Abstract
This article covers the latest contributions of proteomics to the structural and functional characterization of proteasomes and their associated proteins, but also to the detection of proteasomes as clinical biomarkers in diseases. Proteasomes are highly heterogenous supramolecular complexes and constitute important cellular proteases controlling the pool of proteins involved in key cellular functions. The comprehension of the structure/function relationship of proteasomes is therefore of major interest in biology. Numerous biochemical methods have been employed to purify proteasomes, and have led to the identification of complexes of various compositions – depending on the experimental conditions and the type of strategy used. In association with protein separation and enrichment techniques, modern mass spectrometry instruments and mass spectrometry-based quantitative methods, they have led to unprecedented breakthroughs in the in-depth analysis of the diversity and dynamics of proteasome composition and localization under various stimuli or pathological contexts. Proteasome inhibitors are now used in clinics for the treatment of cancer, and recent studies propose that the proteasome should be considered as a predictive biomarker for various pathologies.
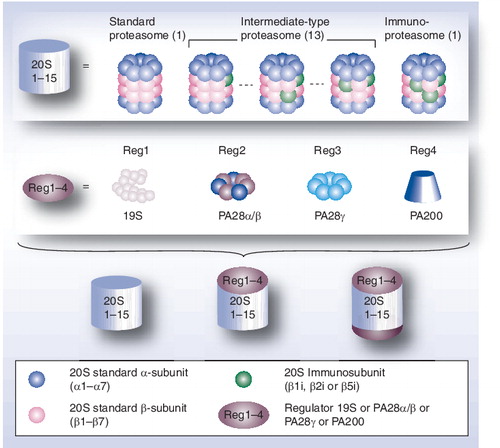
Mammalian proteasome complexes result from the assembly of a 20S core complex, the 20S proteasome, with one or two regulatory particles (RPs) of identical or different protein composition. The 20S core complex is a stable entity assembled in four stacked ring-shape heptamers, α7β7β7α7. Three subunits, β1, β2 and β5, of each β ring exhibit three distinct catalytic activities. These standard β subunits can be completely or partially replaced by three immunosubunits (β1i, β2i and β5i, respectively), which leads to various forms of 20S proteasomes (immunoproteasome and intermediate-type proteasomes). The two α-rings are implicated in the recruitment of RPs at each side of the 20S core complex. In mammals, four activators have been identified: the 19S RPs, PA28αβ, PA28γ and PA200.
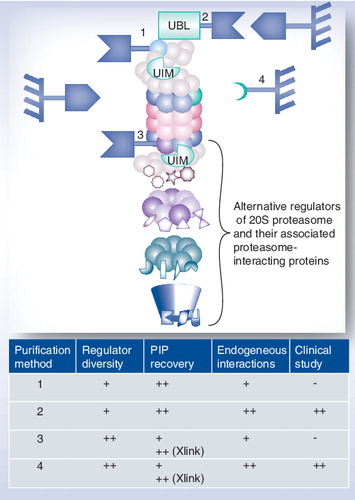
(1) Purification using a tagged 19S regulator subunit, (2) purification using a tagged domain recognizing a subunit of the 19S regulator, (3) purification using a tagged 20S core particle subunit and (4) purification using an antibody specifically directed against a conserved 20S proteasome subunit.
-: Not possible; +: Good; ++: Very good.
PIP: Proteasome-interacting proteins; UBL: Ubiquitin-like; UIM: Ubiquitin-interacting motif; Xlink: Proteins have been in vivo crosslinked prior to purification.
m/z: Mass-to-charge ratio; SRM: Selected reaction monitoring.
It is now clear that proteins mainly act in association with other proteins in a dynamic way to reach a tight control of cellular processes Citation[1]. In order to address these issues, proteomics has turned into functional proteomics, which has become a driving force to develop powerful tools for the study of protein machines, and ultimately, protein networks. Among the eukaryotic cellular protein machineries, the ubiquitin–proteasome system (UPS) is of particular importance since it is involved in the selective degradation of most short-lived intracellular proteins Citation[2,3]. In most cases, the degradation process is initialized through the polyubiquitination of the substrate. The tagged protein is then efficiently recognized by the proteasome, unfolded and degraded. Major biological processes, such as cell cycle progression, apoptosis, DNA repair, epitope generation and cell quality control, are tightly regulated by this system Citation[4]. Many studies have demonstrated that a dysregulation of this machinery is related to various pathologies such as neurodegenerative diseases Citation[5] and cancer Citation[6]; this contributes to the identification of proteasomes as therapeutic targets, especially for some cancers Citation[7].
Proteasome complexes consist of a 20S catalytic core particle (CP) comprising 14–17 different proteins, either alone or associated with one or two regulatory particles (RPs) that can be of identical or different protein composition . In mammals, four activators have been identified: the 19S RP, PA28αβ, PA28γ and PA200. The 26S proteasome is a particular form where the 20S CP is capped by two 19S RPs, forming a 2.4 MDa complex; it is involved in the ubiquitin-dependent degradation of proteins through specialized functions of specific subunits of the 19S, such as polyubiquitinated substrate recognition, ATP-dependent substrate unfolding and ubiquitin recycling. Hybrid proteasomes correspond to forms where the 20S CP is capped with two different regulators, mainly 19S RP and PA28. The eukaryotic 20S proteasome is assembled in four stacked ring-shape heptamers, with seven unique α-subunits in the two outer rings and seven unique β-subunits in the two inner rings. The two β-rings each contain three catalytic subunits – β1, β2 and β5 – which are totally or partially replaced by the so-called immunosubunits – β1i, β2i and β5i – in the immunoproteasome or intermediate-type proteasomes, respectively. The catalytic subunits are responsible for three main proteasome proteolytic activities (trypsin-like, chymotrypsin-like and caspase-like), which can be modulated by the replacement of standard subunits with immunosubunits Citation[8]. The immunoproteasome is induced during the immune response in mammals but, together with intermediate-type proteasomes, also exists as constitutive proteasome complexes – depending on tissues or cell type – and can have distinct proteolytic activities Citation[9–14]. Theoretically, taking into account the rules of cooperative assembly of inducible catalytic subunits, 13 different configurations of intermediate-type proteasomes can be formed, in addition to the standard proteasome and the immunoproteasome Citation[11]. The functional roles of these different proteasome subtypes are largely unresolved, but a specialized function might be associated with each of them. The thymoproteasome is another recently discovered form of 20S proteasome exclusively expressed in cortical thymic epithelial cells of vertebrates and containing the β5t subunit, a novel catalytic site with unusual enzymatic activity, probably involved in the positive selection of developing thymocytes Citation[15]. The association of the 20S CP with its different regulators is ensured through the two α-rings, which also regulate the entry of substrates into the CP.
Proteasome complexes therefore exhibit a high degree of heterogeneity in their overall subunit composition and can, in addition, recruit other so-called proteasome-interacting proteins (PIPs), the identification and role of which are one of the challenges of today’s proteasome research. Proteasomes constitute dynamic structures with respect to the cellular environment, such as inflammation Citation[16], the cell type Citation[9,17,18], its subcellular localization Citation[19–21], the tissue Citation[22–24], the stage of development Citation[25,26] or the pathophysiologic context Citation[10,27–31]. This diversity is probably the result of specialized functions of each individual proteasome form Citation[32], providing varied means to adapt protein degradation pathways to changing conditions in the cell Citation[33]. Therefore, a precise structural characterization, as well as a functional understanding of each proteasome subtype, are needed and still constitute a challenge in the field of proteasomes. Unfortunately, no precise structural determination of the whole 26S proteasome complex is currently available, probably owing to the tremendous complexity, diversity and lability of proteasomes. So far, only subcomplexes have been crystallized for atomic structure determination Citation[34], but recent progress has been made in determining the 26S proteasome structure at subnanometer resolution Citation[35]. Proteomic strategies constitute complementary methods to structure determination approaches for the study of protein machines Citation[36]. The association of modern mass spectrometry (MS) strategies and efficient biochemical approaches, in particular affinity purification (AP) methods, has been critical for the initial success of functional proteomics Citation[37–39]. Today’s challenges for proteomics in the analysis of protein complexes are to determine the composition and stoichiometry of protein complexes and to measure their variations, either in different cell types or after inducing a protein expression change, or over time after applying a stimulus, in order to distinguish stable from dynamic partners and to identify post-translational modifications (PTMs) on interactants.
Detailed proteomic approaches to study proteasomes have recently been reviewed Citation[40,41]. Therefore, this article will mainly cover the developments over the last 4 years in proteasome proteomics, and will envisage future trends for improving our knowledge of this complex machinery and for the consideration of proteasomes as clinical biomarkers in cancer or other pathologies.
Latest developments for the analysis of proteasome complexes by proteomic approaches
Purification of proteasomes
The detailed structural characterization of a given protein complex begins with its purification. When the complex is an enzyme like the proteasome, the challenges are both to preserve its enzymatic activity for further functional studies and to obtain a high purification yield. Given the high heterogeneity of proteasomes and the differences in stability and dynamics of the PIPs, the experimental conditions and the type of strategy used will highly influence the purity, the composition of complexes obtained (20S CP, 26S proteasome or the whole diversity of proteasome complexes), and the nature of the interactants purified (specific, stable and dynamic).
The 20S proteasome is a salt-resistant complex, but its association with the different regulators and partners is much more labile Citation[42,43]. This explains why the number of surveys dealing with 26S proteasomes and the characterization of other existing proteasomes by proteomic strategies is much more limited than those for studying the 20S core complex. Selective salt precipitations, differential or gradient centrifugations and multidimensional liquid chromatographies combined with glycerol gradient ultracentrifugation have been used to purify the 20S CP (see Citation[41] for a review) and the 26S proteasome Citation[44–46] from various species to apparent homogeneity. Recent in-depth characterization of 20S proteasomes subtypes were reported in studies relying on high-resolution anion-exchange chromatography Citation[12] and native isoelectric focusing–free-flow electrophoresis (IEF–FFE) Citation[13] as the final purification step. The different fractions contained reasonably well-separated standard proteasome and immunoproteasomes but also intermediate-type proteasomes. Interestingly, IEF–FFE of intact proteasome complexes can distinguish between differentially phosphorylated 20S proteasome complexes Citation[13]. Intermediate-type proteasomes could be further discriminated into several subtypes, which differed mainly in the composition of their catalytic β-subunits and their enzymatic properties Citation[12,13], but also in their subcellular compartment repartition Citation[11], their tissue/cell distribution Citation[10,12,13], their susceptibility to proteasome inhibitors or with respect to pathologies Citation[10,12]. These classical biochemical methods combined with further subunit identification have therefore improved our knowledge of the 20S proteasome structure/function relationship. However, they remain better suited for the study of 20S than 26S proteasomes, especially when high salt concentrations are used during elution steps Citation[43,47].
The study of native and fully functional forms of proteasomes (20S proteasome in association with its different RPs: single 20S, singly and doubly PA28-capped, singly and doubly 19S-capped, 19S and PA28 hybrid) can be performed using 2D blue native (BN)/sodium dodecyl sulfate polyacrylamide gel electrophoresis (PAGE), which allows for their separation. When combined with MS, the dynamic variations of the different complexes can be analyzed under various stimuli Citation[48,49]. In association with antibody-based gel shift assay, BN PAGE can also constitute an alternative strategy for identifying specific partners from the same multi-protein complex Citation[50], as already demonstrated for proteasome complexes Citation[48,51].
Alternatively, AP methods such as immunoprecipitation, immunochromatography and epitope tagging strategies, although costly and technically challenging, can generate a high yield of pure and functional proteasome complexes from a limited amount of starting material. These methods are also faster and simpler because they can be performed in 1 day and in a one-step procedure. In the past decade, several AP approaches have been developed to purify proteasome complexes. Associated with technological developments in MS-based proteomics, they have clearly enlarged our knowledge concerning protein complex networks Citation[37,39,52]. These approaches can differ in several ways:
• The bait protein employed to catch the complexes;
• The use of an epitope tagging strategy;
• The composition of the buffers used along the purification scheme;
• The sample preparation strategy, for example, the possibility to use an in vivo chemical treatment to strengthen protein–protein interactions.
As will be discussed, these key features influence the diversity of proteasome complexes purified, the efficiency of PIPs recovery, the probability of catching endogenous interactions and the possibility of handling clinical samples .
Purification using a tagged 19S regulator subunit
A fruitful AP–MS approach to analyze protein complexes relies on overexpression of tagged proteins in relevant cell lines. It is particularly suitable for proteins present at low endogenous levels and for labile complexes like 26S proteasomes (, purification method 1). Such a strategy using a tagged 19S subunit and MS analyses of coimmunoprecipitated proteins identified Adrm1, the human orthologue of the yeast Rpn13 subunit Citation[53,54], and confirmed arsenite-inducible RNA-associated protein as an arsenite-inducible subunit of the 19S proteasomal cap Citation[55]. Two-step methods, such as tandem affinity purification (TAP), can also be applied by introducing two different tags, which allows the purification of the complexes with high purity and lowers the number of contaminant proteins Citation[56]. However, TAP strategies are typically multistep procedures involving long incubation times and multiple elutions, which may destabilize labile protein–protein interactions This problem has been successfully overcome by combining TAP and formaldehyde in vivo cross-linking to efficiently capture labile 26S proteasome partners in yeast Citation[57]. In this strategy, a histidine biotin tag, consisting of an in vivo biotinylation signal flanked by two hexahistidine tags, was introduced at the C-terminus of several 19S proteasome subunits. Quantitative MS analysis then allowed the identification of a global map of the yeast 26S proteasome network consisting of 471 PIPs Citation[57]. However, this approach has been performed in denaturing conditions, which prevents further in vitro proteolytic activity measurement of the purified complexes. More generally, the fact that a subunit of the 19S regulator is used as prey might constitute a limitation for two reasons. First, the 19S regulator was recently shown to exert several proteolysis-unrelated biological functions independently of the 20S CP. These include transcription, DNA repair and chromatin remodeling Citation[58,59], and might therefore lead to a specific 19S regulator interactome, which cannot be distinguished from the proteolytic-related proteasome interactome. Second, only proteasome complexes containing at least one 19S particle can be isolated. Free 20S particles and proteasomes involving solely PA28 or PA200 regulators are not purified, even if they are abundant in the entire pool of proteasome complexes Citation[60,61], and might be associated with specific partners. Interestingly, recent surveys describe possible implications of these proteasomes, in particular PA28-associated proteasomes, in various pathologies, such as Parkinson’s disease Citation[30], cancer Citation[31,62,63], viral infection Citation[64,65] and alcoholic liver injury Citation[66].
Purification using a tagged domain recognizing a subunit of the 19S regulator
Targeting wild-type endogenous complexes is an appropriate way of studying protein complexes, since any sample type can be analyzed and no cellular perturbation is expected, as might happen when using overexpression or tagging strategies. Strategies using a tagged domain recognizing the endogenous protein complex of interest as bait have recently been developed for 26S proteasomes purification. The high affinity of the ubiquitin-like (UBL) domain of either hHR23A Citation[67] or hHR23B Citation[68] proteins for the ubiquitin-interacting motif (UIM)-2 domain of S5a (Rpn10), a subunit of the 19S regulator, was used (, purification method 2). These two domains were cloned and produced as recombinant glutathione-S-transferase (GST) fusion proteins. In both approaches, the purification workflow consisted of:
• Binding of proteasomes and other UIM-containing proteins to the GST-UBL;
• Elution with an excess of His-UIM-2;
• Removal of His-UIM-2 using nickel-nitrilotriacetic acid resin.
Further MS experiments allowed the identification of proteasomes and PIPs obtained from rat skeletal muscle Citation[68], rat cortex Citation[60] and human embryonic kidney (HEK-293) cells Citation[67]. Interestingly, together with 20S and 19S proteasome subunits, as well as known PIPs such as proteasome activators, chaperones or ubiquitin conjugates, several proteins belonging to the UPS but not previously known to associate with the 26S proteasome were identified. Among these, some are deubiquitinating enzymes, ubiquitin-conjugating enzymes and ubiquitin ligases. The UBL AP–MS approach is of great interest, since it permits endogenous and functional proteasome complexes from tissues or biological fluids of whatever mammalian origin to be obtained. The UBL domain of hHR23B might be more suited to efficient proteasome complex purification as it presents a higher affinity than the one of hHR23A for S5a Citation[69]. However, the specificity of the proteasome interactome obtained needs further validation, as several of the proteins purified using this approach, in particular all the cellular proteins exhibiting an UIM domain, might be captured through direct binding to the UBL domain. Indeed, the UIM domain occurs in a wide variety of proteins and might constitute a general ubiquitin-binding motif Citation[70]. Moreover, this approach suffers from the same limitations as the purification strategy using a tagged 19S subunit as previously mentioned.
Purification using a tagged or a native 20S CP subunit
Affinity methods using a subunit of the 20S core complex, associated with all of its interactors, are more prone to catch the whole diversity of proteasome complexes (, purification methods 3 and 4). ATP and glycerol preserve the interactions between the 20S CP and the 19S regulators Citation[42,43,49], and are therefore required for a successful purification of 26S proteasomes. On the other hand, ATP is not required to maintain PA200- and PA28-associated proteasome complexes, but these interactions are salt sensitive Citation[49,71]. Strategies using the genetic replacement of β4 Citation[42,43,55,72] in yeast or, more recently, of α7 in plants Citation[73] with an affinity-tagged variant, have been described and led to the efficient and rapid purification of a heterogeneous collection of proteasomes (, purification method 3). These approaches necessitate careful optimization to find the best tag, its best location on the gene and, most importantly, to check the proper integration of the modified subunit in the tightly associated 20S core complex so that the biological and/or catalytic activities are preserved. Simpler strategies using an antibody specifically directed against a conserved 20S proteasome subunit are probably more biologically relevant and also should allow clinical samples to be dealt with (, purification method 4). However, efficient and specific antibodies against one of the protein complex components and resistance to quite harsh washing conditions (to increase specificity) are required. Any constitutive subunit of the 20S CP might be used as prey, as long as the epitope is not hindered when the physiological complex is assembled. The α-subunits of 20S proteasome are excellent candidates since they are theoretically present in all existing subtypes of 20S proteasomes. Strategies using monoclonal antibodies directed against the 20S core α2 subunit Citation[74], α3 subunit Citation[75] and α6 subunit Citation[76] were recently successful to purify proteasome complexes. The mouse IgG1 monoclonal antibody MCP21 Citation[77] is of particular interest, not only because it efficiently and specifically recognizes the 20S core α2 subunit but also because it can easily be produced by a commercially available hybridoma cell line. Our group has reported an efficient workflow for the immunopurification of highly pure 20S proteasomes in a single-step procedure Citation[9,78]. This protocol was then miniaturized to deal with a small number of epithelial colorectal cells (30 × 106 cells) available from patient tissue samples Citation[79]. It allowed the determination and comparison of 20S proteasome complex subunit composition from tumor and corresponding healthy human colorectal cells from clinical samples using 2DE.
This purification strategy was then further optimized to purify the whole pool of proteasome complexes in erythrocytes. ATP and glycerol were added in all buffers to enable the weak interactions between the CP and its regulatory complexes to be maintained Citation[74]. A negative control experiment associated with a label-free differential quantitative proteomic method was conducted to distinguish nonspecific interactors from putative PIPs. In this approach, wild-type endogenous complexes are targeted and can be purified from any human sample as starting material, such as tissues, biopsies, cultured cells and biological fluids. This approach is thus compatible with clinical studies. Successive antibody-based immunoprecipitation directed against two different 20S subunits has also been used to characterize new forms of intermediate-type 20S proteasomes containing either one (β5i) or two (β1i and β5i) immunosubunits, and to show that these proteasomes were able to uniquely process some tumor antigens of clinical interest Citation[14].
To conclude proteasome complexe purification, the previously described approaches represent complementary tools and might be employed depending on which proteasome complex is targeted: a specific 20S subtype, 20S proteasome associated with a specific regulator (like the 19S regulator) or the whole diversity of proteasome complexes. As far as the exhaustive identification of PIPs is concerned, targeting one subunit of the 19S or other 20S proteasome regulators (PA28s or PA200) for the AP might probably be more relevant to efficiently catch labile and dynamic interactors. Indeed, the major known PIPs, such as deubiquitinating enzymes, ubiquitin ligases and multiubiquitin chain-binding proteins, associate transiently or weakly with RPs and not with the 20S core. However, in this case, each RP will have to be targeted independently to identify the exhaustive interactome of all proteasome forms. Moreover, this strategy is not likely to catch some specific 20S proteasome partners such as assembly chaperones or other PIPs implicated in the ubiquitin-independent proteasome degradation (for reviews, see Citation[80]). An alternative approach to purify all PIPs in a single purification scheme would be to target one subunit of the 20S CP while maintaining the labile bonds within entire complexes, so that long-distance linkages could be preserved. As demonstrated recently, in vivo cross-linking of protein–protein interactions can represent a good strategy to strengthen protein interactions Citation[81] and has been used for the identification of proteasome interaction networks Citation[57,74], while chemical cross-linking after proteasome purification has been used for structural studies Citation[35,82,83]. Among several cross-linkers used in such approaches, formaldehyde fulfills several criteria for efficient stabilization of endogenous linkages between nucleic acids or proteins Citation[81,84]. Formaldehyde is also a reversible cross-linker that facilitates MS identification of proteins. Formaldehyde is introduced very early in the proteomic workflow, so that real biological events can be frozen. However, we experienced a detrimental effects of formaldehyde for the recovery of high-molecular-weight PIPs, possibly owing to their precipitation/insolubility during the purification steps Citation[74]. Therefore, both native and cross-linking approaches might be necessary to increase the number of identified proteins. Moreover, careful experimental design with optimization of formaldehyde concentration and inclusion of proper controls is needed to discriminate true interactors from contaminants. Associated with the purification procedure targeting all existing proteasome complexes through the α2 subunit of 20S proteasomes described by our group, in vivo formaldehyde cross-linking has proven to significantly increase the number of PIPs purified compared with a strategy without any cross-linking, while carrying out a rapid and single-step AP experiment and preserving the proteolytic activity of proteasomes Citation[74]. Although the proteasome forms obtained at the end of the purification process are not separated from each other, this strategy could constitute an interesting tool in clinical studies intending to find a possible relationship between the global proteasome profile and a particular cellular state or pathology. It could constitute an interesting approach to improve proteasome complex recovery from tissues, but careful experimental design with optimization of formaldehyde concentration and inclusion of proper controls should be added in order to discriminate true interactors from contaminants.
Proteomic approaches for the study of proteasomes
As previously mentioned in the introduction, proteomic strategies are particularly suited for the study of protein complexes (see reviews Citation[37,38,85,86]). Beyond the identification of the complex interactants, they enable the characterization of PTMs on complex subunits and possibly on partners, PTMs being recognized to tightly regulate protein–protein interactions, and their dynamics upon cellular stimulation Citation[87–89]. More recently, quantitative proteomics has become a valued tool for the analysis of further protein complexes, allowing the identification of transient and labile interactants (for reviews, see Citation[39,52,90]), the measurement of protein complex dynamics in terms of composition and subcellular localization and the determination of protein complex stoichiometry.
Proteomics for the characterization of proteasome subunits
The high-resolving separation power of 2DE associated with MS detection is particularly suited to the analysis of 20S proteasomes because they are easily soluble in common urea buffers, usually employed for the first-dimension separation. In the first reference map of 20S proteasome from human erythrocytes Citation[78], more than 30 spots could be unambiguously detected on the 2DE gel and identified using MS, although only 14–17 distinct subunits were expected. MS results confirmed the proteolytic processing at the N-terminus of β1, β2 and β5 catalytic subunits, thus liberating the threonine residue that is essential for the three catalytic activities of the CP Citation[91]. Numerous other studies have been undertaken and similar 2DE patterns of purified 20S proteasomes from various species, tissues or cell types could be obtained (reviewed in Citation[13,40]), confirming that the 20S proteasome presents a high structural heterogeneity, likely in all types of cells.
19S proteasome subunits exhibit broader isoelectric point (pI) and molecular weight ranges (∼4.7–8.5 and 30,000–106,000 Da, respectively) and, as experienced recently, 2DE does not usually permit the detection of all 19S subunits, possibly owing to stability problems Citation[76,92–95]. Although several studies using 2DE have revealed new PIPs, such as Adrm1 (an orthologue of Rpn13 in yeast) Citation[94] and thioredoxin-related protein 32 (TRP32), a PIP bound to the 19S regulator through Rpn11 Citation[96], shotgun approaches (1D electrophoresis [1DE]–liquid chromatography [LC]–MS/MS or 2D–LC–MS/MS) are more prone to identify proteasome subunits and PIPs Citation[42,57,66–68,73,74,97].
Proteasome subunits exhibit numerous PTMs that might affect the activity of the proteasome but also its stability/assembly, plasticity, subcellular localization and interaction with other molecules Citation[40]. Major limitations for the identification of PTMs on proteins reside in the fact that they are often substoichiometric and dynamic. Recently developed sophisticated strategies, in addition to new MS-based technologies, have enabled progress on the characterization of PTMs on proteasomes. summarizes up-to-date N-terminal processing and endogenous phosphorylation sites identified and localized by MS/MS on human, mouse and yeast proteasomes.
Most 20S proteasome subunits exist as two to four different forms, mostly of different pIs, but also some of different molecular weights, which are separated by 2DE. Standard MALDI-TOF MS/MS or LC–electrospray ionization–MS/MS strategies have permitted explanation of such heterogeneity in some cases only. For example, the phosphorylation of α7 at Ser250 can explain the observed shift in pI for two spots corresponding to this subunit Citation[78]. However, although similar strategies have revealed N-acetylations on several 20S proteasome subunits Citation[17,78,98–100], they have failed to resolve the structural differences implied by the multiple spots observed for most subunits.
A recently described combination of top-down and bottom-up proteomic approaches permitted a deep characterization of the 20S proteasome subunits and their different forms from human erythrocytes Citation[100]. The top-down analysis of each subunit after separation by 2DE and passive elution from the gel could give the experimental mass of each form with a mass accuracy of approximately 0.01%. Comparison with the theoretical mass could then give an indication of a possible PTM occurring on the protein. For example, an increment of mass of 42 and 80 Da is likely to be correlated to an N-acetylation and a phosphorylation, respectively, as obtained in this study for many subunits. This type of MS-based study is particularly powerful since it permits the choice of the most adapted bottom-up strategy to further check the suspected PTM. For instance, if a phosphorylation is suspected, one can decide to use an analysis workflow specifically dedicated to phosphopeptides Citation[101,102], such as immobilized metal ion chromatography (IMAC) or TiO2 enrichment, to enhance the detection sensitivity by the bottom-up experiment. The suspicion of a glycosylation will direct toward other types of enrichment strategies Citation[103,104], such as purification with lectins.
Over the past decade, several sensitive post-electrophoretic stains have been developed as other interesting tools for the specific detection of PTMs, such as phosphorylation, glycosylation and oxidation Citation[105–108]. Importantly, the newly developed dyes are compatible with further MS and can be used before and as a complementary approach to the use of general protein stains dedicated to the complete detection of all proteins in the gel Citation[109]. Workflows associating high-resolution 2DE separation to specific enrichment strategies and multiplex detection systems, for example fluorescent PTM-sensitive dyes, specific chemical-derivation reagents, antibodies and MS, were implemented for the characterization of PTMs including N-acetylation, glycosylation, phosphorylation, oxidation, and nitrosylation on purified proteasomes from murine heart Citation[110] and yeast Citation[93]. Many proteasome 20S and 19S subunits responded positively to at least one of these different detection systems. MS experiments conducted could not confirm all of these. This may be explained by an incomplete protein sequence coverage hampering the detection of some modifications, but possibly also owing to false-positive detections. An additional difficulty of confident PTM confirmation by MS may arise from the use of collision-induced dissociation (CID), which remains the most common ion activation technique employed in laboratories devoted to proteomics today. In CID, activation of the selected ions occurs by collision(s) with neutral gas molecules in a collision cell. For the analysis of PTMs such as phosphorylations and glycosylations, this process typically results in poor informative phosphopeptides and glycopeptides MS/MS spectra, dominated by a neutral loss of the phosphate and glycoside group, respectively. Fragmentation techniques developed more recently, namely electron capture dissociation (ECD) and electron transfer dissociation (ETD), result in more confident identification of peptides modified with a labile moiety (for more details on these techniques, refer to the recent reviews Citation[111–113]). Alternating both fragmentation techniques, ETD and CID, increases the amount of information derived from peptide fragmentation, and the new generation of high-resolution, sensitive and fast-sequencing LTQ Orbitrap mass spectrometers are equipped with both fragmentation modes Citation[114,115]. Therefore, CID/ETD fragmentations, in association with protein fractionation, 2DE separation and/or phosphopeptides enrichment techniques, enabled the identification and assignment of new phosphorylation sites on proteasome complexes from yeast Citation[116] or mammalian tissues Citation[23]. However, none of the five 19S subunits and nine 20S subunits that had been previously determined to be potentially O-Glc-NAc modified in Drosophila melanogaster using immunological and lectin-binding methods Citation[117] could be confirmed by the latest dedicated enrichment and CID/ETD fragmentation methods Citation[118], showing how challenging the MS determination of glycosylation sites still remains. Future developments to improve dedicated tools for the enrichment of specific glycosylated proteins therefore seem needed.
Ubiquitin attachment sites are probably easier to identify because trypsin digestion results in the cleavage at the C-terminal arginine residue in ubiquitin, leaving a characteristic and stable GG or LRGG tag on the lysine residue in the peptide that was covalently linked to ubiquitin Citation[119,120]. Using this signature tryptic footprint, several 20S and 19S proteasome subunits were detected as ubiquitinated in various eukaryotes species after immunopurification of proteasomes Citation[73,121] or after large-scale purification of all cellular ubiquitinated proteins by immunoaffinity chromatographic procedures Citation[122,123]. These results are in agreement with a previous report Citation[124] suggesting that there is an autoregulation of active proteasome levels in cells by autodegradation of nonfunctional 20S proteasome Citation[73,122]. Alternatively, ubiquitination could impart new functions to specific subunits, as demonstrated very recently for Rpn10, one of the ubiquitin receptors of the 26S proteasome Citation[121]. Monoubiquitination of Rpn10 strongly inhibits its UIM-mediated interaction with polyubiquitinated proteins, thereby downregulating substrate recruitment to the proteasome. This event is notably decreased in stress conditions, presumably for more efficient protein degradation.
The recent improvements in the sensitivity of detection using modern mass spectrometers associated with sophisticated enrichment techniques offer an unprecedented breakthrough for the identification of PTMs on proteins. The forthcoming challenges will be to assign a biological function to each of these modifications. As far as proteasomes are concerned, results from the literature highlight a high variability in the PTM pattern between tissues, species and pathophysiological status, probably linked to a regulated proteasome activity, but this has to be confirmed in most cases.
The most relevant approaches will probably consist of linking an identified PTM with a particular biological context. In most cases, the detection system, such as 32P radiolabeling, PTM-directed antibodies, PTM-sensitive dyes or pI/PM shifts on 2DE gels, will not give any information about the precise site of modification. Therefore, the knowledge of potential modification sites reported on a proteasome subunit by global proteomics could help to design targeted experiments, such as targeted MS/MS approaches or directed mutagenesis, to confirm the biological role of a given site of modification. Recently, an observed pI shift of several 20S proteasome subunits from Alzheimer’s disease (AD) brain samples using 2DE pointed out possible variations in PTMs on proteasomes Citation[125]. The previously reported phosphorylation and N-acetylation sites on the α7 subunit were then further investigated using MS/MS as this subunit was one for which a significant pI shift had been observed. Finally moderate N-terminal acetylation and dephosphorylation of subunit α7 were demonstrated in the brain of AD patients. Similarly, 2DE separation of 20S proteasome forms in Jurkat T cells showed a decrease in the phosphorylation status of α7 at the Ser250 site, as a result of 5-fluorouracil-induced apoptosis Citation[27], and this is in accordance with previous work showing a change in phosphorylation of 20S proteasome subunits after induction of apoptosis by doxorubicin in proerythroleukemic K562 cells Citation[126]. The biological incidence of the dephosphorylation of α7, even moderate, might well have an impact on 26S proteasome stability. In order to characterize variations of PTM on proteasome subunits, future developments will require the association of PTM-specific workflows with quantitative proteomic strategies specifically developed for the quantitative analysis of protein complexes, including concurrent identification of interactors and their state and stoichiometry of modification Citation[88]. A recent survey associating proteasome purification, TiO2 enrichment and isobaric tags for relative and absolute quantitation (iTRAQ) labeling demonstrated that the already described phosphorylation of Rpn2 subunit on Thr273 induces the inhibition of proteasome activity in the context of osmotic stress Citation[127]. Sensitive multiple reaction monitoring (MRM) methods will also constitute promising targeted quantitative approaches for the determination of the stoichiometry of PTM-modified sites, as recently described for phosphorylation Citation[128] and ubiquitination Citation[129–131].
Functional analysis of proteasomes by quantitative proteomics
Quantitative proteomics has evolved as an valuable tool for characterization of the biological function of proteins (for a review, see Citation[132]). As reviewed in the next section, recently developed strategies have permitted significant progression in our knowledge of proteasome biology.
Quantitative analysis of the variations in 20S proteasomes subunit composition
2DE associated with image analysis using dedicated software is an attractive technique for the quantification of 20S proteasomes because it enables the separation of all α and β subunits including their numerous forms and their quantification in a single experiment. As already reviewed Citation[41] and more recently published Citation[28,133], it has been used in numerous surveys for the biochemical characterization of 20S proteasomes in different contexts. However, numerous challenges such as spot matching, 2DE gel normalization, and lack of reproducibility of separation or staining have to be overcome for successful quantification. The recently developed DIGE technology has become the method of choice for the detection of differences by image analysis after protein separation by 2DE (see recent reviews Citation[134,135]). In DIGE-based proteomics, the experimental and control samples are derivatized with different fluorophores and the compared sets of proteomes comigrate in a single run, minimizing gel-to-gel variations. The strategy also includes an internal standard, facilitating the normalization step. Recently, this technique enabled a similar 20S proteasome high-resolution 2D pattern as the one previously obtained using commassie blue staining Citation[78] to be obtained, with multiple forms for most proteasomal subunits Citation[125]. pI shifts and changes in relative abundance between healthy and AD brain samples could be observed for several proteasome subunits, possibly due to PTM variations on these subunits. Recently, a study by DIGE of global proteome variation in HL-60 cells, treated with methotrexate, an inhibitor of nucleotide biosynthesis used for the treatment of cancers, pointed out several subunits of the 26S proteasome. Their downregulation might explain the accumulation of ubiquitinated proteins and promotes apoptosis Citation[136]. The catalytic subunit β2 was also identified as one protein associated with multidrug resistance in LoVo human colon cancer cells by DIGE, and this regulation was further validated by immunofluorescence and Western blotting Citation[137]. Numerous other recent global proteome differential analyses using 2DE quantification and MS identification report variations in proteasome subunit abundance. However, measurement of the variations observed using complementary approaches remains necessary to validate the results.
Recently, MS-based quantification techniques have been successfully developed, in association or not with 2DE separation, for the identification of variations in 20S proteasome composition and abundance either in different cell types or in a pathophysiological context. Chemical labeling at the protein level by isotope-coded affinity tag (ICAT) Citation[18,99] or by stable isotope labeling with amino acids in cell culture (SILAC) Citation[27], and at the peptide level after protein digestion Citation[24,138], or even label-free MS quantitation Citation[22,138], have emerged as alternative techniques advantageously replacing the classical post-electrophoretic quantification by image analysis.
When several subunit forms are separated by 2DE prior to quantitation, the variations of each form can be distinguished Citation[18,27,99]. Schmidt et al. recently described a SILAC-based quantitative study of 20S proteasomes separated by 2DE to better understand the observed decreased proteasome-mediated proteolysis of short-lived proteins in apoptotic cells Citation[27]. Significant quantitative changes were observed on the 2DE gels for two forms of the α7 subunit, highlighting a decrease of phosphorylation status, further confirmed by quantitative MS analysis.
Liquid chromatography–MS analysis of isotope-labeled peptides directly after proteasome purification can only detect global variations of each subunit in most cases. However, this can be a sound and straightforward approach for the comparison of the relative abundance of catalytic subunits or PIPs. Raijmakers et al. successfully pointed out the differences in the composition between chromatographically purified bovine liver and spleen 20S core proteasome complexes Citation[24]. In this study, online and on-column sequential derivatization of peptides, by dimethylation using cyanoborohydride and either regular or deuterated formaldehyde, was performed. The results obtained demonstrated that all three catalytic immunosubunits – β1i, β2i and β5i – as well as the PA28 α/β regulator of the 20S proteasome were more abundantly present in the spleen-derived proteasome preparation, as expected from previous observations Citation[139]. In another survey, Gomes et al. compared purified 20S proteasomes from heart and liver using three different quantitative approaches, namely label-free quantitation, chemical labeling using 16O/18O post-digestion labeling and immunoblotting Citation[138]. Again, a clear difference in inducible catalytic subunits, mainly β1i and β5i, was observed in the liver when compared with the heart, although the concentrations of proteasomes per cell/tissue are largely different. Recently, a broader study on heart, kidney, liver, lung, thymus and spleen tissues compared the relative abundance of all α and β subunits from purified 20S proteasomes Citation[22]. As expected, no major change was observed on α1 to α7, β3, β4, β6 and β7 subunits (which are shared by the constitutive and the immunoproteasome); however, differences in the relative abundance of the immunosubunits and their constitutive counterparts could be precisely determined and confirmed the tissue-specific distribution of the immunoproteasome (more abundant in spleen and thymus lymphoid tissues). It also pointed out the existence of immunoproteasome hybrids wherein the subunits β1, β2 and β5 have only been partially replaced by their β1i, β2i or β5i counterparts, and the specific presence in the thymus of the β5t catalytic subunit Citation[15,140].
Identification of specific PIPs & determination of their association stability by quantitative proteomics
Proteomic analysis can be very efficient in identifying interacting proteins. Improvements in MS sensitivity and acquisition speed allow the production of ever larger lists of identified proteins. These, however, may contain contaminating proteins that needs to be distinguished from true partners Citation[141]. These nonspecific co-purified proteins can represent up to 95% of the total pool of proteins identified Citation[142]. More stringent purification methods can be used to decrease the risk of false-positive interactants, but they also restrict the identification of the strongest partners. Modern quantitative proteomics can circumvent this problem by direct quantitative comparison of each identified protein in specific and control pull-downs. These strategies have been recently extensively reviewed Citation[90,141] and essentially rely on isotopic labeling at the protein Citation[143–145] or at the peptide level Citation[88,146], or on label-free quantitative approaches Citation[147,148]. Labeling methods at the protein level usually give a more accurate relative quantification because the label is introduced early in the workflow and labeled samples (usually two or three) are then mixed and analyzed in the same analytical run. By contrast, a label-free approach necessitates parallel sample treatment and analysis. Accurate quantification thus requires a high analytical reproducibility of the workflow and replicate analyses to be performed and handled using dedicated bioinformatics tools. The main advantage of label-free approaches is that they can be used to compare any type and number of samples. High confidence protein–protein interactions are revealed thanks to their high ratios and can be thereby discriminated from a great number of background proteins. These quantitative strategies have successfully been applied to greatly enlarge our current knowledge of the proteasome network. SILAC Citation[57,97] and label-free Citation[74] quantitative approaches have been described in association with AP strategies to characterize proteasome complexes and discriminate true PIPs from nonspecific co-purified proteins. In the classical SILAC strategy, the samples to be compared are mixed very early, at the cellular level, to minimize quantitation errors arising from separate sample handlings. Therefore, after cell lysis, the differentially labeled protein extracts containing either the tagged bait or the control are in contact for a certain time during the AP step. This permits dynamic exchange of the most transient interactors. The more dynamic the interaction is, the faster the exchange is. Therefore, only stable interactions can be detected with high ratios using this approach, termed purification after mixing-SILAC (PAM-SILAC). Lower ratios can be indicative of transient or labile linkages within the complexes and need further investigation using time-controlled PAM-SILAC Citation[97]. An alternative strategy to detect both stable and the most dynamic PIPs was to mix the two protein extracts after purification (mixing after purification [MAP] strategy). Finally, a combination of both strategies is of great interest to obtain a wide interaction network, while gaining information about the nature of the interaction (stable or dynamic). Some well-known PIPs of mammalian proteasomes, such as Usp14 deubiquitinating enzyme or hHR23B multi-ubiqutin chain-binding protein, and even the Rpn13 subunit of 19S regulator, were classified as dynamic, while others were found to be stable PIPs, based on their SILAC ratios Citation[97]. These approaches have been reviewed very recently Citation[39].
Interestingly, a quantitative comparison between a native and a cross-linked protein extract can also give indication about the strength of the interaction of a protein within a complex, as demonstrated recently for PIPs using formaldehyde reticulation Citation[74]. The strategy was to stabilize the most labile and/or dynamic interactions so that the in vitro immunoaffinity-isolated proteasome complex network could be enlarged. The benefit of formaldehyde treatment was evaluated by estimating the relative abundance of PIPs in purified samples from formaldehyde-treated and nontreated erythrocytes using label-free quantification. Interestingly, results indicated that the most significant protein linkage stabilization was observed for the most remote subunits from the α2 subunit of 20S proteasome, used as prey protein. Indeed, subunits from the 19S lid (Rpn3 and Rpn5-12) exhibited high formaldehyde-enrichment ratios compared with subunits from the 20S CP or subunits from the 19S base (Rpts and Rpn1-2). Other PIPs were also only recovered after formaldehyde treatment, which suggested loose interactions within the complex.
Quantitative analysis of the variation in PIPs to elucidate the roles of proteasomes in biological processes & in pathologies
Altered proteasome-mediated degradation is believed to be related to diverse pathologies such as cancer, neurodegenerative disorders, aging or muscle atrophy Citation[7,149–152] through dysfunctions of cellular biological processes such as cell-cycle progression, apoptosis or DNA damage repair, or as a consequence of environmental stresses, such as oxidative stress Citation[153]. Modulation of protein–protein interactions constitutes a new area of research for the development of new drugs and this strategy might be well adapted to proteasomes as increasing evidence demonstrates that some of these disorders might correlate with alterations of PIPs and proteasome regulatory complexes binding to the proteasome Citation[31,66,76,154]. PIPs associate with the proteasome and have an important impact on its structure, assembly, stability and subcellular localization, as well as its substrate recognition and degradation efficiencies. Therefore, the specific modulation of the interaction between the proteasome and some PIPs could constitute new therapeutic strategies. As reviewed in the following section, comparative expression proteomics using modern MS-based quantitative approaches has proven to be of great interest to study dynamic variations of PIPs under normal or pathological contexts.
The proteasome tightly regulates major cellular processes by controlling the cellular pool of key regulatory short-lived proteins. In an effort to better understand how 26S proteasomes regulate cell cycle transitions, the dynamic variations of yeast PIPs along the cellular cycle progress were studied thanks to their SILAC ratio profiles by the QTAX strategy Citation[155]. This enabled categorization of the PIPs and to evaluate whether these groups of proteins were enriched in specific cellular functions, biological pathways or complexes. Interestingly, this analysis showed that a group of PIPs only captured at G1 and S phases was enriched in proteins involved in the yeast mating pheromone-response pathway that induces cell-cycle arrest.
Another very important function of proteasomes is to ensure the quality control of the cell by degrading abnormal and nonfunctional proteins generated under normal and stress conditions Citation[4,16]. Oxidative stress is associated with important pathophysiological events in a variety of diseases, such as neurodegenerative diseases Citation[5,156] or in aging Citation[157,158]. Proteasomes seem to play an important role in preventing the increase of intracellular oxidative stress through the degradation of defective ribosomal products Citation[16] and oxidized proteins, both in the cytosol and the nucleus Citation[159], but also through the regulation of pro-apoptotic proteins level Citation[160]. Inhibition of the proteasome may contribute to the toxicity associated with oxidative stress Citation[161–163]. The mechanism responsible for the decrease in proteasome activity caused by excessive oxidative stress is still not clear. The formation of stable 4-hydroxynonenal adducts on proteins, thereby forming crosslinks that are difficult to be degraded, but also on proteasomes themselves, was reported to inhibit proteasome activity Citation[159,164,165]. Phosphorylation and S-glutathionylation have also been reported on several 20S or 19S proteasome subunits upon exposition to diverse oxidative stresses Citation[165–169]. Another reason for proteasome impairment associated with oxidative stress might be an alteration of the association of PIPs and regulatory complexes with the proteasome, as suggested by the following surveys. An impaired assembly of the 26S proteasome and a decreased expression of Rpn11, the major DUB 19S subunit, were found to be associated with aging Citation[157]. H2O2-induced S-glutathionylation of Rpn2 might induce aberrant interaction of the Rpn1/Rpn2 complex with other proteasomal subunits Citation[168]. Cells lacking arsenite-inducible RNA-associated protein, an arsenic-inducible PIP, accumulate polyubiquitinated substrates when exposed to oxidative stress caused by arsenite Citation[55]. Recently, arsenite-induced changes in proteasome composition were investigated by a SILAC-based quantitative analysis in mammalian cells Citation[76]. TRP32, a PIP with thiol reductase activity primarily detected in 2DE-separated 26S proteasomes Citation[96], was found to be an arsenite-regulated protein by SILAC. The 26S-associated TRP32 level was shown to decrease after oxidative stress exposure of mammalian cells, but the functional significance of this variation could not be elucidated. In another recent survey, alterations in proteasome composition were investigated in the context of alcoholic liver injury Citation[66], where significant inhibition of proteasome activity has been reported. Changes in the amount of PIPs upon chronic ethanol feeding were determined by ICAT and label-free quantitative approaches. PA28α/β proteasome activator and Ecm29, a known PIP also found to be responsible for 26S proteasome disassembly under oxidative stress Citation[170], and three major proteasome-associated deubiquitinases, were significantly decreased in the ethanol-treated purified proteasomes. The lack of these important PIPs within proteasome complexes might be responsible for the observed dramatic decrease of proteasome activity and accumulation of polyubiquitinated proteins.
Quantitative proteomics to elucidate spatial distribution of proteasomes
Proteasomes have been detected in numerous cellular compartments, including the cytosol, nucleus, ER, Golgi apparatus Citation[41] and, to a lesser extent, associated with membranes Citation[171]. Most studies relied on subcellular fractionation followed by immunoblotting or, on in situ observation using immunohistochemistry, immunofluorescence or confocal microscopies Citation[41]. These targeted approaches necessitate the availability of specific antibodies against the proteins of interest.
Quantitative proteomics also constitute a precious tool to assign proteins to their specific organelle or subcellular compartments Citation[172–175] or to profile dynamic changes in the subcellular localization of proteins in cells under different physiological states Citation[19,21]. They permit a global view of the cellular distribution of proteins to be obtained because they are essentially not targeted approaches and can therefore localize previously uncharacterized proteins. Moreover, they can permit the localization of specific protein forms, such as splicing isoforms or forms with PTM, to be assigned, which is of great help for more in-depth biological interpretation. A prerequisite is to optimize an efficient fractionation workflow so that highly pure separated subcellular compartments can be obtained. In a recent study, Boisvert et al. described a successful SILAC-based proteomic strategy, termed ‘spatial proteomics’, to establish a quantitative map of the relative subcellular localization of each protein, and to characterize their dynamic changes induced by DNA damage Citation[19]. Interestingly, while at steady state, the 26S proteasome subunits clustered predominantly in the cytoplasmic fraction, DNA damage was found to induce the relocation of the 20S complex and of all its regulators (PA28s, 19S) in the nucleus. The mechanisms explaining how proteasomes are recruited to the nucleus are not yet known but confirm previous reports linking the function of proteasomes to the DNA repair pathway Citation[176]. Quantitative proteomics on purified proteasomes rather than on whole cellular lysates might permit a more precise distribution of these complexes throughout the cell to be obtained. Indeed, each proteasomal subunit might be found in mature proteasomes but also in premature proteasome forms Citation[177,178] and as free subunits in the cell, where they could have an additional extraproteasomal function, as recently proposed for Rpn10 Citation[179,180].
Quantitative analysis of ubiquitin chain structures and of polyubiquitinated substrates to better understand proteasome-related ubiquitination & its function
Although an increasing number of examples show that structured proteins can be processed by proteasomes independent of ubiquitination (see review Citation[80]), it is widely acknowledged that ubiquitin conjugation constitutes the major signal for 26S proteasome-mediated degradation. Protein ubiquitination is accomplished through a cascade of three consecutive enzymatic reactions involving activating enzymes (E1), conjugating enzymes (E2) and ligases (E3) Citation[2]. It results in the formation of an isopeptide bond between the ε-amino group of a lysine residue within the substrate and the carboxyl group of the C-terminal glycine residue of ubiquitin. Because ubiquitin displays seven accessible lysine residues (K6, K11, K27, K29, K33, K48 and K63) and an N-terminal amino group, which all constitute a possible anchor for polyubiquitination, the diversity of polyubiquitin (polyUb) chains is high Citation[119,181]. Protein ubiquitination is central to many cellular processes Citation[182,183] and the fate of a modified protein depends mainly on the number of ubiquitin moieties attached to it and on the chain topology. Mechanisms of recognition and processing of ubiquitin–protein conjugates by the proteasome have been reviewed recently Citation[184]. PolyUb chains on substrates allow both addressing and binding to the proteasome, the main requirements believed for efficient proteolysis being the particular topology of K48-linked polyUb chains Citation[185] and a minimal chain length of four ubiquitin moieties Citation[186]. Monoubiquitination or polyubiquitination through K63 are generally thought to act in a range of other proteasome-independent cellular processes (for a review, see Citation[187]). However, recent surveys performed, for many, with the aid of quantitative proteomics, raised the question of more complicated mechanisms for substrate recognition and degradation efficiencies by proteasomes complexes. MS is a powerful tool for the study of ubiquitin chain topology as each type of ubiquitin linkage gives a specific tryptic digest: monoubiquitination produces characteristic unbranched ubiquitin tryptic peptides, whereas polyubiquitination generates modified tryptic peptides of ubiquitin where a tag of two G residues is attached to the K which is involved in the chain linkage.
A subset of proteins with K11 linkages have been reported to be degraded by the proteasome Citation[130,188,189]. Moreover, the K11 GG-tagged tryptic peptide of ubiquitin, detected in tryptic digests of whole cell lysates but also of proteasome-bound proteins, is increased, together with its K48 counterpart, upon MG132 inhibition of the proteasome Citation[131] or in yeast strains with mutation in proteasome components Citation[129]. Similarly, all non-K63 polyUb linkages accumulate upon proteasome inhibition Citation[129,190]. Whether K63 polyUb linkages have the ability to target substrates for proteasomal degradation seems to be more a matter of debate. MS-based quantitative analyses of K63 linkages levels upon proteasome inhibition in Saccharomyces cerevisiae or mammalian cells show either a significant increase, suggesting the involvement of K63 as a targeting signal for the 26S proteasome Citation[190–192], no significant changes Citation[129] or a delayed accumulation for long time inhibition Citation[131], possibly in response to stress caused by proteasome inhibition rather than on account of a proteolytic function of K63 linkages. In vitro, however, K63 polyUb chains bind the 26S proteasome as efficiently as K48 chains Citation[131], and are also able to target the degradation of several proteins Citation[130,191,193,194]. Quantitative MS analysis of the polyUb chains K48/K63 ratio showed a higher abundance of K48 chains bound to the proteasome compared with the ratio observed in the whole lysate Citation[131]. Proteasome-bound deubiquitinating activities, which more efficiently degrade long K63 chains than their K48 counterparts, might account for such discrepancies and enable the escape of K63 polyUb substrates from proteasomal degradation Citation[131].
Therefore, even if some specific polyUb linkages are required for proteolysis of particular substrates, most seem competent in addressing proteins to proteasomes, although with different efficiencies. This capacity might constitute a cellular means to more efficiently respond to diverse stresses by an increased and massive proteasome-mediated degradation of proteins. The major limitation of the quantitative proteomic strategies developed to date resides in the difficulty to discriminate a mixture of different homogeneous ubiquitin–ubiquitin linkages from branched mixed-linkage chain topologies. A tryptic peptide of ubiquitin containing two Ub footprints at K29 and K33 could, however, be MS-detected in vivo in yeast Citation[119,195] and in plant Citation[196], suggesting highly complex chain topologies, independently of the species.
Moreover, even if MS is capable of determining global ubiquitin chain topology, it cannot discriminate a free polyUb chain from a protein-linked one. The only way to identify an ubiquitinated protein in a whole cellular lysate is the detection of the mass shift of 114.04 Da, induced by the GG signature on the tryptic peptide triggering the ubiquitin moiety, which is plausible but quite unlikely. Therefore, ubiquitinated proteomes are better studied using affinity-based enrichment methodologies Citation[119,123,190,196–198] and, in association with quantitative proteomics strategies, changes in ubiquitination profiles can be used to better understand ubiquitination and its function. These approaches permitted the identification of hundreds of putative ubiquitinated proteins, but the Ub attachment sites were resolved for only a small subset of these proteins Citation[190,196]. Little sequence consensus was observed, at least in plant Citation[196], and the discovery of new ubiquination sites might therefore rely on the development of ever-sensitive and highly dynamic range MS technologies.
Quantitative analyses comparing the ubiquitinated proteomes of untreated cells and cells treated with proteasome inhibitors constitute interesting tools to identify putative proteasome substrates Citation[190]. As described recently, quantitative proteomics can also help to identify a subset of proteasome-targeted proteins that are substrates of a specific E3 ubiquitin ligase Citation[199].
Role of MS & proteomics for the study of proteasome complexes as therapeutic targets
Proteasome inhibitors are now used in clinics for the treatment of some types of cancer but the cellular pathways that are affected at the molecular level are still not completely defined. Global quantitative proteomic approaches can play an important role in elucidating these pathways. Here, we review such studies that have been undertaken so far, and will also highlight some recent studies that propose consideration of the proteasome as a predictive biomarker for cancer therapies and other diseases, and discuss the growing role of MS in the detection of biomarkers.
Global quantitative proteomic approaches represent an attractive way of deciphering the cellular pathways affected by proteasome inhibition. They can also be valuable to understand resistance mechanisms associated with drug treatment and to help define new therapeutic targets to be used in combination with proteasome inhibition. However, several challenges have to be faced using this approach. A first difficulty comes from the fact that proteasome inhibition will affect many processes and lead to the accumulation of many proteins. Thus, the inhibition time point chosen for the study may be an important parameter. A second difficulty is that the pathways already known to be affected and to play a role in the apoptotic cascade may involve low-abundance proteins, such as, for example, the NF-κB protein Citation[200]. This may explain why few studies have attempted such experiments up to now using various cellular models and several proteasome inhibitors. These studies were based on 2DE separation and differential analysis followed by MALDI–MS or LC–MS/MS protein identification to determine the proteins affected by proteasome inhibition in rat cardiac myocytes Citation[201], in human umbilical vein endothelial cells Citation[202], in colorectal cell lines Citation[203], and in mantle cell lymphoma cell lines Citation[204]. These studies were able to separate more than 1000 protein spots on average and to identify three up to 75 varying proteins. They demonstrate that proteins from the heat-shock protein (HSP) family (HSP70 in most cases) were increased for all cell lines and regardless of the proteasome inhibitor used. Other proteins were then found to be more specific from the cell line studied. Interestingly, sensitive mantle cell lymphoma cell lines differed from resistant ones after inhibition with 25 nM bortezomib and exhibited additional alterations in protein levels of energy metabolism, RNA and transcriptional regulation, and cell division Citation[204]. This opens new perspectives to explore combination therapies targeting complementary pathways for the treatment of lymphoma.
In a very recent study, a global quantitative proteomic approach was used to characterize aggregating proteins in human neuroblastoma cells after proteasome inhibition Citation[205]. The authors performed metabolic labeling using SILAC and identified the proteins affected by proteasome inhibition using MG132 treatment, which is a widely used proteasome inhibitor even though it may inhibit other proteases in the cell. This quantitative strategy was combined with protein fractionation using a sucrose gradient to focus on proteins accumulating in aggregates, which are known to be formed after proteasome inhibition. Moreover, the use of a recent highly sensitive and high acquisition-speed mass spectrometer such as the LTQ Orbitrap instrument provided an efficient tool to identify less abundant proteins. This approach allowed more than 500 proteins to be identified as enriched in aggregates after MG132 treatment, corresponding to 74% of the total number of identified proteins. The identified proteins suggest that aggregates formed upon proteasome inhibition are in part similar to other protein aggregates found in neurodegenerative diseases, but also contain additional proteins associated with the ubiquitin system and also demonstrate an increased level of HSP70.
Another aspect of the analysis of proteasome as a therapeutic target lies in its detection as a diagnostic and/or prognostic biomarker in body fluids. This is supported by the fact that an elevated proteasome amount has been found in several tumor cells Citation[206]. It was then hypothesized that this elevated concentration could also be found in fluids in contact with the tumor cells. Even though the origin of the extracellular proteasome detected in body fluids is still enigmatic Citation[171], it was proposed that it could reflect a pathological state of patients and that it could represent a reliable biomarker for several diseases. Extracellular proteasome concentration has mainly been measured in bronchoalveolar lavage fluid and in blood plasma or serum, where it is referred to as circulating proteasome, using ELISA (for a review, see Citation[171]). In recent years an increasing number of clinical studies have been conducted. They all demonstrate an elevated concentration of 20S proteasome in body fluids of patients with a wide range of pathologies, such as burn and inhalation injury Citation[207,208], acute respiratory distress syndrome Citation[209], melanoma Citation[210,211], malignant transformation of liver cirrhosis Citation[212], sepsis Citation[213] and epithelial ovarian cancer Citation[214]. It can thus be concluded that extracellular and circulating 20S proteasomes, the concentration of which can easily be measured from body fluids, constitute a new class of biomarkers of several pathologies. In some cases, it could efficiently be used to assess efficacy of treatment.
However, the precise composition of circulating 20S complexes is not known when using an ELISA, and various proteasome subtype profiles could exist for an equivalent 20S proteasome amount. In a pathological context one could then imagine using these profiles of a tissue or plasma sample from patients as a more precise clinical tool for diagnosis or prognosis, which could lead to more targeted therapies. Indeed, development of inhibitors targeting a specific proteasome subtype could constitute a sound strategy to reduce side effects of nonspecific inhibitors Citation[215,216]. Immunoproteasome-specific inhibitors(IPSI)-001, the lead compound of a series of recently developed IPSIs, demonstrated a more than 100-fold increased selectivity towards the immunoproteasome. Therefore, tissues expressing predominantly standard proteasomes should not be targeted by this inhibitor – unlike the current generation of proteasome inhibitors, such as bortezomib, which is proteasome specific but not 20S proteasome subtype specific. IPSIs were shown to constitute as valuable alternative drugs for the treatment of multiple myeloma patients who were exhibiting resistance to conventional drugs and nonspecific proteasome inhibitors Citation[217]. Other pathologies where elevated expression of immunoproteasome has been reported in various tissues – such as autoimmune diseases Citation[218], inflammatory bowel disease Citation[29,219,220], major hepatic diseases Citation[221] and degenerative diseases Citation[222] – could also benefit from this new class of proteasome-targeted inhibitors. In the case of inflammatory bowel disease patients, it has recently been demonstrated that the subunit pattern of 20S proteasome from surgical tissue specimens was suitable to differentiate Crohn’s disease and ulcerative colitis: two forms of this disease Citation[28]. It is therefore important to study further proteasome profiles and to develop robust analytical tools to accurately and reproducibly measure proteasome subunit compositions.
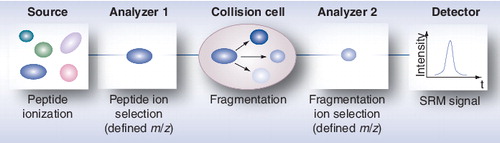
Targeted MS techniques can play a pivotal role in the measurement of all existing subunits of 20S proteasome complexes, including immunosubunits and post-translationaly modified subunits, from patient body fluids. In the studies performed so far, antibodies have mainly been used to measure a few subunits of the standard and the immunoproteasome complexes. An initial attempt to characterize all subunits of circulating proteasome has been reported using 2DE of immunopurified 20S complexes Citation[211], but the low amount of 20S proteasome available and the protein composition of plasma samples were important limiting factors to the success of this approach. Thus, only the composition of proteasome from plasma of patients with stage IV melanoma could be determined and showed a mixture of standard proteasome and immunoproteasome complexes. Targeted MS has been shown to be a sensitive and specific way to measure biomarkers in body fluids Citation[223]. It has limits of detection comparable with those of ELISA for some proteins, as demonstrated for prostate-specific antigen Citation[224,225]. It is based on the definition of proteotypic peptides generated after trypsin digestion, which are specific of the protein to be measured. Although targeted MS analysis does not measure intact proteins, peptides can be carefully chosen when necessary to cover the N- and C-termini of a protein and to detect modified peptides. These peptides are then analyzed by MS using a selected reaction monitoring acquisition mode, which consists of selecting a defined peptide, fragmenting it and specifically analyzing the most abundant fragments formed (; for a review, see Citation[226]). The promising potential of this MS-based approach lies in its sensitivity and high selectivity. Even more importantly, it is fast and it can be multiplexed to measure several tens of proteins in a single LC–MS/MS run Citation[227]. These multiple single reaction monitoring transitions correspond to the MRM mode. Very recent applications have been performed using biological fluids, mainly serum Citation[225,228] and feces Citation[229] from patients, for the detection of several biomarkers in cancer. It is thus expected that targeted proteomics using MRM is going to play a growing role in biomarker validation in the near future.
Expert commentary
Newly developed AP approaches now permit the entire proteasome network to be obtained in a simple procedure, and some of these strategies are compatible with clinical studies, allowing the study of proteasomes in real pathological contexts. Meanwhile, the latest improvements in protein/peptide separation, enrichment and labeling techniques in MS and in bioinformatics enabled proteomics to tackle functional studies. These advances contributed to significant progress in proteasome biology knowledge. In particular, the understanding of the variations of proteasome quantities, subunit composition, subunit PTM modification or subcellular localization in response to various stimuli or in disease states could be improved. Further studies are needed to fully characterize the dynamics of proteasome diversity, and the major challenge will probably be to understand the interplay between the structure of a specific proteasome form and its cellular function in a given physiological or pathological context. Future development in targeted quantitative proteomics could then help to validate such a relationship in a large cohort of patient samples.
Five-year view
Proteomics has evolved as a valuable tool for the study of proteasomes for both fundamental and clinical purposes. Indeed, while 2DE separation of purified proteasome complexes and global proteomic approaches can play an important role in understanding the structure/function relationship of proteasome subtypes and in elucidating the cellular pathways affected during proteasome inhibition, targeted strategies such as MRM are on the way to providing promising therapeutic approaches for the quantitative analysis of proteasomes in biological fluids or for the detection of pathology-related UPS components. Indeed, sensitive and specific analytical mass spectrometric strategies will be required to access the quantification of each low-abundance proteasome form in complex patient samples. Nevertheless, recent progress in the field of structural biology has been made using MS for the analysis of intact protein complexes. Applied to proteasome complexes, these approaches have contributed, for example, to the determination of the stoichiometry and the architecture of the 19S regulatory complex. One can foresee that this field, which brings complementary information, will combine efficiently with proteomics data in the near future to provide a more comprehensive view of proteasome complexes in the cell.
Table 1. Summary of the N-terminal processing and endogenous phosphorylation sites identified and localized by MS/MS on human, mouse and yeast proteasomes.
Key issues
• Proteasomes constitute highly heterogenous complexes and are regulated in a dynamic way.
• The different 20S proteasome subtypes act as platforms recruiting regulatory complexes and proteasome-interacting proteins of diverse structures and of unknown functions.
• The latest affinity purification strategies can target the whole diversity of endogenous proteasomes and proteasome-interacting proteins, and are compatible with clinical studies.
• 2D electrophoresis and shotgun approaches, associated with mass spectrometry, are complementary tools for the characterization of the structural diversities and networks of proteasomes.
• The new generation of high-resolution, sensitive and fast-sequencing LTQ Orbitrap mass spectrometers equipped with complementary collision-induced dissociation and electron transfer dissociation fragmentation modes results in a more confident identification of post-translational modifications on proteasome subunits. In particular, in association with protein fractionation and specific enrichment techniques, they enabled significant increases in our knowledge on the proteasome phosphorylation status.
• Quantitative proteomics has evolved as a valuable tool for the analysis of the biological function of proteasomes through characterization of the proteasome network and its dynamics, and of proteasome subcellular localization, in response to various stimuli.
• K48-linked polyubiquitin chains are probably the most efficient proteasome-targeting signals. Most of the other polyubiquitin linkages probably also address, although less efficiently, proteins to proteasomes, and this might help to modulate protein degradation in response to the cellular demand.
• Targeted proteomics using multiple reaction monitoring will probably play a growing role in biomarker validation in the near future. Multiple reaction monitoring could constitute an efficient complementary technique to ELISA for the detection of proteasome as a diagnostic and/or prognostic biomarker in body fluids.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell92, 291–294 (1998).
- Hershko A, Ciechanover A. The ubiquitin system. Annu. Rev. Biochem.67, 425–479 (1998).
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev.82, 373–428 (2002).
- Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays22, 442–451 (2000).
- Goldberg AL. On prions, proteasomes, and mad cows. N. Engl. J. Med.357, 1150–1152 (2007).
- Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol.23, 4776–4789 (2005).
- Wu WK, Cho CH, Lee CW et al. Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett.293, 15–22 (2010).
- Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch. Biochem. Biophys.383, 1–16 (2000).
- Bousquet-Dubouch MP, Uttenweiler-Joseph S, Ducoux-Petit M, Matondo M, Monsarrat B, Burlet-Schiltz O. Purification and proteomic analysis of 20S proteasomes from human cells. Methods Mol. Biol.432, 301–320 (2008).
- Zoeger A, Blau M, Egerer K, Feist E, Dahlmann B. Circulating proteasomes are functional and have a subtype pattern distinct from 20S proteasomes in major blood cells. Clin. Chem.52, 2079–2086 (2006).
- Klare N, Seeger M, Janek K, Jungblut PR, Dahlmann B. Intermediate-type 20 S proteasomes in HeLa cells: “asymmetric” subunit composition, diversity and adaptation. J. Mol. Biol.373, 1–10 (2007).
- Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J. Mol. Biol.303, 643–653 (2000).
- Drews O, Wildgruber R, Zong C et al. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol. Cell Proteomics6, 2021–2031 (2007).
- Guillaume B, Chapiro J, Stroobant V et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl Acad. Sci. USA107(43), 18599–18604 (2010) .
- Murata S, Sasaki K, Kishimoto T et al. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science316, 1349–1353 (2007).
- Seifert U, Bialy LP, Ebstein F et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell142, 613–624 (2010).
- Gomes AV, Zong C, Edmondson RD et al. Mapping the murine cardiac 26S proteasome complexes. Circ. Res.99, 362–371 (2006).
- Froment C, Uttenweiler-Joseph S, Bousquet-Dubouch MP et al. A quantitative proteomic approach using two-dimensional gel electrophoresis and isotope-coded affinity tag labeling for studying human 20S proteasome heterogeneity. Proteomics5, 2351–2363 (2005).
- Boisvert FM, Lam YW, Lamont D, Lamont AI. A quantitative proteomic analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell Proteomics9, 457–470 (2010).
- Baldin V, Militello M, Thomas Y et al. A novel role for PA28γ-proteasome in nuclear speckle organization and SR protein trafficking. Mol. Biol. Cell19, 1706–1716 (2008).
- Yan W, Hwang D, Aebersold R. Quantitative proteomic analysis to profile dynamic changes in the spatial distribution of cellular proteins. Methods Mol. Biol.432, 389–401 (2008).
- Pelletier S, Schuurman KG, Berkers CR, Ovaa H, Heck AJ, Raijmakers R. Quantifying cross-tissue diversity in proteasome complexes by mass spectrometry. Mol. Biosyst.6, 1450–1453 (2010).
- Lu H, Zong C, Wang Y et al. Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol. Cell Proteomics7, 2073–2089 (2008).
- Raijmakers R, Berkers CR, de Jong A, Ovaa H, Heck AJ, Mohammed S. Automated online sequential isotope labeling for protein quantitation applied to proteasome tissue-specific diversity. Mol. Cell Proteomics7, 1755–1762 (2008).
- Haass C, Kloetzel PM. The Drosophila proteasome undergoes changes in its subunit pattern during development. Exp. Cell Res.180, 243–252 (1989).
- Hutson MR, Rhodes MR, Kirby ML. Differential expression of a proteasomal subunit during chick development. Biochem. Biophys. Res. Commun.234, 216–223 (1997).
- Schmidt F, Dahlmann B, Hustoft HK et al. Quantitative proteome analysis of the 20S proteasome of apoptotic Jurkat T cells. Amino Acids41(2), 351–361 (2010).
- Visekruna A, Joeris T, Schmidt N et al. Comparative expression analysis and characterization of 20S proteasomes in human intestinal tissues: the proteasome pattern as diagnostic tool for IBD patients. Inflamm. Bowel Dis.15, 526–533 (2009).
- Visekruna A, Slavova N, Dullat S et al. Expression of catalytic proteasome subunits in the gut of patients with Crohn’s disease. Int. J. Colorectal Dis.24, 1133–1139 (2009).
- McNaught KS, Jnobaptiste R, Jackson T, Jengelley TA. The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson’s disease. Synapse64, 241–250 (2010).
- Lemaire R, Menguellet SA, Stauber J et al. Specific MALDI imaging and profiling for biomarker hunting and validation: fragment of the 11S proteasome activator complex, Reg α fragment, is a new potential ovary cancer biomarker. J. Proteome Res.6, 4127–4134 (2007).
- Glickman MH, Raveh D. Proteasome plasticity. FEBS Lett.579, 3214–3223 (2005).
- Hanna J, Finley D. A proteasome for all occasions. FEBS Lett.581, 2854–2861 (2007).
- Kim HM, Yu Y, Cheng Y. Structure characterization of the 26S proteasome. Biochim. Biophys. Acta1809, 67–79 (2011).
- Bohn S, Beck F, Sakata E et al. Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. Proc. Natl Acad. Sci. USA107, 20992–20997 (2010).
- Nickell S, Beck F, Scheres SH et al. Insights into the molecular architecture of the 26S proteasome. Proc. Natl Acad. Sci. USA106, 11943–11947 (2009).
- Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol.8, 645–654 (2007).
- Kocher T, Superti-Furga G. Mass spectrometry-based functional proteomics: from molecular machines to protein networks. Nat. Methods4, 807–815 (2007).
- Kaake RM, Wang X, Huang L. Profiling of protein interaction networks of protein complexes using affinity purification and quantitative mass spectrometry. Mol. Cell Proteomics9, 1650–1665 (2010).
- Wang X, Guerrero C, Kaiser P, Huang L. Proteomics of proteasome complexes and ubiquitinated proteins. Expert Rev. Proteomics4, 649–665 (2007).
- Drews O, Zong C, Ping P. Exploring proteasome complexes by proteomic approaches. Proteomics7, 1047–1058 (2007).
- Verma R, Chen S, Feldman R et al. Proteasomal proteomics: identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell11, 3425–3439 (2000).
- Leggett DS, Hanna J, Borodovsky A et al. Multiple associated proteins regulate proteasome structure and function. Mol. Cell10, 495–507 (2002).
- Yang P, Fu H, Walker J et al. Purification of the Arabidopsis 26 S proteasome: biochemical and molecular analyses revealed the presence of multiple isoforms. J. Biol. Chem.279, 6401–6413 (2004).
- Hirano Y, Murata S, Tanaka K. Large- and small-scale purification of mammalian 26S proteasomes. Methods Enzymol.399, 227–240 (2005).
- Horiguchi R, Dohra H, Tokumoto T. Comparative proteome analysis of changes in the 26S proteasome during oocyte maturation in goldfish. Proteomics6, 4195–4202 (2006).
- Leggett DS, Glickman MH, Finley D. Purification of proteasomes, proteasome subcomplexes, and proteasome-associated proteins from budding yeast. Methods Mol. Biol.301, 57–70 (2005).
- Camacho-Carvajal MM, Wollscheid B, Aebersold R, Steimle V, Schamel WW. Two-dimensional Blue native/SDS gel electrophoresis of multi-protein complexes from whole cellular lysates: a proteomics approach. Mol. Cell Proteomics3, 176–182 (2004).
- Shibatani T, Carlson EJ, Larabee F, McCormack AL, Fruh K, Skach WR. Global organization and function of mammalian cytosolic proteasome pools: Implications for PA28 and 19S regulatory complexes. Mol. Biol. Cell17, 4962–4971 (2006).
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat. Protoc.1, 418–428 (2006).
- Elsasser S, Chandler-Militello D, Muller B, Hanna J, Finley D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem.279, 26817–26822 (2004).
- Oeljeklaus S, Meyer HE, Warscheid B. New dimensions in the study of protein complexes using quantitative mass spectrometry. FEBS Lett.583, 1674–1683 (2009).
- Hamazaki J, Iemura S, Natsume T, Yashiroda H, Tanaka K, Murata S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J.25, 4524–4536 (2006).
- Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, Goldberg AL. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J.25, 5742–5753 (2006).
- Stanhill A, Haynes CM, Zhang Y et al. An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol. Cell23, 875–885 (2006).
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol.17, 1030–1032 (1999).
- Guerrero C, Milenkovic T, Przulj N, Kaiser P, Huang L. Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl Acad. Sci. USA105, 13333–13338 (2008).
- Sikder D, Johnston SA, Kodadek T. Widespread, but non-identical, association of proteasomal 19 and 20 S proteins with yeast chromatin. J. Biol. Chem.281, 27346–27355 (2006).
- Collins GA, Tansey WP. The proteasome: a utility tool for transcription? Curr. Opin. Genet. Dev.16, 197–202 (2006).
- Tai HC, Besche H, Goldberg AL, Schuman EM. Characterization of the brain 26S proteasome and its interacting proteins. Front Mol. Neurosci.3, pii12 (2010).
- Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-γ and contribution to ATP-dependent proteolysis. J. Biol. Chem.275, 14336–14345 (2000).
- Giusti L, Iacconi P, Ciregia F et al. Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J. Proteome Res.7, 4079–4088 (2008).
- Wang Z, Feng X, Liu X et al. Involvement of potential pathways in malignant transformation from oral leukoplakia to oral squamous cell carcinoma revealed by proteomic analysis. BMC Genomics10, 383 (2009).
- Moriishi K, Shoji I, Mori Y et al. Involvement of PA28γ in the propagation of hepatitis C virus. Hepatology52, 411–420 (2010).
- Gao G, Wong J, Zhang J et al. Proteasome activator REG{γ} enhances coxsackieviral infection via facilitating p53 degradation. J. Virol.84(21), 11056–11066 (2010).
- Bousquet-Dubouch MP, Nguen S, Bouyssie D et al. Chronic ethanol feeding affects proteasome-interacting proteins. Proteomics9, 3609–3622 (2009).
- Scanlon TC, Gottlieb B, Durcan TM, Fon EA, Beitel LK, Trifiro MA. Isolation of human proteasomes and putative proteasome-interacting proteins using a novel affinity chromatography method. Exp. Cell Res.315(2), 176–189 (2008).
- Besche H, Haas W, Gygi S, Goldberg A. Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry48(11), 2538–2549 (2009).
- Chen L, Madura K. Evidence for distinct functions for human DNA repair factors hHR23A and hHR23B. FEBS Lett.580, 3401–3408 (2006).
- Hofmann K, Falquet L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem. Sci.26, 347–350 (2001).
- Rechsteiner M, Hill CP. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol.15, 27–33 (2005).
- Babbitt SE, Kiss A, Deffenbaugh AE et al. ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell121, 553–565 (2005).
- Book AJ, Gladman NP, Lee SS, Scalf M, Smith LM, Vierstra RD. Affinity purification of the Arabidopsis 26S proteasome reveals a diverse array of plant proteolytic complexes. J. Biol. Chem.285(33), 25554–25569 (2010).
- Bousquet-Dubouch MP, Baudelet E, Guerin F et al. Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol. Cell Proteomics8, 1150–1164 (2009).
- Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ. Res.107, 1094–1101 (2010).
- Wiseman RL, Chin KT, Haynes CM et al. Thioredoxin-related protein 32 is an arsenite-regulated thiol reductase of the proteasome 19 S particle. J. Biol. Chem.284, 15233–15245 (2009).
- Hendil KB. Development and use of antiproteasome monoclonal antibodies. Methods Enzymol.398, 439–453 (2005).
- Claverol S, Burlet-Schiltz O, Girbal-Neuhauser E, Gairin JE, Monsarrat B. Mapping and structural dissection of human 20 S proteasome using proteomic approaches. Mol. Cell Proteomics1, 567–578 (2002).
- Ducoux-Petit M, Uttenweiler-Joseph S, Brichory F et al. Scaled-down purification protocol to access proteomic analysis of 20S proteasome from human tissue samples: comparison of normal and tumor colorectal cells. J. Proteome Res.7, 2852–2859 (2008).
- Jariel-Encontre I, Bossis G, Piechaczyk M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta1786, 153–177 (2008).
- Vasilescu J, Guo X, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics4, 3845–3854 (2004).
- Hartmann-Petersen R, Tanaka K, Hendil KB. Quaternary structure of the ATPase complex of human 26S proteasomes determined by chemical cross-linking. Arch. Biochem. Biophys.386, 89–94 (2001).
- Kao A, Chiu CL, Vellucci D et al. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteomics10(1), M110.002212 (2010).
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci.25, 99–104 (2000).
- Wodak SJ, Pu S, Vlasblom J, Seraphin B. Challenges and rewards of interaction proteomics. Mol. Cell Proteomics8, 3–18 (2009).
- Yang W, Steen H, Freeman MR. Proteomic approaches to the analysis of multiprotein signaling complexes. Proteomics8, 832–851 (2008).
- Matsuoka S, Ballif BA, Smogorzewska A et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science316, 1160–1166 (2007).
- Pflieger D, Junger MA, Muller M et al. Quantitative proteomic analysis of protein complexes: concurrent identification of interactors and their state of phosphorylation. Mol. Cell Proteomics7, 326–346 (2008).
- Olsen JV, Blagoev B, Gnad F et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell127, 635–648 (2006).
- Vermeulen M, Hubner NC, Mann M. High confidence determination of specific protein-protein interactions using quantitative mass spectrometry. Curr. Opin. Biotechnol.19, 331–337 (2008).
- Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell86, 961–972 (1996).
- Kautto L, Grinyer J, Birch D et al. Rapid purification method for the 26S proteasome from the filamentous fungus Trichoderma reesei. Protein Expr. Purif.67, 156–163 (2009).
- Kikuchi J, Iwafune Y, Akiyama T et al. Co- and post-translational modifications of the 26S proteasome in yeast. Proteomics10(15), 2769–2779 (2010).
- Jorgensen JP, Lauridsen AM, Kristensen P et al. Adrm1, a putative cell adhesion regulating protein, is a novel proteasome-associated factor. J. Mol. Biol.360, 1043–1052 (2006).
- Wang D, Zong C, Koag MC et al. Proteome dynamics and proteome function of cardiac 19S proteasomes. Mol. Cell Proteomics10, M110006122 (2011).
- Andersen KM, Madsen L, Prag S et al. Thioredoxin Txnl1/TRP32 is a redox-active cofactor of the 26 S proteasome. J. Biol. Chem.284, 15246–15254 (2009).
- Wang X, Huang L. Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell Proteomics7, 46–57 (2008).
- Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, Huang L. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry46, 3553–3565 (2007).
- Schmidt F, Dahlmann B, Janek K et al. Comprehensive quantitative proteome analysis of 20S proteasome subtypes from rat liver by isotope coded affinity tag and 2-D gel-based approaches. Proteomics6, 4622–4632 (2006).
- Uttenweiler-Joseph S, Claverol S, Sylvius L, Bousquet-Dubouch MP, Burlet-Schiltz O, Monsarrat B. Toward a full characterization of the human 20S proteasome subunits and their isoforms by a combination of proteomic approaches. Methods Mol. Biol.484, 111–130 (2008).
- Marcantonio M, Trost M, Courcelles M, Desjardins M, Thibault P. Combined enzymatic and data mining approaches for comprehensive phosphoproteome analyses: application to cell signaling events of interferon-γ-stimulated macrophages. Mol. Cell Proteomics7, 645–660 (2008).
- Ishihama Y, Wei FY, Aoshima K, Sato T, Kuromitsu J, Oda Y. Enhancement of the efficiency of phosphoproteomic identification by removing phosphates after phosphopeptide enrichment. J. Proteome Res.6, 1139–1144 (2007).
- Wei X, Li L. Comparative glycoproteomics: approaches and applications. Brief Funct. Genomic Proteomic8, 104–113 (2009).
- Dai Z, Zhou J, Qiu SJ, Liu YK, Fan J. Lectin-based glycoproteomics to explore and analyze hepatocellular carcinoma-related glycoprotein markers. Electrophoresis30, 2957–2966 (2009).
- Steinberg TH, Pretty On Top K, Berggren KN et al. Rapid and simple single nanogram detection of glycoproteins in polyacrylamide gels and on electroblots. Proteomics1, 841–855 (2001).
- Steinberg TH, Agnew BJ, Gee KR et al. Global quantitative phosphoprotein analysis using multiplexed proteomics technology. Proteomics3, 1128–1144 (2003).
- Riederer BM. Non-covalent and covalent protein labeling in two-dimensional gel electrophoresis. J. Proteomics71, 231–244 (2008).
- Riederer IM, Herrero RM, Leuba G, Riederer BM. Serial protein labeling with infrared maleimide dyes to identify cysteine modifications. J. Proteomics71, 222–230 (2008).
- Wu J, Lenchik NJ, Pabst MJ, Solomon SS, Shull J, Gerling IC. Functional characterization of two-dimensional gel-separated proteins using sequential staining. Electrophoresis26, 225–237 (2005).
- Zong C, Young GW, Wang Y et al. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics8, 5025–5037 (2008).
- Wiesner J, Premsler T, Sickmann A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics8, 4466–4483 (2008).
- Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell Proteomics6, 1942–1951 (2007).
- Boersema PJ, Mohammed S, Heck AJ. Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom.44, 861–878 (2009).
- Jung HR, Pasini D, Helin K, Jensen ON. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell Proteomics9, 838–850 (2010).
- Mischerikow N, Altelaar AF, Navarro JD, Mohammed S, Heck A. Comparative assessment of site assignments in CID and ETD spectra of phosphopeptides discloses limited relocation of phosphate groups. Mol. Cell Proteomics9(10), 2140–2148(2010).
- Chi A, Huttenhower C, Geer LY et al. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl Acad. Sci. USA104, 2193–2198 (2007).
- Sumegi M, Hunyadi-Gulyas E, Medzihradszky KF, Udvardy A. 26S proteasome subunits are O-linked N-acetylglucosamine-modified in Drosophila melanogaster. Biochem. Biophys. Res. Commun.312, 1284–1289 (2003).
- Klement E, Lipinszki Z, Kupihar Z, Udvardy A, Medzihradszky KF. Enrichment of O-GlcNAc modified proteins by the periodate oxidation-hydrazide resin capture approach. J. Proteome Res.9, 2200–2206 (2010).
- Peng J, Schwartz D, Elias JE et al. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol.21, 921–926 (2003).
- Denis NJ, Vasilescu J, Lambert JP, Smith JC, Figeys D. Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics7, 868–874 (2007).
- Isasa M, Katz EJ, Kim W et al. Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol. Cell38, 733–745 (2010).
- Ventadour S, Jarzaguet M, Wing SS et al. A new method of purification of proteasome substrates reveals polyubiquitination of 20 S proteasome subunits. J. Biol. Chem.282, 5302–5309 (2007).
- Matsumoto M, Hatakeyama S, Oyamada K, Oda Y, Nishimura T, Nakayama KI. Large-scale analysis of the human ubiquitin-related proteome. Proteomics5, 4145–4151 (2005).
- Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of proteasome β5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem.280, 11840–11850 (2005).
- Gillardon F, Kloss A, Berg M et al. The 20S proteasome isolated from Alzheimer’s disease brain shows post-translational modifications but unchanged proteolytic activity. J. Neurochem.101, 1483–1490 (2007).
- Tsimokha AS, Mittenberg AG, Kulichkova VA, Kozhukharova IV, Gause LN, Konstantinova IM. Changes in composition and activities of 26S proteasomes under the action of doxorubicin--apoptosis inductor of erythroleukemic K562 cells. Cell Biol. Int.31, 338–348 (2007).
- Lee SH, Park Y, Yoon SK, Yoon JB. Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. J. Biol. Chem.285(53), 41280–41289 (2010).
- Domanski D, Murphy LC, Borchers CH. Assay development for the determination of phosphorylation stoichiometry using multiple reaction monitoring methods with and without phosphatase treatment: application to breast cancer signaling pathways. Anal. Chem.82, 5610–5620 (2010).
- Xu P, Duong DM, Seyfried NT et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell137, 133–145 (2009).
- Kirkpatrick DS, Hathaway NA, Hanna J et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol.8, 700–710 (2006).
- Jacobson AD, Zhang NY, Xu P et al. The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. J. Biol. Chem.284, 35485–35494 (2009).
- Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol.190, 491–500 (2010).
- Chouduri AU, Tokumoto T, Dohra H, Ushimaru T, Yamada S. Functional and biochemical characterization of the 20S proteasome in a yeast temperature-sensitive mutant, rpt6-1. BMC Biochem.9, 20 (2008).
- Minden JS, Dowd SR, Meyer HE, Stuhler K. Difference gel electrophoresis. Electrophoresis30(Suppl. 1), S156–S161 (2009).
- Timms JF, Cramer R. Difference gel electrophoresis. Proteomics8, 4886–4897 (2008).
- Agarwal NK, Mueller GA, Mueller C, Streich JH, Asif AR, Dihazi H. Expression proteomics of acute promyelocytic leukaemia cells treated with methotrexate. Biochim. Biophys. Acta1804, 918–928 (2010).
- Visentin M, Simula MP, Sartor F, Petrucco A, De Re V, Toffoli G. Identification of proteins associated to multi-drug resistance in LoVo human colon cancer cells. Int. J. Oncol.34, 1281–1289 (2009).
- Gomes AV, Young GW, Wang Y et al. Contrasting proteome biology and functional heterogeneity of the 20 S proteasome complexes in mammalian tissues. Mol. Cell Proteomics8, 302–315 (2009).
- Macagno A, Gilliet M, Sallusto F, Lanzavecchia A, Nestle FO, Groettrup M. Dendritic cells up-regulate immunoproteasomes and the proteasome regulator PA28 during maturation. Eur. J. Immunol.29, 4037–4042 (1999).
- Murata S, Takahama Y, Tanaka K. Thymoproteasome: probable role in generating positively selecting peptides. Curr. Opin. Immunol.20, 192–196 (2008).
- Trinkle-Mulcahy L, Boulon S, Lam YW et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol.183, 223–239 (2008).
- Selbach M, Mann M. Protein interaction screening by quantitative immunoprecipitation combined with knockdown (QUICK). Nat. Methods3, 981–983 (2006).
- Ong SE, Blagoev B, Kratchmarova I et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics1, 376–386 (2002).
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol.17, 994–999 (1999).
- Schmidt A, Kellermann J, Lottspeich F. A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics5, 4–15 (2005).
- Ross PL, Huang YN, Marchese JN et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics3, 1154–1169 (2004).
- Old WM, Meyer-Arendt K, Aveline-Wolf L et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell Proteomics4, 1487–1502 (2005).
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res.5, 2339–2347 (2006).
- Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol.17, 1807–1819 (2006).
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp. Gerontol.40, 622–633 (2005).
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature443, 780–786 (2006).
- Lopez-Otin C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat. Rev. Cancer10, 278–292 (2010).
- Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell Proteomics10(5), R110.006924 (2011).
- Matondo M, Bousquet-Dubouch MP, Gallay N et al. Proteasome inhibitor-induced apoptosis in acute myeloid leukemia: a correlation with the proteasome status. Leuk. Res.34(4), 498–506 (2009).
- Kaake RM, Milenkovic T, Przulj N, Kaiser P, Huang L. Characterization of cell cycle specific protein interaction networks of the yeast 26S proteasome complex by the QTAX strategy. J. Proteome Res.9, 2016–2029 (2010).
- Bonda DJ, Wang X, Perry G et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology59, 290–294 (2010).
- Tonoki A, Kuranaga E, Tomioka T et al. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell Biol.29, 1095–1106 (2009).
- Zabel C, Nguyen HP, Hin SC, Hartl D, Mao L, Klose J. Proteasome and oxidative phoshorylation changes may explain why aging is a risk factor for neurodegenerative disorders. J. Proteomics73, 2230–2238 (2010).
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature426, 895–899 (2003).
- Powell SR, Wang P, Divald A et al. Aggregates of oxidized proteins (lipofuscin) induce apoptosis through proteasome inhibition and dysregulation of proapoptotic proteins. Free Radic. Biol. Med.38, 1093–1101 (2005).
- Keller JN, Huang FF, Zhu H, Yu J, Ho YS, Kindy TS. Oxidative stress-associated impairment of proteasome activity during ischemia-reperfusion injury. J. Cereb. Blood Flow Metab.20, 1467–1473 (2000).
- Farout L, Friguet B. Proteasome function in aging and oxidative stress: implications in protein maintenance failure. Antioxid. Redox. Signal.8, 205–216 (2006).
- Fataccioli V, Andraud E, Gentil M, French SW, Rouach H. Effects of chronic ethanol administration on rat liver proteasome activities: relationship with oxidative stress. Hepatology29, 14–20 (1999).
- Bulteau AL, Lundberg KC, Humphries KM et al. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem.276, 30057–30063 (2001).
- Bardag-Gorce F, Li J, French BA, French SW. The effect of ethanol-induced CYP2E1 on proteasome activity: the role of 4-hydroxynonenal. Exp. Mol. Pathol.78, 109–115 (2005).
- Bardag-Gorce F, Venkatesh R, Li J, French BA, French SW. Hyperphosphorylation of rat liver proteasome subunits: the effects of ethanol and okadaic acid are compared. Life Sci.75, 585–597 (2004).
- Demasi M, Silva GM, Netto LE. 20 S proteasome from Saccharomyces cerevisiae is responsive to redox modifications and is S-glutathionylated. J. Biol. Chem.278, 679–685 (2003).
- Zmijewski JW, Banerjee S, Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J. Biol. Chem.284, 22213–22221 (2009).
- Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry44, 13893–13901 (2005).
- Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal.3, ra88 (2010).
- Sixt SU, Dahlmann B. Extracellular, circulating proteasomes and ubiquitin – incidence and relevance. Biochim. Biophys. Acta1782, 817–823 (2008).
- Andreyev AY, Shen Z, Guan Z et al. Application of proteomic marker ensembles to subcellular organelle identification. Mol. Cell Proteomics9, 388–402 (2010).
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature426, 570–574 (2003).
- Foster LJ, de Hoog CL, Zhang Y, Xie X, Mootha VK, Mann M. A mammalian organelle map by protein correlation profiling. Cell125, 187–199 (2006).
- Dunkley TP, Hester S, Shadforth IP et al. Mapping the Arabidopsis organelle proteome. Proc. Natl Acad. Sci. USA103, 6518–6523 (2006).
- Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J.21, 3516–3525 (2002).
- Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol.20, 391–401 (2010).
- Gallastegui N, Groll M. The 26S proteasome: assembly and function of a destructive machine. Trends Biochem. Sci.35, 634–642 (2010).
- Matiuhin Y, Kirkpatrick DS, Ziv I et al. Extraproteasomal Rpn10 restricts access of the polyubiquitin-binding protein Dsk2 to proteasome. Mol. Cell32, 415–425 (2008).
- Zhang D, Chen T, Ziv I et al. Together, Rpn10 and Dsk2 can serve as a polyubiquitin chain-length sensor. Mol. Cell36, 1018–1033 (2009).
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol.8, 610–616 (2004).
- Wickliffe K, Williamson A, Jin L, Rape M. The multiple layers of ubiquitin-dependent cell cycle control. Chem. Rev.109, 1537–1548 (2009).
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature458, 422–429 (2009).
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem.78, 477–513 (2009).
- Finley D, Sadis S, Monia BP et al. Inhibition of proteolysis and cell cycle progression in a multiubiquitination-deficient yeast mutant. Mol. Cell Biol.14, 5501–5509 (1994).
- Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J. Biol. Chem.269, 7059–7061 (1994).
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell33, 275–286 (2009).
- Baboshina OV, Haas AL. Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2EPF and RAD6 are recognized by 26 S proteasome subunit 5. J. Biol. Chem.271, 2823–2831 (1996).
- Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell133, 653–665 (2008).
- Meierhofer D, Wang X, Huang L, Kaiser P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome Res.7, 4566–4576 (2008).
- Saeki Y, Kudo T, Sone T et al. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J.28, 359–371 (2009).
- Bennett EJ, Shaler TA, Woodman B et al. Global changes to the ubiquitin system in Huntington’s disease. Nature448, 704–708 (2007).
- Kim HT, Kim KP, Lledias F et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem.282, 17375–17386 (2007).
- Hofmann RM, Pickart CM. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J. Biol. Chem.276, 27936–27943 (2001).
- Tagwerker C, Flick K, Cui M et al. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol. Cell Proteomics5, 737–748 (2006).
- Saracco SA, Hansson M, Scalf M, Walker JM, Smith LM, Vierstra RD. Tandem affinity purification and mass spectrometric analysis of ubiquitylated proteins in Arabidopsis. Plant J.59, 344–358 (2009).
- Kirkpatrick DS, Weldon SF, Tsaprailis G, Liebler DC, Gandolfi AJ. Proteomic identification of ubiquitinated proteins from human cells expressing His-tagged ubiquitin. Proteomics5, 2104–2111 (2005).
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep.10, 1250–1258 (2009).
- Burande CF, Heuze ML, Lamsoul I, Monsarrat B, Uttenweiler-Joseph S, Lutz PG. A label-free quantitative proteomics strategy to identify E3 ubiquitin ligase substrates targeted to proteasome degradation. Mol. Cell Proteomics8, 1719–1727 (2009).
- Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia21, 30–36 (2007).
- Doll D, Sarikas A, Krajcik R, Zolk O. Proteomic expression analysis of cardiomyocytes subjected to proteasome inhibition. Biochem. Biophys. Res. Commun.353, 436–442 (2007).
- Bieler S, Meiners S, Stangl V, Pohl T, Stangl K. Comprehensive proteomic and transcriptomic analysis reveals early induction of a protective anti-oxidative stress response by low-dose proteasome inhibition. Proteomics9, 3257–3267 (2009).
- Loeffler-Ragg J, Mueller D, Gamerith G et al. Proteomic identification of aldo-keto reductase AKR1B10 induction after treatment of colorectal cancer cells with the proteasome inhibitor bortezomib. Mol.Cancer Ther.8, 1995–2006 (2009).
- Weinkauf M, Zimmermann Y, Hartmann E et al. 2-D PAGE-based comparison of proteasome inhibitor bortezomib in sensitive and resistant mantle cell lymphoma. Electrophoresis30, 974–986 (2009).
- Wilde IB, Brack M, Winget JM, Mayor T. Proteomic characterization of aggregating proteins after the inhibition of the ubiquitin proteasome system. J. Proteome Res.10(3), 1062–1072 (2011).
- Kumatori A, Tanaka K, Inamura N et al. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl Acad. Sci. USA87, 7071–7075 (1990).
- Albright JM, Romero J, Saini V et al. Proteasomes in human bronchoalveolar lavage fluid after burn and inhalation injury. J. Burn Care Res.30, 948–956 (2009).
- Majetschak M, Zedler S, Romero J et al. Circulating proteasomes after burn injury. J. Burn Care Res.31, 243–250 (2010).
- Sixt SU, Adamzik M, Spyrka D et al. Alveolar extracellular 20S proteasome in patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med.179, 1098–1106 (2009).
- Stoebner PE, Lavabre-Bertrand T, Henry L et al. High plasma proteasome levels are detected in patients with metastatic malignant melanoma. Br. J. Dermatol.152, 948–953 (2005).
- Henry L, Lavabre-Bertrand T, Douche T et al. Diagnostic value and prognostic significance of plasmatic proteasome level in patients with melanoma. Exp. Dermatol.19, 1054–1059 (2010).
- Henry L, Lavabre-Bertrand T, Vercambre L et al. Plasma proteasome level is a reliable early marker of malignant transformation of liver cirrhosis. Gut58, 833–838 (2009).
- Yousef AA, Suliman GA, Mabrouk MM. The value of correlation of serum 20S proteasome concentration and percentage of lymphocytic apoptosis in critically ill patients: a prospective observational study. Crit. Care14, R215 (2010).
- Heubner M, Wimberger P, Dahlmann B et al. The prognostic impact of circulating proteasome concentrations in patients with epithelial ovarian cancer. Gynecol. Oncol.120, 233–238 (2011).
- Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: an unresolved issue. Neurologia25, 116–131 (2010).
- Colson K, Doss DS, Swift R, Tariman J. Expanding role of bortezomib in multiple myeloma: nursing implications. Cancer Nurs.31, 239–249 (2008).
- Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood113, 4667–4676 (2009).
- Egerer T, Martinez-Gamboa L, Dankof A et al. Tissue-specific up-regulation of the proteasome subunit β5i (LMP7) in Sjogren's syndrome. Arthritis Rheum.54, 1501–1508 (2006).
- Visekruna A, Joeris T, Seidel D et al. Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Invest.116, 3195–3203 (2006).
- Fitzpatrick LR, Small JS, Poritz LS, McKenna KJ, Koltun WA. Enhanced intestinal expression of the proteasome subunit low molecular mass polypeptide 2 in patients with inflammatory bowel disease. Dis. Colon Rectum50, 337–348; discussion 348–350 (2007).
- Vasuri F, Capizzi E, Bellavista E et al. Studies on immunoproteasome in human liver. Part I: absence in fetuses, presence in normal subjects, and increased levels in chronic active hepatitis and cirrhosis. Biochem. Biophys. Res. Commun.397, 301–306 (2010).
- Mishto M, Bellavista E, Santoro A et al. Immunoproteasome and LMP2 polymorphism in aged and Alzheimer’s disease brains. Neurobiol. Aging27, 54–66 (2006).
- Huttenhain R, Malmstrom J, Picotti P, Aebersold R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr. Opin. Chem. Biol.13, 518–525 (2009).
- Fortin T, Salvador A, Charrier JP et al. Multiple reaction monitoring cubed for protein quantification at the low nanogram/milliliter level in nondepleted human serum. Anal. Chem.81, 9343–9352 (2009).
- Fortin T, Salvador A, Charrier JP et al. Clinical quantitation of prostate-specific antigen biomarker in the low nanogram/milliliter range by conventional bore liquid chromatography-tandem mass spectrometry (multiple reaction monitoring) coupling and correlation with ELISA tests. Mol. Cell Proteomics8, 1006–1015 (2009).
- Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol.4, 222 (2008).
- Picotti P, Rinner O, Stallmach R et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods7, 43–46 (2010).
- Makawita S, Diamandis EP. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin. Chem.56, 212–222 (2010).
- Ang CS, Phung J, Nice EC. The discovery and validation of colorectal cancer biomarkers. Biomed. Chromatogr.25, 82–99 (2011).
- Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell92, 367–380 (1998).
- Groll M, Ditzel L, Lowe J et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature386, 463–471 (1997).
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell Proteomics7, 1389–1396 (2008).
- Li X, Gerber SA, Rudner AD et al. Large-scale phosphorylation analysis of α-factor-arrested Saccharomyces cerevisiae. J. Proteome Res.6, 1190–1197 (2007).
- Zong C, Gomes AV, Drews O et al. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ. Res.99, 372–380 (2006).
- Iwafune Y, Kawasaki H, Hirano H. Identification of three phosphorylation sites in the α7 subunit of the yeast 20S proteasome in vivo using mass spectrometry. Arch. Biochem. Biophys.431, 9–15 (2004).
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl Acad. Sci. USA104, 10364–10369 (2007).
- Dubiel W, Ferrell K, Rechsteiner M. Subunits of the regulatory complex of the 26S protease. Mol. Biol. Rep.21, 27–34 (1995).
- Coux O, Nothwang HG, Silva Pereira I, Recillas Targa F, Bey F, Scherrer K. Phylogenic relationships of the amino acid sequences of prosome (proteasome, MCP) subunits. Mol. Gen. Genet.245, 769–780 (1994).
- Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J. Biol. Chem.282, 22460–22471 (2007).
- Beausoleil SA, Jedrychowski M, Schwartz D et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl Acad. Sci. USA101, 12130–12135 (2004).