Abstract
A number of studies in murine models have suggested that the immune system may edit different tumors by forcing their expression profiles so that they escape immune reactions and proliferate. Glioblastoma (GB), the most frequent and aggressive primary brain tumor, provides a good example of this, thanks to the production of numerous immunosuppressive molecules (with TGF-β being of paramount importance), downregulation of the MHC complex and deregulation of the potential for antigen presentation by the surrounding microglia. Given that surgery, radiotherapy and chemotherapy with available protocols have limited effects on the survival of GB patients, different immunotherapy strategies have been developed, based on the use of dendritic cells, antibodies and peptide vaccination. Presently, bevacizumab, a humanized anti-VEGF antibody, provides the most successful example for immune-based treatment of GB, however, its action is limited in time, as the often tumor relapses due to still undefined immunoediting mechanisms. Altered function of EGF receptor-driven pathways is common in GB and is most frequently due to the presence of a deleted form named EGFRvIII, providing a unique cancer epitope that has been targeted by immunotherapy. A recent trial of GB immunotherapy based on vaccination with the EGFRvIII peptide has shown clinical benefit: interestingly most GBs at relapse were negative for EGFRvIII expression, a relevant, direct example of cancer immunoediting. Investigations on the mechanisms of GB immunoediting will lead to an increased understanding of the biology of this malignancy and hopefully provide novel therapeutic targets.
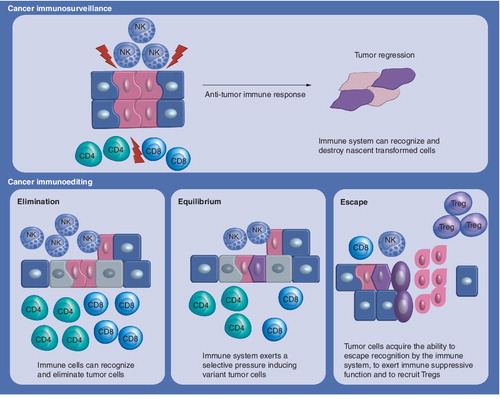
Immunoediting is a process triggered after the encounter between immune components and tumor cells and implies three different outcomes: elimination of cancer cells; equilibrium or immune selection of less immunogenic cancer arising during anti-tumor immune response; escape or expansion of tumor variants resistant to immune reaction.
NK: Natural killer; Treg: Regulatory T cell.
Due to the subsequent lack of antigen presentation, a T-cell-mediated immune response is evaded and tumor may become more advanced. Tumor cells can resist apoptosis by downregulating the death receptor. Apart from the defensive mechanisms that tumors use to avoid elimination, several ‘immunosubversion’ (active immunosuppressive) mechanisms against the immune system have also been reported. Tumors may inhibit CD8+ T-cell proliferation and promote CD4+ T-cell apoptosis by immunosuppressive cytokines and other factors, including TGF-β, IL-10 and prostaglandin E2, which have been reported to inhibit immune function and induce the activation or conversion of immunosuppressive cells, as well as CD4+ CD25+ Tregs and myeloid-derived suppressive cells.
BBB: Blood–brain barrier; CTL: Cytotoxic T lymphocyte; FasL: Fas ligand; HLA: Human leukocyte antigen; NK: Natural killer; TCR: T-cell receptor.
As shown recently by Sampson et al.Citation[156], the large majority of glioblastomas treated by vaccination were immunoedited and did not show EGFRvIII-positive cells.
DC: Dendritic cell; EGFR: EGF receptor.
![Figure 3. Different steps of the immune response generated by vaccination, with a peptide representing the specific epitope generated by the most frequent mutation of epidermal growth factor receptor (called EGFRvIII ΔEGFR in the figure).As shown recently by Sampson et al.Citation[156], the large majority of glioblastomas treated by vaccination were immunoedited and did not show EGFRvIII-positive cells.DC: Dendritic cell; EGFR: EGF receptor.](/cms/asset/5d9a0923-4992-4108-a621-5930ecf44a61/iery_a_11219632_f0003_b.jpg)
Cancer immunoediting: the concept
In 1909 Paul Ehrlich postulated that cancer occurs spontaneously in vivo and that the immune system is able to recognize neoplastic cells and provide protection against them Citation[1]. In 1950 Lewis Thomas introduced the theory of immunosurveillance Citation[2], subsequently developed by Sir MacFarlane Burnet Citation[3]. The theory states that immunosurveillance is a physiological function of the immune system. Cancerous cells are seen as foreign and thus can be constantly eliminated by immune surveillance. Evidence of immunosurveillance in humans are reported, including an increased incidence of virally induced tumors in immunosuppressed patients and reverse correlations between tumor-infiltrating lymphocytes and tumor survival Citation[4].
The capacity of the immune system to control and ‘sculpt’ cancer, defined as cancer immunoediting, is the result of three processes that may take place independently or in sequence: elimination (in which the immune system may erase the tumor in ‘naive’ hosts), equilibrium (proliferation of transformed cells is controlled by the immune system), and escape (tumor cell variants with decreased immunogenicity or with the capacity to evade immune responses become responsible for clinically evaluable cancers) Citation[5].
Elimination (cancer immunosurveillance)
The immune system provides one of the body’s main defenses against cancer. In cancer cells some of the surface antigens change and cause the formation of new immune defenders, including cytotoxic T lymphocytes (CTLs), natural killer (NK) cells and macrophages. Cells of the immune system involved in tumor responses are responsible for different functions: phagocytosis, antigen presentation to T cells, killing of tumor cells and regulation of humoral and cell-mediated immune responses. The presence of tumor cells and tumor antigens may initiate the release of danger signals, such as cytokines and heat-shock proteins, and induce activation and maturation of dendritic cells (DCs). Although conventional lymphatics are absent in the CNS, access across perivascular spaces and cerebrospinal space allows antigen-loaded DCs to migrate to cervical lymph nodes where they present tumor antigens to CD4+ and CD8+ T cells Citation[6,7]. After clonal expansion of T cells and migration from the lymph nodes, T cells mediate elimination of tumor cells. The effector T cells, CD4+ T cells and CD8+ CTLs, leave cervical lymph nodes after activation and migrate to the brain tumor site. The specific homing of T cells, including expression of CNS homing-specific integrin and chemokine receptors Citation[8], is probably imprinted by brain tumor antigen DCs Citation[9,10]. At the tumor site, the CD8+ T cells recognize the tumor cells and induce killing mediated by cytotoxic effector molecules; for example, perforin, granzyme B and IFN-γ. The tumor cell-killing action of the CD8+ CTLs is supported by the concurrent production of IL-2 by the CD4+ T cells, promoting intratumoral proliferation and expansion of the T cells.
Equilibrium
During the elimination step variants of tumor cells arise with different features, in particular they are more resistant to killing. It is possible that a variant may escape the elimination mechanism or recruit other cells with regulatory functions for self-protection and thus proliferate unchallenged. This process leads to an equilibrium, where mutations and genetic instability coupled with the selective pressure exerted by the immune system select for less immunogenic tumor cells, which will acquire the ability to escape recognition by the immune system.
One interesting example of immunoediting is provided by vaccination studies in breast cancer. Increased expression of HER-2/neu (c-erbB2) is associated with increased risk of recurrence in ductal carcinoma in situ (DCIS) and poorer prognosis in node-positive breast cancer. Before surgery, HER-2/neu-positive DCIS patients were vaccinated with DCs pulsed with HER-2/neu HLA class I and II peptides Citation[11]. Notably, the ratio between low levels of expression of CD28 and high levels of the inhibitory B7 ligand CTLA-4 was inverted after vaccination in CD8+ T cells of many patients, showing HER-2/neu-HLA-A2 tetramer staining. These changes may have contributed to breaking tumor-induced tolerance and were associated with increased production of IFN-γ by CD8+ T cells, as demonstrated by ELISPOT and other techniques. Interestingly, increased number of T cells infiltrating the tumor were detected after vaccination, mostly represented by CD4+ T cells and CD20+ B cells: postvaccine serum of six out of nine patients showed elevated complement-dependent lysis of HER-2/neu-positive cells. Seven out of 11 evaluable patients showed markedly decreased HER-2/neu expression in surgical specimens, suggesting an active process of ‘immunoediting’ for HER-2/neu-expressing tumor cells following vaccination Citation[11]. Thus, antigen or peptide loss is another way by which cancer cells avoid recognition by the immune system. When tumor cells acquire traits that enable survival, they enter a state of equilibrium with the immune system (the immune system suppresses tumor growth but is unable to eradicate tumor mass).
Escape
Tumor cells have been found to escape elimination by the immune system, thus being immunoedited, through a variety of mechanisms.
Escape from antigenic recognition
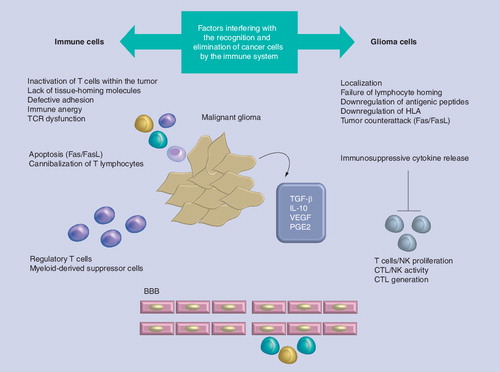
Immune evasion strategies that prevent recognition of cancer cells as foreign, defined as antigenicity, include (; ):
• Tumor localization in the CNS, which has been considered inaccessible to the immune system, is per se considered a factor that can interfere with immunosurveillance. In the healthy individual leukocyte trafficking into the CNS is very low and highly controlled by the blood–brain barrier (BBB). In general, resting lymphocytes fail to enter the CNS, whereas under inflammatory conditions, such as in multiple sclerosis or experimental autoimmune encephalomyelitis, they can readily cross the BBB. The observations that upon adoptive transfer of autoimmune encephalomyelitis into syngeneic healthy mice specific CD4+ T cells migrate into the CNS across the healthy BBB changed the concept of CNS immune privilege. These observations imply that the BBB may actively select lymphocyte entry into the CNS by allowing activated but not resting T cells to cross the BBB Citation[12,13]; and activated but not resting T cells have the receptors to bind corresponding ligands on the BBB. It was reported that brain tumors at earliest stages of formation in such areas are invisible to immune surveillance and thus cannot be targeted by immune reactions Citation[8,14]. Only when tissue damage is massive and in the presence of the destruction of the BBB, brain tumors are accessible to peripheral immune components Citation[14–16]. However, Tran Thang et al. have recently shown in a transgenic mouse model of astrocytoma (GFAP–V12HA–Ras mice) that the immune system is alerted at an early stage of gliomagenesis Citation[17]. In addition, other environments may constitute immune-privileged sites; for example, the eyes and gonads, where an inflammatory response would have detrimental consequences for organ homeostasis Citation[18];
• Failure of lymphocyte homing due to lack of adhesion molecules: in particular, T-cell homing in the CNS is regulated by α4β1 integrins and by their interaction with VCAM-1 on cerebral vascular endothelium Citation[10]. TGF-β may repress expression of these adhesion molecules causing failure of lymphocyte homing to the tumor Citation[19]. T-lymphocyte priming takes place in draining cervical lymph nodes and is characterized by an upregulation of α4 and β1 integrins involved in mediating lymphocyte migration Citation[10]. These α4-β1 integrins interact with VCAM-1 on cerebral vascular endothelium, facilitating the entry of activated T lymphocytes across the BBB. After CNS entry, antigen-specific reactivation of T lymphocytes is fundamental for T-cell persistence and amplification of T-cell-specific responses in the CNS Citation[20,21]. In particular, the CNS entry of CD4+ T cells is determined by the activation stage, whereas the CNS recruitment of CD8+ T cells is regulated by ‘antigen specificity’, which seems dependent on constitutive luminal expression of MHC class I by cerebral endothelium, but independent of antigen presentation by perivascular APC Citation[22];
• Antigen modulation, also defined as immunoselection, is a characteristic of cancer cells to evade attack by the immune system: indeed, they are able to generate variants lacking features that make them available for T-cell recognition and destruction Citation[23].
Cancer cells can evade recognition by T cells through mutations or gene deletions, leading to downregulation of key proteins involved in the antigen presentation machinery. In particular, it has been shown that the transporter associated with antigen presentation (TAP), responsible for loading peptides on MHC class I, and essential subunits in the immune proteasome (i.e., the low-molecular-mass polypeptide LMP-2 and LMP-7) are defective in cancer cells Citation[24,25]. Furthermore, Satoh et al. found that malignant gliomas express undetectable or very low levels of TAP1 protein and did not express TAP1 mRNA whereas LMP2 protein was not altered. They also found an upregulated expression of TAP1 and MHC class I molecule by IFN-γ and IFN-β, suggesting that malignant glioma cells may be less immunogenic due to low levels of TAP1 expression Citation[26].
Microglia are a relevant fraction of the CNS cell population, with a critical role in generating significant innate and adaptive immune responses. Effective antigen presentation requires interaction between the TCR and antigen peptides presented by the MHC of antigen-presenting cells (APCs). Different studies in gliomas have confirmed the presence of high levels of infiltrating microglia that, however, does not show MHC class II upregulation following stimulation Citation[27,28]. Furthermore, stimulated microglia in the presence of glioma cells downregulates the production of TNF-α and increases that of IL-10 Citation[29]. Also contributing to local immune suppression is the expression in glioma-associated microglia of B7-H1 and FASL, both inducing T-cell apoptosis Citation[30,31].
Escape from immunogenic response
Immune evasion strategies that prevent the induction of an effective immune response are outlined here (; ). For an overview also see Citation[201].
Release of immunosuppressive cytokines
Release of immunosuppressive cytokines, including TGF-β, IL-10, VEGF, prostaglandin E2, and other factors such as indoleamin 2,3 dioxygenase (IDO) and nitric-oxide synthase-2 (NOS-2), have been reported to inhibit immune function Citation[32–37]. TGF-β has been considered the most potent immunosuppressive cytokine in a variety of cancer types including malignant glioma. It is responsible for affecting proliferation, activation and differentiation of immune components in both innate and adaptive immune response. In particular, TGF-β is able to inhibit the proliferation of T cells, NK cells, B cells, monocytes and macrophages. In addition TGF-β has been shown to induce apoptosis in B and T cells Citation[38–40]. Glioma cells also secrete IL-10: IL-10 shares many of the same immunosuppressive effects on T lymphocytes as TGF-β, and, in addition, secretion of IL-10 in the context of a tumor can render the tumor totally insensitive to CTL-mediated lysis Citation[41–43].
Downregulation of MHC class I & expression of costimulatory molecules
Downregulation of MHC class I Citation[44] and expression of costimulatory inhibitory molecules such as B7-H1 Citation[45–47], HLA-E and HLA-G Citation[48], have also been reported to evade the immune system.
Dunn et al. summarized and compared all studies performed on human malignant gliomas concerning tumor infiltration of lymphocytes: no correlation between lymphocyte infiltration in gliomas and a good clinical outcome has been found Citation[9]. The immunoediting phenomenon naturally occurs in brain tumors during tumor progression, as suggested by Mittelbronn and colleagues Citation[49]. This study has been addressed to elucidate the correlation between immune infiltration and expression of immunoinhibitory molecule HLA-E in human astrocytomas grade I–III and GB in vivo. In particular, for GB a strong correlation between high-level expression of HLA-E and CD8+ T-cell infiltration has been found: HLA-E, which is already overexpressed in GB, might be stimulated by the presence of CTL and NK. More precisely, CTL tumor infiltration may correlate with the IFN-γ production, which is responsible for increased HLA-E expression. Upon immune selection, tumor cells can evolve to subvert the immune system by upregulating the immunoinhibitory molecule HLA-E to counteract cytotoxic T- and NK-cell responses. This process may relate with downregulation of MHC class I expression Citation[50] and impaired antigen presenting ability in high-grade gliomas Citation[51].
Recently, Wei and colleagues Citation[52] have suggested that a subpopulation of cells derived from GB, defined as cancer-initiating cells, play an important role in mediating immunosuppression by direct cell-to-cell contact by expressing B7-H1 and by secreted factors such as Galectin-3 Citation[53], resulting in the inhibition of T-cell activation and proliferation, induction of regulatory T cells Citation[54,55], and initiation of T-cell apoptosis Citation[56,57]. Similar results concerning low immunogenicity and high suppressive activity of GB initiating cells have been found by Di Tomaso and colleagues Citation[58].
Escape from apoptosis
It has been reported that glioma cells may express both Fas and FasL. Glioma cells are able to activate Fas-mediated apoptosis of tumor-infiltrating T cells by expressing FasL and there is a direct effect on tumor proliferation in glioma cells expressing Fas Citation[59–61].
Cannibalization of T-lymphocytes
Cell cannibalism was detected in malignant tumors of different histology such as breast cancer, bladder cancer and medulloblastoma Citation[62–64], and found to be related to a poor prognosis Citation[65,66]. Lugini et al. described that metastatic melanoma cells are able to phagocytose apoptotic cells Citation[67]. Starting from this observation and using a model of co-cultivation of metastatic melanoma cells with autologous T lymphocytes, they have shown that live T lymphocytes are endocytosed by tumor cells Citation[68]. Cannibalism of live lymphocytes occurs via a peculiar mechanism: in early stages, large vacuoles are formed where the lymphocyte remains alive; at later time points engulfed cells undergo necrosis and degeneration. This mechanism is also considered as a sort of feeding activity in order to sustain survival and progression of malignant cancer cells in an adverse microenvironment. Although not investigated in GB, cannibalism could be a potential mechanism of immune escape in these tumors as well.
Tregs
The suppressive mechanisms of Tregs have been studied in vitro and are considered to be mediated by soluble factors (e.g., TGF-β, IL-10, perforin and granzyme B) or direct cell–cell contact (e.g., CTLA-4 and B7-H4), suppressing the functions of T cells, APCs and NK cells Citation[69–71], and preventing the development of T helper cells as well as CTLs Citation[72]. Immunosuppressive factors, such as TGF-β and IL-10, promote proliferation, differentiation and also conversion of Tregs Citation[70]. The induction of anti-tumor immune responses can be prevented, directly affecting the differentiation, expansion and or recruitment of Tregs from the periphery. Tregs can also home into the tumor under the influence of CCL17 and CCL22 chemokines Citation[73,74]. An increased tumor infiltration of Tregs during tumor progression has been observed in experimental glioma models Citation[75,76] and several studies inversely correlated tumor infiltration of Tregs with clinical outcome Citation[70,74]. El Andaloussi and colleagues suggested a direct correlation of Treg tumor infiltration and human brain tumor grade, and supported the role of Tregs for suppressing anti-tumor responses and for causing increased tumor growth Citation[77,78]. In a mouse model of astrocytoma, Tran Thang et al. found that a significant proportion of Tregs are present at early stages, when mice are still asymptomatic Citation[17]. In other studies where Tregs were depleted through administration of antibodies specific for either CD25, CTLA-4 or GITR, tumors were more efficiently rejected and enhanced the specific anti-tumor responses induced with immunotherapy Citation[55,75,76]. Tregs are thus defined as crucial for the immune escape of brain tumors, and have also been considered to be responsible for immunotherapy failure.
Recently we suggested that intratumoral vaccination using mature lysate-loaded DCs in a murine glioma model potentiate the anti-tumor response primed by peripheral DC injection through the modulation of tumor microenvironment and the decrease in tumor-infiltrating Tregs Citation[79].
Regulatory T cells within the tumor microenvironment are known to play a significant role in the suppression of local anti-tumor immune responses in different human cancers.
The presence of a high number of intratumoral Tregs has been associated with higher risk of recurrent and poor overall survival of patients with breast cancer Citation[80], colorectal cancer Citation[81], gastric cancer Citation[82,83], leukemia Citation[84,85], lung cancer Citation[85,86], lymphoma Citation[87,88], melanoma Citation[88,89], ovarian cancer Citation[85], pancreatic cancer Citation[80] and gliomas Citation[54,90]. In all of these cancers, Tregs are under investigation as a potential prognostic factor and they may represent a novel therapeutic target Citation[91].
T-cell receptor dysfunction
Signaling defects in T cells, which specifically impair T-cell receptor (TCR) functions, were identified at the tumor site and in the peripheral blood of patients with cancer Citation[92]. TCR – or Fcγ receptor III-associated signal-transducing ζ chain – are important for the functional integrity of immune cells and responses. Correlations between clinical and pathologic data, and ζ expression in immune cells, suggest that low levels of ζ expression are predictive of poor prognosis in cancer patients Citation[93–95]. Several data have shown that anergy in T cells from patients with malignant gliomas is correlated to defects in early transmembrane pathways through the TCR CD3 complex Citation[96].
Tumor-released exosomes
Tumor cells are able to secrete into the host environment vesicular structures with pleiotropic effect defined as exosomes. These exosomes can be involved in suppressing and deviating immune responses at different levels. They can induce apoptosis in activated anti-tumor cells through FasL and TRAIL, and can skew monocyte differentiation into myeloid-derived suppressor cells or defective DCs, blocking T-cell proliferation and function through TGF-β secretion Citation[97–99]. Their involvement in melanoma and carcinoma chemoresistance is also known Citation[100,101]. Graner and coworkers identified exosomes derived from brain tumor cell lines and investigated their heat-shock protein (HSP) content and potential source of extracellular HSPs, indicating that brain tumor exosomes may be immunogenic and able to modify anti-tumor immunity Citation[102]. In addition, exosomes are able to serve as a means by which GB can modify their environment, inducing tumor growth and invasion Citation[103], and they can also exert systemic effects owing to their ability to escape the BBB. Many observations demonstrated the presence of exosomes in serum from GB patients containing TGF-β, EGF receptor (EGFR), EGFRvIII Citation[104,105].
Brain cancer immunotherapy: an overview
The median survival for patients with GB is only 12–15 months. The current standard of care for patients with newly diagnosed GB consists of surgical resection, fractionated radiotherapy with concurrent temozolomide (an oral alkylating drug) and subsequent treatment with six cycles of adjuvant temozolomide Citation[106–108].
Active immunization of patients with malignant gliomas is regarded as a promising strategy to induce anti-tumor response in vivo. One relevant immunotherapeutic approach to amplify tumor-specific T-cell responses is the use of ex vivo-generated, autologous tumor antigen-loaded DCs as a vaccine Citation[109]. DC- or peptide-based immunotherapy has been considered as considerably promising in recent years in preclinical models and many clinical trials for malignant gliomas. The role of DCs in eliciting efficient antigen-specific immunity is well recognized. Their use in generating anti-tumor effects in mouse models is still an important area of research, as the results gained from these studies can be useful in developing strategies for cancer patients. Many studies using rodent models have shown that cancer cells can be targeted and eliminated. In the clinical setting, several Phase I/II studies have been based on this strategy Citation[110–119]: even though different DC preparations, antigens and routes of application were employed in these trials, different levels of intratumoral cytotoxic T-cell infiltration could be detected in the patients undergoing surgery after vaccination. Moreover, anti-tumoral immunity was detected in the periphery in approximately 40% of patients and prolonged survival has been suggested for immunological responders or compared with historical controls. DC vaccinations appeared to be more effective in patients with younger age, small residual tumor and low expression of TGF-β2.
Several studies suggest that the induction of effective immune responses is dependent upon proper DC maturation Citation[120,121]: in most clinical trials the ‘maturation cocktail’ includes TNF-α, IL-1β, IL-6 and PGE2 Citation[122]. To increase the immunogenicity of a vaccine, different strategies have been used in murine models to eliminate Tregs, including CD25-specific monoclonal antibodies Citation[123]. In humans, anti-CD25 antibodies such as daclizumab have been developed, but their capacity to deplete human Tregs is questioned by transplantation studies. Recent data suggest that in a mouse model of glioma monoclonal antibodies against IL-2α receptor CD25, when administered during lymphodepletion caused by temozolomide, decrease the number of Tregs and increase the anti-tumor efficacy of vaccination Citation[124]. A reduction in Treg frequency was also verified in patients receiving anti-CD25 antibodies after temozolomide-induced lymphodepletion Citation[124].
Another interesting approach to reduce the number of tumor-infiltrating Tregs is the selective block of their trafficking to the tumor site. Upregulated CXCR4 expression has been found in glioma-infiltrating Tregs Citation[76,125]: implementation of CXCR4 inhibitors may thus improve immunotherapeutic strategies against malignant gliomas Citation[126].
Chemotherapy is presently being investigated as a tool for Treg depletion. In rats, administration of metronomic temozolomide (TMZ) reduced circulating Tregs and their inhibitory functions on effector T cells Citation[127]. Furthermore, TMZ appears to interfere with Treg trafficking to the tumor Citation[128]. Thus, the combination with TMZ chemotherapy could be an attractive strategy to improve efficacy of DC immunotherapy of GB. Other considerations encourage the combination of immunotherapeutic strategies with conventional therapeutic methods such as radiotherapy and chemotherapy. Recent data show that tumor cell death caused by chemotherapy or radiotherapy may favor more effective, cytotoxic immune responses Citation[129,130]. Vice versa, there are also reports on increased sensitivity to TMZ chemotherapy after immunotherapy Citation[131]. Thus, combination therapies integrating vaccination strategies into the standard chemoradiotherapy protocol appear to act in a synergistic manner. There is preliminary evidence from a case report of successful combination of immunotherapy and TMZ Citation[132]. However, the individual therapeutics components have to be carefully timed to optimize the immune response.
Targeting single antigens is unlikely to be successful, considering the genetic instability of such cancers eventually leading to immunoediting. Thus, an important step to increase the efficiacy of future immunotherapy trials is the identification of multiple glioma antigens. Ideally, glioma antigens should be highly expressed by the majority of glioma cells, have greater expression in tumor than in normal cells, strongly immunogenic, and present in multiples in order to avoid immune escape. Expression in CSCs could be a further requisite Citation[133]: SOX2 and SOX6 provide interesting examples of candidate glioma antigens Citation[134,135].
In the near future, genomic profiling of surgical specimens may provide further therapeutic targets dictating selection of immunotherapy, chemotherapy or agents to target particular pathways based on specific genetic signatures. The first advancement was made in September 2008 when Parsons et al. sequenced 20,661 protein-coding genes, determined the presence of amplifications and deletions using high-density oligonucleotide arrays, and performed gene-expression analyses using next-generation sequencing technologies in 22 human tumor samples Citation[136]. This extensive analysis led to the discovery of a number of genes not yet known to be altered in GBs. Of particular interest was the appearance of mutations in the active site of isocitrate dehydrogenase 1 (IDH1), detected in 12% of GB. Mutations in IDH1 were more frequent in young patients and in patients with secondary GBs, and were associated with increased survival. One month later the Cancer Genome Atlas (TCGA) consortium (a pilot project aiming to assess the value of large-scale multi-dimensional analysis of molecular characteristics in human cancer) reported the interim integrative analysis of DNA copy number, gene expression and DNA methylation aberrations in 206 GB and nucleotide sequence changes in 91 of them Citation[137]. This analysis provided new insights into the roles of ERBB2, NF1 and TP53, uncovered frequent mutations of the phosphatidylinositol-3-OH kinase regulatory subunit gene PIK3R1, and provided a network view of the pathways altered in GB development. More recently, Verhaak et al. described a gene expression-based molecular classification of GB into proneural, neural, classical and mesenchymal subtypes that were associated with specific patterns of somatic mutations and copy number alterations. In particular, mutations or altered expression of EGFR, NF1 and PDGFRA/IDH1 appeared linked to the classical, mesenchymal and proneural profiles, respectively Citation[138]. Of interest, recent data have shown that patients whose tumors had mesenchymal gene-expression signatures exhibited increased survival following DC vaccination compared with historic controls of the same genetic subtype. Furthermore, tumor samples with the mesenchymal gene expression signature had a higher number of CD3+ and CD8+ tumor-infiltrating lymphocytes compared with glioblastomas of other gene-expression signatures Citation[139].
The EGFRvIII paradigm
Amplification of the EGFR and subsequent overexpression of the EGFR protein is the most common genetic alteration in primary GB, with a frequency of approximately 40% Citation[140]. EGFR (also referred to as ErbB1 or Her1) is a member of the ErbB family of receptors including ErbB2 (p185Neu or Her2), ErbB3 (Her3) and ErbB4 (Her4). All of them have been found to be overexpressed, amplified or altered in a wide range of human epithelial tumors, and share a similar structure including cytoplasmic domain-containing tyrosine residues phosphorylated upon ligand binding. Receptor activation results in further activation of downstream substrates and the transcription of genes relevant for cell division, cell death, migration and invasion, a cascade of molecular events actively investigated by different genomic and proteomic approaches Citation[141].
Of GB that overexpress EGFR, 63–75% also have rearrangements of the EGFR gene, resulting in tumors expressing both wild-type EGFR and mutated EGFR: the most common of these EGFR mutations is the EGFRvIII. This mutation has not been observed in normal tissue but has an overall prevalence of 20–30% in all patients with GB and 50–60% in patients whose tumors show amplification of wild-type EGFRCitation[140]. The mutation causes the loss of exons 2–7 of the EGFR gene, resulting in an in-frame deletion of 267 amino acids in the extracellular domain.
The use of EGFRvIII expression as a prognostic marker is controversial Citation[142]. The use of combination markers appears more promising. Shinojima et al. found that EGFRvIII was only a prognostic marker for GB patients if EGFR gene amplification was also present Citation[143], whereas Pelloski et al. found that the absence of both EGFRvIII and YKL-40 expression (a tumor-promoting factor often secreted by cancer and inflammatory cells) was associated with a better prognosis (median time: 85 vs 47–56 weeks) Citation[144].
Although the truncated extracellular domain cannot bind any known ligand, the receptor shows constitutive tyrosine kinase activity, and its impaired endocytosis and degradation may also contribute to its oncogenic activity. As mentioned earlier, another mechanism of EGFRvIII tumorigenicity is possibly based on glioma cell secretion of microvesicles expressing EGFRvIII Citation[103,105]. Furthermore, glioma cells carrying the EGFRvIII deletion also express IL-6 and/or leukemia inhibitory factor (LIF) cytokines. These cytokines activate gp130, which in turn activates wild-type EGFR in neighboring cells, leading to enhanced rates of tumor growth, supporting the view that a minor tumor cell population can drive accelerated growth of the entire tumor mass Citation[145].
The GB cell line U87MG retrovirally transfected with EGFRvIII (U87MG.EGFRvIII) has a significant growth advantage in vivo and in vitro when compared with the parental cell line Citation[146]. Several signaling pathways appear important in achieving these effects. A large-scale analysis of phosphorylated proteins mediated by EGFRvIII pointed to PI3K signaling is the dominant signaling pathway activated as a result of EGFRvIII expression in GB cells Citation[147].
EGFRvIII may also have a role in promoting cancer stem cell renewal: in particular experiments by Horvath et al. suggested that abnormal spindle-like microcephaly (ASPM) -associated expression, a promoter of neural stem cell self-proliferation, is upregulated in U87MG.EGFRvIII cells compared with parental cells Citation[148]. The proliferative effects of EGFRvIII are potentiated by its antiapoptotic effects. The reduced apoptotic rate of EGFRvIII-expressing cells is due to upregulation of Bcl-XL expression Citation[149].
The EGFRvIII variant has also been shown to have a role in tumor invasion by upregulation of genes that promote an invasive phenotype such as matrix metalloproteinases Citation[150]. Interestingly, recent data showed that as a result of EGFRvIII-dependent transformation, GB cells become hypersensitive to signaling mediated by tissue factor and protease-activated receptors, producing angiogenic factors (e.g., VEGF and IL-8) Citation[151].
Approaches to EGFR inhibition relevant in GB include monoclonal antibodies, tyrosine kinase inhibitors and vaccines.
The unique EGFRvIII junctional peptide has been utilized as a peptide vaccine to promote anti-tumor host immune responses against EGFRvIII-positive tumors Citation[152]. Rodents challenged with EGFRvIII-positive cells subcutaneously or intracerebrally displayed significant tumor reduction and increased survival rates following administration of DCs loaded with an EGFRvIII synthetic peptide conjugated to keyhole limpet hemocyanin (KLH) Citation[153] or with EGFRvIII–KLH only Citation[154].
Only a few trials have reported on EGFRvIII-specific therapy in glioma patients. Two clinical trials using vaccines against EGFRvIII have enrolled newly diagnosed malignant glioma and GB patients, and encouraging results have been reported Citation[155,156].
The first study evaluated the safety and immunogenicity of a DC-based vaccine targeting the EGFRvIII antigen. Patients with newly diagnosed GB after surgery and radiotherapy received intradermal vaccinations with autologous mature DCs pulsed with an EGFRvIII-specific peptide conjugated to KLH. No allergic reactions or serious adverse events were encountered. EGFRvIII-specific immune responses were evident in most patients. Median time to progression from vaccination was 6.8 months (95% CI: 2.5–8.8), and median survival time was 18.7 months from vaccination and 22.8 months from time of histologic diagnosis.
The second study was a Phase II trial that investigated the immunogenicity of an EGFRvIII peptide vaccine and the survival in patients with newly diagnosed, EGFRvIII-expressing GB and minimal tumor burden. Intradermal vaccinations were administered until tumor progression. The 6-month progression-free survival (PFS-6) rate after vaccination was 67 and 94% after diagnosis. The median OS was 26.0 months greater than that observed in a control group matched for eligibility criteria, prognostic factors, and temozolomide treatment (p = 0.0013). The development of specific antibody (p = 0.025) or delayed-type hypersensitivity (p = 0.03) responses to EGFRvIII had a significant effect on OS. Strikingly, at recurrence, 82% of patients had lost EGFRvIII expression (p = 0.001).
Thus, the results of this trial provide the first evidence of the relevance of tumor immunoediting in shaping immune responses to glioma . The disappearance of EGFRvIII-positive cells after immunotherapy indicates that the immune system primed against cells expressing the EGFRvIII peptide may kill them effectively. In spite of this the tumor may survive: this may be due to continued EGR signaling due to EGFR amplification in the absence of EGFRvIII. Data on maintained amplification of EGFR were not available in the manuscript by Sampson et al. Thus, investigation on the EGFR pathway of relapsing tumors after EGFRvIII peptide vaccination will provide important hints on the relevance, redundancy and flexibility of this critical pathway of tumorigenesis. Recent data have shown a deletion of the NFKBIA gene in approximately 22% of GB Citation[157]. This deletion is usually absent in GB with EGFR amplification, suggesting that NF-κB deletion can substitute for EGFR amplification in the pathogenesis of GB. The observation prompts the study of the NF-κB status in GB before and EGFRvIII peptide immunotherapy treatment and consequent immunoediting.
Resistance to bevacizumab: an example of immunoediting?
Glioblastoma is at the same time both a hypoxic and highly vascularized tumor. A growing debate has been fuelled by data gained over the last 6 years on the presence of a subpopulation of glioma cells with stem-like features (cancer stem cell-like cells [CSCs]), such as enhanced self-renewal capacity, undifferentiated state and multipotency that are tumorigenic when orthotopically transplanted into immunodeficient animals. One stimulating area of research that allows the combination of traditional cancer biology and the CSC field is based on evidence suggesting that hypoxia may promote CSC development Citation[158]. GBs are hypoxic tumors and hypoxia is a poor prognostic factor. Reports from different groups support the idea that hypoxia is a critical driver of CSC maintenance and proliferation. This observation suggests that CSCs may activate different metabolic programs and highlights the importance of the microenvironment in tumor development and progression. In order to target the hypoxic niche, inhibitors of hypoxia-inducible factors and hypoxia-specific cytotoxic agents could be considered. These agents may have clinical relevance because disease progression during bevacizumab therapy is often associated with the development of an invasive character, possibly driven by tumor hypoxia. Thus, combined treatments with antihypoxic agents may represent a new therapeutic approach to overcome the resistance to antiangiogenic therapy.
Several studies at the preclinical and clinical level have been developed to evaluate the therapeutic efficacy of bevacizumab, an antibody that targets VEGF. Bevacizumab in combination with irinotecan in patients with recurrent GB demonstrated an objective response rate of 57% and a PFS-6 of 46% Citation[159,160]. A subsequent large Phase II trial of bevacizumab alone or in association with irinotecan in patients with recurrent GB confirmed the clinical efficacy of bevacizumab Citation[161], and led to its approval by the US FDA in May 2009. The PFS-6 rates were 42.6 and 50.3%, the objective response rates were 28.2 and 37.8%, and median overall survival times were 9.2 months and 8.7 months with bevacizumab and bevacizumab plus irinotecan, respectively Citation[161]. VEGF pathways can also be targeted at the level of the receptor.
Most patients with GB, however, developed disease progression during the first year of treatment with bevacizuamb. In a fraction of these patients a diffuse infiltrative pattern of recurrence was detected. Recent pilot data in vitro suggest that resistance to bevacizumab is based on activation of other proangiogenic factors and of invasion programs Citation[162]. These changes in expression profiles are modulated by bevacizumab and may therefore be considered as a novel form of immunoediting. As a consequence, therapeutic strategies that target both angiogenesis and invasion will be needed in attempts to overcoming resistance to antiangiogenic agents.
Furthermore, it should be considered that VEGF inhibition may also influence immune system–tumor interactions by favoring DC maturation and T-cell activation, as proposed by previous studies Citation[163–166].
Expert commentary
Brain tumor immunotherapy is an evolving field. Evolution is low, unfortunately, despite great advances in the understanding of brain tumor biology and molecular immunology Citation[167]. Part of this difficulty is due to the poorness of preclinical models, still mostly based on injection of tumor cells into the mouse brain. A second issue is the difficulty in translating experimental data into clinical practice. Good Manufactory Practice, albeit necessary, requires considerable investment, and development of clinical trials require approval by local and central authorities. Overall this slows down the pace of translational research.
Biologically we believe that one central point to be addressed by future research is local microenvironment. Tumor immune suppression has been targeted in a recent Phase IIb trial based on the intratumoral delivery of trabedersen, a synthetic antisense phosphorothioate oligodeoxynucleotide complementary to the mRNA of the human TGF-β2 gene Citation[168]. Results showed limited efficacy in anaplastic astrocytomas and in glioblastomas. The approach is interesting, though, as it may be coupled with peripheral immunotherapy, trying to counteract immune anergy determined by tumoral production of TGF-β. Other small molecules are also showing promising results in preclinical experiments and may be thought of in the near future as coupled to systemic immunotherapy Citation[169].
A second point raised by multiple experiences of melanoma immunotherapy and, more recently, by the results of EGFRvIII vaccination in GB Citation[156], relates to the necessity to design vaccination strategies based on multiple antigens or on cellular subpopulations that appear critical to tumor development (e.g., CSCs). Initial preclinical data seem to support this approach Citation[170].
Five-year view
Novel molecular targets
During the next 5 years it is expected that the clinical experience based on EGFRvIII immunotherapy will become stronger. A larger clinical study may confirm the efficacy of peptide-based immunotherapy and the molecular consequences of GB immunoediting will be investigated further. As the TCGA project on human GB gets closer to completion more tumor epitopes will become available for targeting. This, as well as other large-scale projects of high-throughput screening of cancer genomes and matched normal tissues, may also provide novel clues on polymorphisms underlying the heterogeneity of immune responses and immunoediting of cancer: a relevant example has been provided by studies on the role of different TLR4 alleles of breast cancer patients in responses to radio- and chemo-therapy Citation[129].
The molecular subclassification of GB that is derived by large-scale sequencing may also help to define tumor responses to immunotherapy as well as appropriate subpopulations that may be used for DC loading. In this context, the increased appreciation of DC crosstalk with NK cells Citation[171] and of the NK role in the immunotherapy of solid tumors Citation[172,173] may stimulate further research on their role in glioma immunotherapy and immunoediting.
Reshaping the tumor microenvironment
It is also likely that more data will be gained on the clinical efficacy of perturbing the tumor microenvironment by direct injection of small molecules including miRNA. More research is indeed expected to develop the role of miRNA in tumor modulation of the immune environment. Recent data, for instance, have shown that miR29, which is downregulated in brain tumors, plays an important role in downregulation of the expression of B7-H3, a surface immunomodulatory glycoprotein, inhibiting NK cells and T cells Citation[174]. Along a similar line, other data have shown that Dicer by causing maturation of miR-222 and -339 (two suppressors of ICAM-1 expression on tumor cells), decreases the susceptibility of tumor cells to CTL-mediated cytolysis Citation[175]. Furthermore, recent studies underline the potency of local TLR treatment in antiglioma therapy, showing in a murine glioma model that local CpG-ODN treatment restores anti-tumor immunity Citation[176].
Targeting immunological checkpoints
It is also expected that therapeutic strategies aimed at blocking different immunological checkpoints will gain momentum Citation[177]. Recent results obtained with the anti-CTLA-4 antibody ipilimumab in the treatment of advanced melanoma Citation[178] may pave the way to similar clinical approaches in neuro-oncology, as preclinical observations may suggest Citation[179].
Table 1. Factors interfering with recognition and elimination of cancer cells by immune components.
Key issues
• Editing of tumors by the immune system can cause tumor eradication or result in significant molecular reshaping.
• An increasing number of clinical results support further research on the role of the immune system as a (potentially) powerful tool to combat brain tumors, and glioblastoma (GB) in particular.
• The identification of common genetic alterations in GB may provide important targets for immunotherapy. EGFRvIII provides a relevant paradigm in this sense, as preclinical and clinical studies have shown that such targeting by antibody, dendritic cell presentation or direct injection of the EGFRvIII peptide sequence can be effective.
• The recent conclusion of the first clinical study based on EGFRvIII vaccination has shown that more than 80% of relapsing GB fail to show EGFRvIII expression, providing direct evidence for the existence of tumor immunoediting.
• Overall, the previous experience of GB immunotherapy and immunoediting will give us new tools to improve the efficacy of immunotherapy.
• It is expected that local perturbation of the tumor immunosuppressive environment will play a major role in increasing the efficacy of brain tumor immunotherapy.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Ehrlich P. The Collected Papers of Paul Ehrlich. 3, 302 (1957).
- Thomas L. Cellular and Humoral Aspects of Hypersensitivity (1959).
- Burnet M. Cancer; a biological approach. I. The processes of control. Br. Med. J.1(5022), 779–786 (1957).
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol.6(11), 836–848 (2006).
- Koebel CM, Vermi W, Swann JB et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature450(7171), 903–907 (2007).
- Hochmeister S, Zeitelhofer M, Bauer J et al. After injection into the striatum, in vitro-differentiated microglia- and bone marrow-derived dendritic cells can leave the central nervous system via the blood stream. Am. J. Pathol.173(6), 1669–1681 (2008).
- Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol.3(7), 569–581 (2003).
- Grauer OM, Wesseling P, Adema GJ. Immunotherapy of diffuse gliomas: biological background, current status and future developments. Brain Pathol.19(4), 674–693 (2009).
- Dunn GP, Dunn IF, Curry WT. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immunol.7, 12 (2007).
- Calzascia T, Masson F, Di Berardino-Besson W et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity22(2), 175–184 (2005).
- Czerniecki BJ, Koski GK, Koldovsky U et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res.67(4), 1842–1852 (2007).
- Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci.9, 271–277 (1986).
- Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res.28(2), 254–260 (1991).
- Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol.8(3), 261–279 (2006).
- Perrin G, Schnuriger V, Quiquerez AL et al. Astrocytoma infiltrating lymphocytes include major T cell clonal expansions confined to the CD8 subset. Int. Immunol.11(8), 1337–1350 (1999).
- Von Hanwehr RI, Hofman FM, Taylor CR, Apuzzo ML. Mononuclear lymphoid populations infiltrating the microenvironment of primary CNS tumors. Characterization of cell subsets with monoclonal antibodies. J. Neurosurg.60(6), 1138–1147 (1984).
- Tran Thang NN, Derouazi M, Philippin G et al. Immune infiltration of spontaneous mouse astrocytomas is dominated by immunosuppressive cells from early stages of tumor development. Cancer Res.70(12), 4829–4839 (2010).
- Goldschneider I, Cone RE. A central role for peripheral dendritic cells in the induction of acquired thymic tolerance. Trends Immunol.24(2), 77–81 (2003).
- Lohr J, Ratliff T, Huppertz A et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived transforming growth factor-betas. Clin. Cancer Res.17(13), 4296–4308 (2011).
- Masson F, Calzascia T, Di Berardino-Besson W, De Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J. Immunol.179(2), 845–853 (2007).
- Calzascia T, Di Berardino-Besson W, Wilmotte R et al. Cutting edge: cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J. Immunol.171(5), 2187–2191 (2003).
- Galea I, Bernardes-Silva M, Forse PA, Van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood–brain barrier. J. Exp. Med.204(9), 2023–2030 (2007).
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat. Rev. Immunol.6(10), 715–727 (2006).
- Cresswell AC, Sisley K, Laws D, Parsons MA, Rennie IG, Murray AK. Reduced expression of TAP-1 and TAP-2 in posterior uveal melanoma is associated with progression to metastatic disease. Melanoma Res11(3), 275–281 (2001).
- Romero JM, Jimenez P, Cabrera T et al. Coordinated downregulation of the antigen presentation machinery and HLA class I/beta2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer113(4), 605–610 (2005).
- Satoh E, Mabuchi T, Satoh H, Asahara T, Nukui H, Naganuma H. Reduced expression of the transporter associated with antigen processing 1 molecule in malignant glioma cells, and its restoration by interferon-γ and -β. J. Neurosurg.104(2), 264–271 (2006).
- Schartner JM, Hagar AR, Van Handel M, Zhang L, Nadkarni N, Badie B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia51(4), 279–285 (2005).
- Badie B, Bartley B, Schartner J. Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J. Neuroimmunol.133(1–2), 39–45 (2002).
- Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE. Astrocytic regulation of human monocytic/microglial activation. J. Immunol.181(8), 5425–5432 (2008).
- Magnus T, Schreiner B, Korn T et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J. Neurosci.25(10), 2537–2546 (2005).
- Badie B, Schartner J, Prabakaran S, Paul J, Vorpahl J. Expression of Fas ligand by microglia: possible role in glioma immune evasion. J. Neuroimmunol.120(1–2), 19–24 (2001).
- Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol. Today12(10), 370–374 (1991).
- Bodmer S, Strommer K, Frei K et al. Immunosuppression and transforming growth factor-β in glioblastoma. Preferential production of transforming growth factor-β 2. J. Immunol.143(10), 3222–3229 (1989).
- Buccoliero AM, Caldarella A, Gheri CF et al. Inducible cyclooxygenase (COX-2) in glioblastoma – clinical and immunohistochemical (COX-2-VEGF) correlations. Clin. Neuropathol.25(2), 59–66 (2006).
- Castelli MG, Chiabrando C, Fanelli R et al. Prostaglandin and thromboxane synthesis by human intracranial tumors. Cancer Res.49(6), 1505–1508 (1989).
- Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res.55(4), 727–730 (1995).
- Nitta T, Hishii M, Sato K, Okumura K. Selective expression of interleukin-10 gene within glioblastoma multiforme. Brain Res.649(1–2), 122–128 (1994).
- Constam DB, Philipp J, Malipiero UV, Ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol.148(5), 1404–1410 (1992).
- Weller M, Constam DB, Malipiero U, Fontana A. Transforming growth factor-beta 2 induces apoptosis of murine T cell clones without down-regulating bcl-2 mRNA expression. Eur. J. Immunol.24(6), 1293–1300 (1994).
- Weller M, Fontana A. The failure of current immunotherapy for malignant glioma. Tumor-derived TGF-β, T-cell apoptosis, and the immune privilege of the brain. Brain Res. Rev.21(2), 128–151 (1995).
- Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res.17(5A), 3217–3224 (1997).
- Hishii M, Nitta T, Ishida H et al. Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery37(6), 1160–1166; discussion 1166–1167 (1995).
- De Vleeschouwer S, Spencer Lopes I, Ceuppens JL, Van Gool SW. Persistent IL-10 production is required for glioma growth suppressive activity by Th1-directed effector cells after stimulation with tumor lysate-loaded dendritic cells. J. Neurooncol.84(2), 131–140 (2007).
- Zagzag D, Salnikow K, Chiriboga L et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab. Invest.85(3), 328–341 (2005).
- Wilmotte R, Burkhardt K, Kindler V et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport16(10), 1081–1085 (2005).
- Wintterle S, Schreiner B, Mitsdoerffer M et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res.63(21), 7462–7467 (2003).
- Parsa AT, Waldron JS, Panner A et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med.13(1), 84–88 (2007).
- Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J. Neuropathol. Exp. Neurol.64(6), 523–528 (2005).
- Mittelbronn M, Simon P, Loffler C et al. Elevated HLA-E levels in human glioblastomas but not in grade I to III astrocytomas correlate with infiltrating CD8+ cells. J. Neuroimmunol.189(1–2), 50–58 (2007).
- Facoetti A, Nano R, Zelini P et al. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin. Cancer Res.11(23), 8304–8311 (2005).
- Mehling M, Simon P, Mittelbronn M et al. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol.114(2), 111–119 (2007).
- Wei J, Barr J, Kong LY et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin. Cancer Res.16(2), 461–473 (2010).
- Fukumori T, Takenaka Y, Yoshii T et al. CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res.63(23), 8302–8311 (2003).
- Heimberger AB, Abou-Ghazal M, Reina-Ortiz C et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res.14(16), 5166–5172 (2008).
- Fecci PE, Sweeney AE, Grossi PM et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res.12(14 Pt 1), 4294–4305 (2006).
- Kim R, Emi M, Tanabe K, Uchida Y, Toge T. The role of Fas ligand and transforming growth factor beta in tumor progression: molecular mechanisms of immune privilege via Fas-mediated apoptosis and potential targets for cancer therapy. Cancer100(11), 2281–2291 (2004).
- Hegardt P, Widegren B, Sjogren HO. Nitric-oxide-dependent systemic immunosuppression in animals with progressively growing malignant gliomas. Cell Immunol.200(2), 116–127 (2000).
- Di Tomaso T, Mazzoleni S, Wang E et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res.16(3), 800–813 (2010).
- Weller M, Frei K, Groscurth P, Krammer PH, Yonekawa Y, Fontana A. Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J. Clin. Invest.94(3), 954–964 (1994).
- Saas P, Walker PR, Hahne M et al. Fas ligand expression by astrocytoma in vivo: maintaining immune privilege in the brain? J. Clin. Invest.99(6), 1173–1178 (1997).
- Shinohara H, Yagita H, Ikawa Y, Oyaizu N. Fas drives cell cycle progression in glioma cells via extracellular signal-regulated kinase activation. Cancer Res.60(6), 1766–1772 (2000).
- Fujii M, Ishii Y, Wakabayashi T et al. Cytologic diagnosis of male breast cancer with nipple discharge. A case report. Acta Cytol.30(1), 21–24 (1986).
- Kojima S, Sekine H, Fukui I, Ohshima H. Clinical significance of ‘cannibalism’ in urinary cytology of bladder cancer. Acta Cytol.42(6), 1365–1369 (1998).
- Kumar PV, Hosseinzadeh M, Bedayat GR. Cytologic findings of medulloblastoma in crush smears. Acta Cytol.45(4), 542–546 (2001).
- Desimone PA, East R, Powell RD, Jr. Phagocytic tumor cell activity in oat cell carcinoma of the lung. Hum. Pathol.11(5 Suppl.), 535–539 (1980).
- Caruso RA, Muda AO, Bersiga A, Rigoli L, Inferrera C. Morphological evidence of neutrophil–tumor cell phagocytosis (cannibalism) in human gastric adenocarcinomas. Ultrastruct. Pathol.26(5), 315–321 (2002).
- Lugini L, Lozupone F, Matarrese P et al. Potent phagocytic activity discriminates metastatic and primary human malignant melanomas: a key role of ezrin. Lab. Invest.83(11), 1555–1567 (2003).
- Lugini L, Matarrese P, Tinari A et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res.66(7), 3629–3638 (2006).
- Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity21(4), 589–601 (2004).
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer5(4), 263–274 (2005).
- Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+ CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol.174(4), 1783–1786 (2005).
- Chaput N, Darrasse-Jeze G, Bergot AS et al. Regulatory T cells prevent CD8 T cell maturation by inhibiting CD4 Th cells at tumor sites. J. Immunol.179(8), 4969–4978 (2007).
- Mizukami Y, Kono K, Kawaguchi Y et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int. J. Cancer122(10), 2286–2293 (2008).
- Curiel TJ, Coukos G, Zou L et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med.10(9), 942–949 (2004).
- El Andaloussi A, Han Y, Lesniak MS. Prolongation of survival following depletion of CD4+ CD25+ regulatory T cells in mice with experimental brain tumors. J. Neurosurg.105(3), 430–437 (2006).
- Grauer OM, Nierkens S, Bennink E et al. CD4+ FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int. J. Cancer121(1), 95–105 (2007).
- El Andaloussi A, Lesniak MS. An increase in CD4+ CD25+ FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol.8(3), 234–243 (2006).
- El Andaloussi A, Lesniak MS. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J. Neurooncol.83(2), 145–152 (2007).
- Pellegatta S, Poliani PL, Stucchi E et al. Intra-tumoral dendritic cells increase efficacy of peripheral vaccination by modulation of glioma microenvironment. Neuro Oncol.12(4), 377–388 (2010).
- Liyanage UK, Moore TT, Joo HG et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol.169(5), 2756–2761 (2002).
- Somasundaram R, Jacob L, Swoboda R et al. Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-beta. Cancer Res.62(18), 5267–5272 (2002).
- Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin. Cancer Res.9(12), 4404–4408 (2003).
- Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+ CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer98(5), 1089–1099 (2003).
- Karube K, Ohshima K, Tsuchiya T et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br. J. Haematol.126(1), 81–84 (2004).
- Woo EY, Chu CS, Goletz TJ et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res.61(12), 4766–4772 (2001).
- Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin. Cancer Res.9(2), 606–612 (2003).
- Marshall NA, Christie LE, Munro LR et al. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood103(5), 1755–1762 (2004).
- Viguier M, Lemaitre F, Verola O et al. Foxp3 expressing CD4+ CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol.173(2), 1444–1453 (2004).
- Gray CP, Arosio P, Hersey P. Association of increased levels of heavy-chain ferritin with increased CD4+ CD25+ regulatory T-cell levels in patients with melanoma. Clin. Cancer Res.9(7), 2551–2559 (2003).
- Jacobs JF, Idema AJ, Bol KF et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol.11(4), 394–402 (2009).
- Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat. Rev. Cancer7(11), 880–887 (2007).
- Whiteside TL. Signaling defects in T lymphocytes of patients with malignancy. Cancer Immunol. Immunother.48(7), 346–352 (1999).
- Dworacki G, Meidenbauer N, Kuss I et al. Decreased zeta chain expression and apoptosis in CD3+ peripheral blood T lymphocytes of patients with melanoma. Clin. Cancer Res.7(3 Suppl.), 947s–957s (2001).
- Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin. Cancer Res.8(10), 3137–3145 (2002).
- Whiteside TL. Down-regulation of ζ-chain expression in T cells: a biomarker of prognosis in cancer? Cancer Immunol. Immunother.53(10), 865–878 (2004).
- Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol.100(1–2), 216–232 (1999).
- Soderberg A, Barral AM, Soderstrom M, Sander B, Rosen A. Redox-signaling transmitted in trans to neighboring cells by melanoma-derived TNF-containing exosomes. Free Radic. Biol. Med.43(1), 90–99 (2007).
- Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res.67(15), 7458–7466 (2007).
- Andreola G, Rivoltini L, Castelli C et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med.195(10), 1303–1316 (2002).
- Safaei R, Larson BJ, Cheng TC et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther.4(10), 1595–1604 (2005).
- Luciani F, Spada M, De Milito A et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl Cancer Inst.96(22), 1702–1713 (2004).
- Graner MW, Cumming RI, Bigner DD. The heat shock response and chaperones/heat shock proteins in brain tumors: surface expression, release, and possible immune consequences. J. Neurosci.27(42), 11214–11227 (2007).
- Skog J, Wurdinger T, Van Rijn S et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol.10(12), 1470–1476 (2008).
- Al-Nedawi K, Meehan B, Micallef J et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol.10(5), 619–624 (2008).
- Graner MW, Alzate O, Dechkovskaia AM et al. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J.23(5), 1541–1557 (2009).
- Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med.352(10), 987–996 (2005).
- Stupp R, Hegi ME, Mason WP et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised Phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol.10(5), 459–466 (2009).
- Park DM, Sathornsumetee S, Rich JN. Medical oncology: treatment and management of malignant gliomas. Nat. Rev. Clin. Oncol.7(2), 75–77 (2010).
- Gilboa E. DC-based cancer vaccines. J. Clin. Invest.117(5), 1195–1203 (2007).
- De Vleeschouwer S, Fieuws S, Rutkowski S et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res.14(10), 3098–3104 (2008).
- Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a Phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother.50(7), 337–344 (2001).
- Kikuchi T, Akasaki Y, Abe T et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother.27(6), 452–459 (2004).
- Liau LM, Prins RM, Kiertscher SM et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res.11(15), 5515–5525 (2005).
- Rutkowski S, De Vleeschouwer S, Kaempgen E et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br. J. Cancer91(9), 1656–1662 (2004).
- Wheeler CJ, Black KL, Liu G et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res.68(14), 5955–5964 (2008).
- Yamanaka R, Abe T, Yajima N et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical Phase I/II trial. Br. J. Cancer89(7), 1172–1179 (2003).
- Yamanaka R, Homma J, Yajima N et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical Phase I/II trial. Clin. Cancer Res.11(11), 4160–4167 (2005).
- Yu JS, Wheeler CJ, Zeltzer PM et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res.61(3), 842–847 (2001).
- Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res.64(14), 4973–4979 (2004).
- De Vries IJ, Lesterhuis WJ, Scharenborg NM et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin. Cancer Res.9(14), 5091–5100 (2003).
- De Vries IJ, Krooshoop DJ, Scharenborg NM et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res.63(1), 12–17 (2003).
- Jonuleit H, Kuhn U, Muller G et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol.27(12), 3135–3142 (1997).
- Grauer OM, Sutmuller RP, Van Maren W et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int. J. Cancer122(8), 1794–1802 (2008).
- Mitchell DA, Cui X, Schmittling RJ et al. Monoclonal antibody blockade of IL-2 receptor {alpha} during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood118(11), 3003–3012 (2011).
- Jacobs JF, Idema AJ, Bol KF et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol.225(1–2), 195–199 (2010).
- Redjal N, Chan JA, Segal RA, Kung AL. CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin. Cancer Res.12(22), 6765–6771 (2006).
- Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother.58(10), 1627–1634 (2009).
- Jordan JT, Sun W, Hussain SF, Deangulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother.57(1), 123–131 (2008).
- Apetoh L, Ghiringhelli F, Tesniere A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med.13(9), 1050–1059 (2007).
- Haynes NM, Van Der Most RG, Lake RA, Smyth MJ. Immunogenic anti-cancer chemotherapy as an emerging concept. Curr. Opin. Immunol.20(5), 545–557 (2008).
- Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin. Cancer Res.10(16), 5316–5326 (2004).
- Heimberger AB, Sun W, Hussain SF et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol.10(1), 98–103 (2008).
- Dhodapkar MV. Immunity to stemness genes in human cancer. Curr. Opin. Immunol.22(2), 245–250 (2010).
- Schmitz M, Temme A, Senner V et al. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br. J. Cancer96(8), 1293–1301 (2007).
- Ueda R, Ohkusu-Tsukada K, Fusaki N et al. Identification of HLA-A2- and A24-restricted T-cell epitopes derived from SOX6 expressed in glioma stem cells for immunotherapy. Int. J. Cancer126(4), 919–929 (2010).
- Parsons DW, Jones S, Zhang X et al. An integrated genomic analysis of human glioblastoma multiforme. Science321(5897), 1807–1812 (2008).
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature455(7216), 1061–1068 (2008).
- Verhaak RG, Hoadley KA, Purdom E et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell17(1), 98–110 (2010).
- Prins RM, Soto H, Konkankit V et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res.17(6), 1603–1615 (2011).
- Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci.16(6), 748–754 (2009).
- Sahin O, Wiemann S. Functional genomics and proteomics approaches to study the ERBB network in cancer. FEBS Lett.583(11), 1766–1771 (2009).
- Omuro AM. What is the relevance of determining EGFR-variant-III status in glioblastomas? Nat. Clin. Pract. Oncol.5(4), 188–189 (2008).
- Shinojima N, Tada K, Shiraishi S et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res.63(20), 6962–6970 (2003).
- Pelloski CE, Ballman KV, Furth AF et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol.25(16), 2288–2294 (2007).
- Inda MM, Bonavia R, Mukasa A et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev.24(16), 1731–1745 (2010).
- Nishikawa R, Ji XD, Harmon RC et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl Acad. Sci. USA91(16), 7727–7731 (1994).
- Huang PH, Mukasa A, Bonavia R et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl Acad. Sci. USA104(31), 12867–12872 (2007).
- Horvath S, Zhang B, Carlson M et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl Acad. Sci. USA103(46), 17402–17407 (2006).
- Mishima K, Johns TG, Luwor RB et al. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res.61(14), 5349–5354 (2001).
- Lal A, Glazer CA, Martinson HM et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res.62(12), 3335–3339 (2002).
- Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood116(5), 815–818 (2010).
- Choi BD, Archer GE, Mitchell DA et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol.19(4), 713–723 (2009).
- Heimberger AB, Archer GE, Crotty LE et al. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery50(1), 158–164; discussion 164–165 (2002).
- Heimberger AB, Crotty LE, Archer GE et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res.9(11), 4247–4254 (2003).
- Sampson JH, Archer GE, Mitchell DA et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther.8(10), 2773–2779 (2009).
- Sampson JH, Heimberger AB, Archer GE et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol.28(31), 4722–4729 (2010).
- Bredel M, Scholtens DM, Yadav AK et al.NFKBIA deletion in glioblastomas. N. Engl. J. Med.364(7), 627–637 (2010).
- Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer102(5), 789–795 (2010).
- Vredenburgh JJ, Desjardins A, Herndon JE, 2nd et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res.13(4), 1253–1259 (2007).
- Vredenburgh JJ, Desjardins A, Herndon JE, 2nd et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol.25(30), 4722–4729 (2007).
- Friedman HS, Prados MD, Wen PY et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol.27(28), 4733–4740 (2009).
- Lucio-Eterovic AK, Piao Y, De Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin. Cancer Res.15(14), 4589–4599 (2009).
- Gabrilovich D, Ishida T, Oyama T et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood92(11), 4150–4166 (1998).
- Oyama T, Ran S, Ishida T et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol.160(3), 1224–1232 (1998).
- Gabrilovich DI, Chen HL, Girgis KR et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med.2(10), 1096–1103 (1996).
- Ohm JE, Gabrilovich DI, Sempowski GD et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood101(12), 4878–4886 (2003).
- Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol.13(1), 3–13 (2010).
- Bogdahn U, Hau P, Stockhammer G et al. Targeted therapy for high-grade glioma with the TGF-{beta}2 inhibitor trabedersen: results of a randomized and controlled Phase IIb study. Neuro Oncol.13(1), 132–142 (2011).
- Anido J, Saez-Borderias A, Gonzalez-Junca A et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-Initiating cell population in human glioblastoma. Cancer Cell18(6), 655–668 (2010).
- Pellegatta S, Poliani PL, Corno D et al. Neurospheres enriched in cancer stem–like cells are highly effective in eliciting a dendritic cell–mediated immune response against malignant gliomas. Cancer Res.earch66(21), 10247–10252 (2006).
- Kalinski P, Mailliard RB, Giermasz A et al. Natural killer-dendritic cell cross-talk in cancer immunotherapy. Expert Opin. Biol. Ther.5(10), 1303–1315 (2005).
- Burke S, Lakshmikanth T, Colucci F, Carbone E. New views on natural killer cell-based immunotherapy for melanoma treatment. Trends Immunol.31(9), 339–345 (2010).
- Cho D, Shook DR, Shimasaki N, Chang YH, Fujisaki H, Campana D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin. Cancer Res.16(15), 3901–3909 (2010).
- Xu H, Cheung IY, Guo HF, Cheung NK. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer Res.69(15), 6275–6281 (2009).
- Ueda R, Kohanbash G, Sasaki K et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc. Natl Acad. Sci. USA106(26), 10746–10751 (2009).
- Grauer OM, Molling JW, Bennink E et al. TLR ligands in the local treatment of established intracerebral murine gliomas. J. Immunol.181(10), 6720–6729 (2008).
- Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv. Immunol.90, 297–339 (2006).
- Hodi FS, O’day SJ, Mcdermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med.363(8), 711–723 (2010).
- Fecci PE, Ochiai H, Mitchell DA et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res.13(7), 2158–2167 (2007).
- Engelhardt B. Molecular mechanisms involved in T cell migration across the blood–brain barrier. J. Neural Transm.113(4), 477–485 (2006).
Website
- Immune Evasion Strategies of Cancers www.brown.edu/Courses/Bio_160/Projects1999/cancer/imevstca.html