research. Br. J. Haematol. 137(5), 479–485 (2007).
- Walters MC, Sullivan KM, Bernaudin F et al. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood 85(4), 879–884 (1995).
- Walters MC, Hardy K, Edwards S et al.; Multicenter Study of Bone Marrow Transplantation for Sickle Cell Disease. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 16(2), 263–272 (2010).
- Bernaudin F, Socie G, Kuentz M et al.; SFGM-TC. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 110(7), 2749–2756 (2007).
- Walters MC, Patience M, Leisenring W et al. Barriers to bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 2(2), 100–104 (1996).
- Kodish E, Lantos J, Stocking C, Singer PA, Siegler M, Johnson FL. Bone marrow transplantation for sickle cell disease. A study of parents’ decisions. N. Engl. J. Med. 325(19), 1349–1353 (1991).
- Wu CJ, Gladwin M, Tisdale J et al. Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. Br. J. Haematol. 139(3), 504–507 (2007).
- Krishnamurti L, Kharbanda S, Biernacki MA et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 14(11), 1270–1278 (2008).
- Horwitz ME, Spasojevic I, Morris A et al. Fludarabine-based nonmyeloablative stem cell transplantation for sickle cell disease with and without renal failure: clinical outcome and pharmacokinetics. Biol. Blood Marrow Transplant. 13(12), 1422–1426 (2007).
- Brichard B, Vermylen C, Ninane J, Cornu G. Persistence of fetal hemoglobin production after successful transplantation of cord blood stem cells in a patient with sickle cell anemia. J. Pediatr. 128(2), 241–243 (1996).
- Miniero R, Rocha V, Saracco P et al. Cord blood transplantation (CBT) in hemoglobinopathies. Eurocord. Bone Marrow Transplant. 22(Suppl. 1), S78–S79 (1998).
- Gore L, Lane PA, Quinones RR, Giller RH. Successful cord blood transplantation for sickle cell anemia from a sibling who is human leukocyte antigen-identical: implications for comprehensive care. J. Pediatr. Hematol. Oncol. 22(5), 437–440 (2000).
- Locatelli F, Rocha V, Reed W et al.; Eurocord Transplant Group. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 101(6), 2137–2143 (2003).
- Adamkiewicz TV, Szabolcs P, Haight A et al. Unrelated cord blood transplantation in children with sickle cell disease: review of four-center experience. Pediatr. Transplant. 11(6), 641–644 (2007).
- Mazur M, Kurtzberg J, Halperin E, Ciocci G, Szabolcs P. Transplantation of a child with sickle cell anemia with an unrelated cord blood unit after reduced intensity conditioning. J. Pediatr. Hematol. Oncol. 28(12), 840–844 (2006).
- Sauter C, Rausen AR, Barker JN. Successful unrelated donor cord blood transplantation for adult sickle cell disease and Hodgkin lymphoma. Bone Marrow Transplant. 45(7), 1252 (2010).
- Kamani NR, Walters MC, Carter S et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the Phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol. Blood Marrow Transplant. 18(8), 1265–1272 (2012).
- Raj A, Bertolone S, Cheerva A. Successful treatment of refractory autoimmune hemolytic anemia with monthly rituximab following nonmyeloablative stem cell transplantation for sickle cell disease. J. Pediatr. Hematol. Oncol. 26(5), 312–314 (2004).
- Brodsky RA, Luznik L, Bolaños-Meade J, Leffell MS, Jones RJ, Fuchs EJ. Reduced intensity HLA-haploidentical BMT with post transplantation cyclophosphamide in nonmalignant hematologic diseases. Bone Marrow Transplant. 42(8), 523–527 (2008).
- Bolaños-Meade J, Fuchs EJ, Luznik L et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 120(22), 4285–4291 (2012).
- Machado RF, Martyr S, Kato GJ et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br. J. Haematol. 130(3), 445–453 (2005).
- Machado RF, Barst RJ, Yovetich NA et al.; Walk-PHaSST Investigators and Patients. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 118(4), 855–864 (2011).
- Kaul DK, Zhang X, Dasgupta T, Fabry ME. Arginine therapy of transgenic-knockout sickle mice improves microvascular function by reducing non-nitric oxide vasodilators, hemolysis, and oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 295(1), H39–H47 (2008).
- Morris CR, Kuypers FA, Larkin S et al. Arginine therapy: a novel strategy to induce nitric oxide production in sickle cell disease. Br. J. Haematol. 111(2), 498–500 (2000).
- Kato GJ. Novel small molecule therapeutics for sickle cell disease: nitric oxide, carbon monoxide, nitrite, and apolipoprotein A-I. Hematology Am. Soc. Hematol. Educ. Program 186–192 (2008).
- Hoppe C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br. J. Haematol. 153(5), 655–663 (2011).
- Ataga KI, Reid M, Ballas SK et al.; ICA-17043-10 Study Investigators. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a Phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br. J. Haematol. 153(1), 92–104 (2011).
- Adams RJ, McKie VC, Hsu L et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 339(1), 5–11 (1998).
- Adams RJ, Brambilla DJ, Granger S et al.; STOP Study. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 103(10), 3689–3694 (2004).
- Nichols FT, Jones AM, Adams RJ. Stroke prevention in sickle cell disease (STOP) study guidelines for transcranial Doppler testing. J. Neuroimaging 11(4), 354–362 (2001).
- Pavlakis SG, Rees RC, Huang X et al.; BABY HUG Investigators. Transcranial doppler ultrasonography (TCD) in infants with sickle cell anemia: baseline data from the BABY HUG trial. Pediatr. Blood Cancer 54(2), 256–259 (2010).
- Wang WC, Ware RE, Miller ST et al.; BABY HUG Investigators. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 377(9778), 1663–1672 (2011).
- Lebensburger JD, Miller ST, Howard TH et al.; BABY HUG Investigators. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatr. Blood Cancer 59(4), 675–678 (2012).
- Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood 104(2), 336–339 (2004).
- McCarville MB, Goodin GS, Fortner G et al. Evaluation of a comprehensive transcranial doppler screening program for children with sickle cell anemia. Pediatr. Blood Cancer 50(4), 818–821 (2008).
- Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial Doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. J. Pediatr. 157(3), 479–484 (2010).
- McCavit TL, Gilbert M, Buchanan GR. Prophylactic penicillin after 5 years of age in patients with sickle cell disease: A survey of sickle cell disease experts. Pediatr. Blood Cancer 60(6), 935–939 (2012).
- Krejza J, Mariak Z, Melhem ER, Bert RJ. A guide to the identification of major cerebral arteries with transcranial color Doppler sonography. AJR. Am. J. Roentgenol. 174(5), 1297–1303 (2000).
- Bazzocchi M, Quaia E, Zuiani C, Moroldo M. Transcranial Doppler: state of the art. Eur. J. Radiol. 27(Suppl. 2), S141–S148 (1998).
- Bulas D. Screening children for sickle cell vasculopathy: guidelines for transcranial Doppler evaluation. Pediatr. Radiol. 35(3), 235–241 (2005).
- Neish AS, Blews DE, Simms CA, Merritt RK, Spinks AJ. Screening for stroke in sickle cell anemia: comparison of transcranial Doppler imaging and nonimaging US techniques. Radiology 222(3), 709–714 (2002).
- Malouf AJ Jr, Hamrick-Turner JE, Doherty MC, Dhillon GS, Iyer RV, Smith MG. Implementation of the STOP protocol for stroke prevention in sickle cell anemia by using duplex power Doppler imaging. Radiology 219(2), 359–365 (2001).
- Jones AM, Seibert JJ, Nichols FT et al. Comparison of transcranial color Doppler imaging (TCDI) and transcranial Doppler (TCD) in children with sickle-cell anemia. Pediatr. Radiol. 31(7), 461–469 (2001).
- Jones A, Granger S, Brambilla D et al. Can peak systolic velocities be used for prediction of stroke in sickle cell anemia? Pediatr. Radiol. 35(1), 66–72 (2005).
- Zimmerman RA. MRI/MRA evaluation of sickle cell disease of the brain. Pediatr. Radiol. 35(3), 249–257 (2005).
- Kandeel AY, Zimmerman RA, Ohene-Frempong K. Comparison of magnetic resonance angiography and conventional angiography in sickle cell disease: clinical significance and reliability. Neuroradiology 38(5), 409–416 (1996).
- Al-Kandari FA, Owunwanne A, Syed GM et al. Regional cerebral blood flow in patients with sickle cell disease: study with single photon emission computed tomography. Ann. Nucl. Med. 21(8), 439–445 (2007).
- Christen T, Bolar DS, Zaharchuk G. Imaging brain oxygenation with MRI using blood oxygenation approaches: methods, validation, and clinical applications. AJNR. Am. J. Neuroradiol. (2012).
- Adams RJ, Brambilla D; Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N. Engl. J. Med. 353(26), 2769–2778 (2005).
- Gulbis B, Haberman D, Dufour D et al. Hydroxyurea for sickle cell disease in children and for prevention of cerebrovascular events: the Belgian experience. Blood 105(7), 2685–2690 (2005).
- Kratovil T, Bulas D, Driscoll MC, Speller-Brown B, McCarter R, Minniti CP. Hydroxyurea therapy lowers TCD velocities in children with sickle cell disease. Pediatr. Blood Cancer 47(7), 894–900 (2006).
- Zimmerman SA, Schultz WH, Burgett S, Mortier NA, Ware RE. Hydroxyurea therapy lowers transcranial Doppler flow velocities in children with sickle cell anemia. Blood 110(3), 1043–1047 (2007).
- Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am. J. Med. 122(6), 507–512 (2009).
- Steen RG, Hankins GM, Xiong X et al. Prospective brain imaging evaluation of children with sickle cell trait: initial observations. Radiology 228(1), 208–215 (2003).
Websites
- ClinicalTrials.gov. www.clinicaltrials.gov
Stroke in patients with sickle cell disease
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/experthematology. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association's Physician's Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. You are seeing a 2-month-old male infant with sickle cell disease (SCD) and an SS genotype. His father also has SCD and has a history of 2 prior strokes. The parents are concerned regarding their son’s risk of stroke. What can you tell them regarding the epidemiology of stroke in SCD?
□ A The overall prevalence of stroke among all patients with SCD exceeds 25%
□ B The highest incidence of stroke in childhood is between the ages of 2 and 5 years who have SS disease
□ C Genotype has little influence on the risk of stroke among patients with SCD
□ D Silent infarcts generally do not occur among children under 2 years of age
2. What can you tell these parents regarding risk factors for stroke among patients with SCD?
□ A A recent episode of acute chest syndrome is a risk factor for stroke
□ B A time-averaged mean of the maximum (TAMM) velocity on transcranial Doppler ultrasound (TCD) of 160 cm/s is an important risk factor for stroke
□ C Higher hemoglobin levels can predict an increased risk of hemorrhagic stroke
□ D Abnormal TCD velocity is most effective in predicting the risk of silent infarcts
3. Which of the following options should be part of your primary stroke prevention plan for this child?
□ A The child should be evaluated with MRI now
□ B The child should be evaluated with TCD now
□ C The child should be evaluated with imaging TCD (TCDi) now
□ D TCD should be initiated at age 2 years
4. The patient does well for years but presents with vertigo and fatigue at age 16 years. A workup reveals probable ischemic stroke. Which of the following statements regarding the acute and ongoing treatment of this patient is most accurate?
□ A He should receive an evaluation for acute treatment with tissue plasminogen activator
□ B Acute treatment should include clopidogrel and heparin
□ C Chronic transfusion therapy (CTT) should be considered as a lifelong treatment
□ D Hydroxyurea has replaced chronic transfusion therapy as the best secondary prevention measure against recurrent stroke