MFH: Malignant fibrous histiocytoma; MSC: Mesenchymal stem cell; RMS: Rhabdomyosarcoma.
Reproduced with permission from Citation[33].
![Figure 1. Mesenchymal stem cells, by virtue of their broad plasticity, may be at the origin of different histological subtypes of human sarcomas.MFH: Malignant fibrous histiocytoma; MSC: Mesenchymal stem cell; RMS: Rhabdomyosarcoma.Reproduced with permission from Citation[33].](/cms/asset/1f2c94ed-d45f-46cd-a477-04d42a1e7cca/iery_a_11221280_f0001_b.jpg)
CSC: Cancer stem cell; ESFT: Ewing’s sarcoma family tumor; MSC: Mesenchymal stem cell.
Sarcomas are rare malignant tumors that comprise less than 5% of all malignancies but approximately 15% of pediatric cancers. They arise in mesenchymal tissues and often display highly aggressive behavior with proclivity toward early hematogenous metastasis. Surgery, when applied early, can be curative, and preoperative chemotherapy has been shown to be effective in the management of localized chemosensitive tumors, significantly augmenting the proportion of long-term survivors. However, the tendency toward relapse is relatively high and the prognosis of chemoresistant and disseminated sarcomas remains excessively poor despite multimodal therapeutic approaches. Genetic analysis distinguishes two classes of sarcomas. One comprises tumors that display multiple complex karyotypic abnormalities with no specific pattern. The other is composed of tumors that harbor defined genetic mutations believed to underlie their pathogenesis, principally represented by single reciprocal signature chromosomal translocations. Identification of recurrent genetic abnormalities associated with specific sarcoma subtypes has not only facilitated their diagnosis but has greatly contributed toward understanding their biology Citation[1–6]. The fusion genes resulting from the translocations encode, for the most part, aberrant transcription factors believed to play an essential role in the pathogenesis of the corresponding sarcoma subtype by altering the gene-expression repertoire, which, in appropriately permissive cells, is believed to trigger transformation either alone or in combination with other genetic events.
Origin of sarcomas
Sarcomas are widely believed to develop as a result of genetic mutations in mesenchymal progenitor/stem cells, but their precise cellular origin remains unknown. Putative cells of origin of some sarcomas have been proposed based on their histological features, including, for example, osteoblasts for osteosarcoma and smooth muscle cells for leiomyosarcoma . However, for many of these malignancies that do not display a specific histological phenotype, such as synovial sarcoma or Ewing’s sarcoma, the cell of origin is still under debate. Several strategies have been employed in a effort to identify the putative cell of origin of sarcomas:
• Generation of transgenic mice expressing specific sarcoma-associated translocations driven by tissue-specific promoters; expression of the fusion genes in different primary cell populations in an effort to identify the cellular environment that is permissive for their expression and function;
• Gene-expression profile analysis of sarcoma-derived cell lines in which the specific chromosomal translocation-generated fusion genes had been knocked down to determine the molecular phenotype of the putative cell of origin prior to fusion protein expression;
• Comparison of sarcoma gene-expression profiles to those of a broad panel of normal cells or tissues.
Development of transgenic mouse models of defined human sarcomas has been moderately successful, due, at least in part, to the toxicity of sarcoma-associated fusion genes in most primary cells. However, at least three mouse models of sarcoma associated with specific chromosomal translocations have been generated that recapitulate many of the features of their human counterparts and provide clues as to their cell of origin. They include the conditional PAX3–FKHR knock-in model of alveolar rhabdomyosarcoma, where the fusion gene is expressed in terminally differentiated skeletal muscle cells Citation[7]; the conditional SYT–SSX-2 knock-in model where expression of the oncoprotein in immature myoblasts leads to induction of synovial sarcoma with 100% penetrance (while its expression in more differentiated cells induces myopathy without tumor initiation) Citation[8]; and the TLS/FUS–CHOP transgenic model, where ubiquitous expression of the TLS/FUS–CHOP transgene resulted in the exclusive generation of myxoid liposarcoma-like tumors in their classical anatomical locations Citation[9]. The additional strategies mentioned earlier have been useful in determining the origin of other sarcomas, and particularly that of Ewing’s sarcoma.
The quest for the ESFT cell of origin
Originally described in 1921 by James Ewing as an ‘endothelioma of the bone’, Ewing’s sarcoma, often referred to as Ewing’s sarcoma family tumors (ESFTs), is the second most common bone malignancy after osteosarcoma, arising in children and young adults with a peak incidence at age 15 years. Similar to other sarcomas, ESFTs are aggressive tumors with a tendency toward recurrence following resection and pronounced proclivity toward early hematogenous metastasis. In 85% of cases, ESFT cells harbor the translocation t(11;22)(q24;q12) that generates the EWSR1–FLI-1 fusion gene Citation[10]. In another 10–15% of cases the translocation t(21;12)(22;12) generates the EWSR1–ERG fusion, whereas the remaining 1–5% of cases may contain one of several possible translocations, each resulting in a fusion gene composed of a portion of the EWSR1 gene and a member of the ets transcription factor family.
All EWSR1–ETS fusion proteins contain the transactivation domain of EWSR1 and the DNA binding domain (DBD) of the corresponding ETS fusion partner, suggesting that enhanced and/or inappropriate expression of ETS fusion partner target genes resulting from potent transactivation by EWSR1 could lead to cellular transformation. However, there is evidence that the target gene specificity of the fusion protein is not identical to that of its component ETS factor Citation[11–13]. Several potential EWSR1–FLI-1 target genes whose induction may be implicated in transformation and/or tumor progression have been identified, including MYC, ID2, CCND1, PDGFC, IGF1, CAV1 and GLI1 (reviewed in Citation[14]), which are induced, and p21, p57kip, TGFβRII and IGFBP-3, which are repressed by the fusion protein. Growth inhibitory effects of antisense EWSR1–FLI-1 and EWSR1–FLI-1 siRNA expression in human Ewing sarcoma cell lines further support the notion that EWSR1–FLI-1 is implicated in ESFT development Citation[15], and EWSR1–FLI-1 appears to play a critical role in inducing the ESFT small round cell phenotype.
Interestingly, although most Ewing sarcomas occur in bone, 15% of primary ESFTs may arise in a variety of extraosseous sites Citation[16]. Ewing sarcoma cells express the lymphoid cell adhesion receptor CD99, several neural cell markers, including neural-specific enolase (NSE), S-100, synaptophysin and CD56 Citation[16], and mesenchymal markers, including vimentin. The wide range of organs and tissues from which ESFTs can arise, together with histological features that suggest poor differentiation with both mesenchymal and neuroectodermal features Citation[16], has fueled an as yet unresolved debate as to their neuroectodermal or mesenchymal origin. A major impediment toward the understanding ESFT biology, in general, and its cellular origin, in particular, is the lack of a transgenic animal model. Three alternative approaches have therefore been used to address the potential origins of ESFTs .
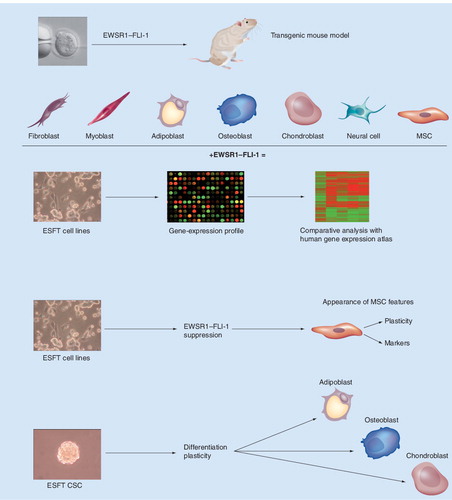
The first consisted of expression of EWSR1–FLI-1 in different primary or immortalized cellular backgrounds, in an attempt to define the permissive cellular environment for the oncogenic properties of the fusion protein. Expression of EWSR1–FLI-1 in murine NIH-3T3 cells resulted in anchorage-independent growth Citation[17] and accelerated tumorigenesis in immunocompromised mice with a tumor phenotype reminiscent of that of human Ewing sarcoma Citation[18]. These observations are consistent with the notion that EWSR1–FLI-1 can enhance oncogenesis and that it is largely responsible for the histological characteristics associated with ESFT. By contrast, the same approach using Rat-1 cells Citation[17], primary mouse embryonic fibroblasts (MEFs) Citation[19] and hTERT-immortalized human primary fibroblasts Citation[20] not only failed to induce transformation but resulted in growth arrest and apoptosis, underscoring the importance of the cellular environment for EWSR1–FLI-1-mediated oncogenesis. Work from our own and other laboratories has shown that expression of EWSR1–FLI-1 in primary mouse bone marrow-derived mesenchymal progenitor cells generates tumors that display hallmarks of Ewing’s sarcoma, including a small round cell phenotype, expression of ESFT-associated markers, IGF-1 dependence and induction or repression of numerous EWSR1–FLI-1 target genes Citation[21,22].
These observations were supported by subsequent studies where expression of EWSR1–FLI-1 in primary human mesenchymal stem cells (MSCs) resulted in the induction of numerous genes encoding neural cell markers and implicated in neural crest development and neuronal differentiation, in addition to most of the known EWSR1–FLI-1 target genes Citation[23,24]. Moreover, hMSCs expressing EWSR1–FLI-1 displayed a gene-expression profile bearing striking similarity to that of primary ESFTs.
The second strategy used to identify the normal counterpart of ESFT cells consisted of a bioinformatic comparison of primary Ewing’s sarcoma gene-expression profiles with those of a wide range of normal human cells and tissues. Two independent studies using this approach came to different conclusions. One study showed that Ewing’s sarcoma cells displayed the most significant similarity to endothelial and neural cells Citation[25], whereas the other revealed that among primary cells the highest similarity to ESFT cells was displayed by MSCs Citation[26].
Finally, a third strategy consisted of the gene-expression profile analysis of Ewing’s sarcoma cell lines subjected to stable shRNA-mediated EWSR1–FLI-1 suppression Citation[27]. This approach revealed that ESFT cells depleted of EWSR1–FLI-1 display hMSC features, including induction of the classical MSCs markers CD44 and CD73, and the typical trilineage differentiation plasticity displayed by primary MSCs. Consistent with these observations, we have recently identified cancer stem cells in ESFTs and found that they display marked mesenchymal trilineage plasticity, despite retaining EWSR1–FLI-1 expression. ESFT-initiating cells may, therefore, conserve the functional plasticity of their putative physiological precursors Citation[28].
Together, these independent studies provide solid evidence that MSCs display intrinsic permissiveness for the transforming potential of EWSR1–FLI-1 and constitute the right cells in the right location for EWSR1–FLI-1-mediated oncogenesis. Moreover, the reported migration of MSCs to all body compartements during the embryonic development could explain the broad tissue distribution of ESFT. The observed induction of genes implicated in neuronal differentiation and neural crest development indicates that EWSR1–FLI-1 can direct partial neuroectodermal differentiation of primary MSC, indicating that ESFTs need not arise from a neuroectodermal precursor to explain their primitive neuroectodermal phenotype. MSCs were originally defined by two properties that they display in culture: the ability to proliferate as clones and to differentiate into mesenchymal lineages, including osteocytes, chondrocytes and adipocytes Citation[29]. Subsequent work defined markers that enabled their prospective isolation, enrichment, and characterization. However, little was known regarding their in vivo biology, particularly with respect to their developmental derivation. Recent work has suggested that hematopoietic stem cells can generate MSCs Citation[30], potentially explaining the similarity between ESFTs and endothelial cells.
Two subsequent studies have shed further light onto MSC properties, suggesting that a significant portion of MSCs may originate from the neuroepithelium and neural crest stem cells (NCSCs) in vitro and in vivo. The first study showed that the earliest wave of mouse MSCs in the embryonic trunk is generated from Sox1+ neuroepithelium but not from mesoderm. The numbers of these neural-derived MSCs declined in the bone marrow over time such that they represent only a very small fraction of the MSC population by adulthood Citation[31]. The second study reported the derivation of NCSCs from human embryonic stem cells, and their cell culture condition-dependent propagation toward PNS and mesenchymal lineages, including MSCs Citation[32].
Taken together, these observations suggest that ESFTs may derive from a MSC of hematopoietic or neuroectodermal origin, capable of maintaining dual plasticity, and providing a convincing biological rationale for the phenotypic duality displayed by these tumors. They may thereby help lay to rest the duel between supporters of MSCs and NCSCs for the title of the ESFT cell of origin.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Bennicelli JL, Barr FG. Genetics and the biologic basis of sarcomas. Curr. Opin. Oncol.11(4), 267–274 (1999).
- Bennicelli JL, Barr FG. Chromosomal translocations and sarcomas. Curr. Opin. Oncol.14(4), 412–419 (2002).
- Busam KJ, Fletcher CD. The clinical role of molecular genetics in soft tissue tumor pathology. Cancer Metastasis Rev.16(1–2), 207–227 (1997).
- Dockhorn-Dworniczak B, Schafer KL, Blasius S et al. Assessment of molecular genetic detection of chromosome translocations in the differential diagnosis of pediatric sarcomas. Klin. Padiatr.209(4), 156–641 (1997).
- Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat. Rev. Cancer3(9), 685–694 (2003).
- Shipley J, Crew J, Gusterson B. The molecular biology of soft tissue sarcomas. Eur. J. Cancer29A(14), 2054–2058 (1993).
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev.18(21), 2614–2626 (2004).
- Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell.11(4), 375–388 (2007).
- Perez-Losada J, Pintado B, Gutierrez-Adan A et al. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene19(20), 2413–2422 (2000).
- Delattre O, Zucman J, Plougastel B et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature359(6391), 162–165 (1992).
- Arvand A, Bastians H, Welford SM, Thompson AD, Ruderman JV, Denny CT. EWS/FLI1 up regulates mE2-C, a cyclin-selective ubiquitin conjugating enzyme involved in cyclin B destruction. Oncogene17(16), 2039–2045 (1998).
- May WA, Arvand A, Thompson AD, Braun BS, Wright M, Denny CT. EWS/FLI1-induced manic fringe renders NIH 3T3 cells tumorigenic. Nat. Genet.17(4), 495–497 (1997).
- Thompson AD, Braun BS, Arvand A, et al. EAT-2 is a novel SH2 domain containing protein that is up regulated by Ewing’s sarcoma EWS/FLI1 fusion gene. Oncogene13(12), 2649–2658 (1996).
- Riggi N, Cironi L, Suva ML, Stamenkovic I. Sarcomas: genetics, signalling, and cellular origins. Part 1: the fellowship of TET. J. Pathol.213(1), 4–20 (2007).
- Kovar H, Aryee DN, Jug G, et al. EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro . Cell Growth Differ.7(4), 429–437 (1996).
- Ushigome S, Machinami R, Sorensen PH. Ewing sarcoma/primitive neuroectodermal tumor. In: WHO Classification of Tumors, Pathology and Genetics, Tumors of Soft Tissue and Bone. Fletcher CDM, Unni KK, Mertens F (Eds). 298–300 (2003).
- May WA, Gishizky ML, Lessnick SL et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl Acad. Sci. USA90(12), 5752–5756 (1993).
- Thompson AD, Teitell MA, Arvand A, Denny CT. Divergent Ewing’s sarcoma EWS/ETS fusions confer a common tumorigenic phenotype on NIH3T3 cells. Oncogene18(40), 5506–5513 (1999).
- Deneen B, Denny CT. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene20(46), 6731–6741 (2001).
- Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell.1(4), 393–401 (2002).
- Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA, Ilaria RL Jr. Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells results in EWS/FLI-1-dependent, ewing sarcoma-like tumors. Cancer Res.65(19), 8698–8705 (2005).
- Riggi N, Cironi L, Provero P, et al. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res.65(24), 11459–11468 (2005).
- Miyagawa Y, Okita H, Nakaijima H, et al. Inducible expression of chimeric EWS/ETS proteins confers Ewing’s family tumor-like phenotypes to human mesenchymal progenitor cells. Mol. Cell. Biol.28(7), 2125–2137 (2008).
- Riggi N, Suva ML, Suva D, et al. EWS–FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res.68(7), 2176–2185 (2008).
- Staege MS, Hutter C, Neumann I, et al. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res.64(22), 8213–8221 (2004).
- Kauer M, Ban J, Kofler R et al. A molecular function map of Ewing’s sarcoma. PLoS ONE4(4), e5415 (2009).
- Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell.11(5), 421–429 (2007).
- Suva ML, Riggi N, Stehle JC, et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res.69(5), 1776–1781 (2009).
- Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science276(5309), 71–74 (1997).
- Ogawa M, LaRue AC, Drake CJ. Hematopoietic origin of fibroblasts/myofibroblasts: its pathophysiologic implications. Blood108(9), 2893–2896 (2006).
- Takashima Y, Era T, Nakao K, et al. Neuroepithelial cells supply an initial transient wave of MSC differentiation. Cell129(7), 1377–1388 (2007).
- Lee G, Kim H, Elkabetz Y et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol.25(12), 1468–1475 (2007).
- Charytonowicz E. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell?. Cancer Lett.279(2), 126–136 (2008).