Abstract
Obstructive sleep apnea (OSA), the most common form of sleep-disordered breathing, is prevalent and frequently underdiagnosed in our community. Although presenting with predominantly respiratory symptoms, the most serious complications from OSA are cardiovascular, including arrhythmias, disease of the sinus node and conducting system, and sudden cardiac death. The acute and chronic effects of OSA on the cardiovascular system, which include major effects on autonomic function during sleep and wakefulness, are potent contributors to the development and persistence of cardiac arrhythmias. Although large randomized studies are currently lacking, treatment of OSA may be an important primary or additional therapy to supplement the use of drugs or devices in the treatment of cardiac arrhythmias.
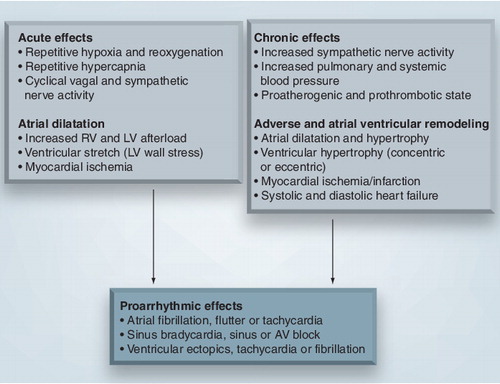
AV: Atrioventricular; LV: Left ventricle; RV: Right ventricle.
AF: Atrial fibrillation; AHI: Apnea–hypopnea index.
Reproduced with permission from Citation[108]. Oxford University Press.
![Figure 2. Atrial fibrillation burden increases with sleep apnea severity.AF: Atrial fibrillation; AHI: Apnea–hypopnea index.Reproduced with permission from Citation[108]. Oxford University Press.](/cms/asset/8dce9ba0-5a6d-46a4-8502-6fe59ffb6d01/ierk_a_11210808_f0002_b.jpg)
Time of (A) AF onset and (B) reversal to sinus rhythm is shown inset.
ABD: Abdominal movement; AF: Atrial fibrillation; HR: Heart rate; NAF: Nasal air flow; SaO2: Oxygen saturation; SR: Sinus rhythm; SVES: Supraventricular ectopics; THO: Thoracic movement; VES: Ventricular ectopics.
Reproduced with permission from Citation[116]. BMJ Publishing Group Ltd.
![Figure 3. Paroxysmal atrial fibrillation occurring during sleep after a prolonged apnea with significant oxygen desaturation.Time of (A) AF onset and (B) reversal to sinus rhythm is shown inset.ABD: Abdominal movement; AF: Atrial fibrillation; HR: Heart rate; NAF: Nasal air flow; SaO2: Oxygen saturation; SR: Sinus rhythm; SVES: Supraventricular ectopics; THO: Thoracic movement; VES: Ventricular ectopics.Reproduced with permission from Citation[116]. BMJ Publishing Group Ltd.](/cms/asset/c0df7d5c-315d-44a6-b10a-d1b0d4787cce/ierk_a_11210808_f0003_b.jpg)
AHI: Apnea–hypopnea index; OSA: Obstructive sleep apnea.
Reproduced with permission from Citation[7]. Copyright © 2005 Massachusetts Medical Society. All rights reserved.
![Figure 4. Reversed pattern of sudden death in sleep apnea, with a nocturnal predilection.AHI: Apnea–hypopnea index; OSA: Obstructive sleep apnea.Reproduced with permission from Citation[7]. Copyright © 2005 Massachusetts Medical Society. All rights reserved.](/cms/asset/1519b6b8-b2b8-4f07-a467-2d7031eb6d4a/ierk_a_11210808_f0004_b.jpg)
Obstructive sleep apnea (OSA) is a common disorder characterized by multiple transient episodes of pharyngeal obstruction, resulting in repeated breathing interruptions, either partial (hypopneas) or complete (apneas) during sleep. Although presenting predominantly with respiratory symptoms (snoring, choking and witnessed breathing interruptions) with or without tiredness or sleepiness, it is now well recognized that the most serious complications of OSA are cardiovascular, including arrhythmias, systemic and pulmonary hypertension, coronary disease, heart failure, sudden death and stroke Citation[1–6].
The finding of increased risk of sudden death during sleep in habitual snorers Citation[5] was assumed to be due to the increased risk of OSA in such patients, and while the association between arrhythmias and sudden death in OSA is not clearly established, there is an increasing body of evidence suggesting that this may be the case Citation[7]. This article focuses on our current understanding of OSA with regard to its pathophysiology, associations with cardiac arrhythmias and effects of treatment, and aims to identify current controversies and gaps in our knowledge of OSA and arrhythmias.
OSA: definitions & scope of the problem
Sleep-disordered breathing (SDB) consists of a spectrum of clinical disorders ranging from simple snoring and upper airway resistance syndrome (increased respiratory effort without apnea or hypopnea) to its more serious forms, including OSA and central sleep apnea (apnea or hypopnea without respiratory effort). OSA is the most common form of SDB, and is currently diagnosed by the ‘gold standard’, overnight polysomnography. Polysomnography was developed in the 1950s to study the physiology of sleep and its disorders Citation[8]. The physiological parameters measured during polysomnography include sleep stages (by EEG), respiratory effort (as evidenced by chest and abdominal wall movement), nasal and oral airflow, arterial oxygen saturation, heart rate and rhythm, body position and limb movements. These parameters allow us to determine the severity of OSA using the apnea–hypopnea index (AHI), which is defined empirically as the number of obstructive respiratory events per hour of sleep (mild OSA: AHI 5–15/h; moderate: 15–30/h; severe: >30/h). The ‘severity’ of OSA may vary during the night, being more marked during rapid eye movement (REM) sleep in some patients, and may vary with body position during sleep. The presence and type of breathing disturbance during sleep is also affected by the onset of congestive heart failure. These traditional or accepted definitions of SDB are inherently arbitrary, almost certainly oversimplistic, and may not be appropriate for the cardiovascular consequences of OSA and cardiac arrhythmias in particular.
Obstructive sleep apnea is a common condition, with estimates from several large-scale population studies suggesting prevalence rates of approximately 20–25% and 10–28% in middle-aged men and women, respectively Citation[9–11]. In addition, the prevalence rates increase with age in both sexes, such that with each 10-year increase in age, the odds ratio of having OSA is 2.2 Citation[11]. Moreover, studies have indicated that approximately 75–80% of severe OSA patients remain undiagnosed Citation[12], and this may actually be due to, among other things, the fact that most patients with OSA are not sleepy Citation[9,10]. The prevalence rates are also expected to increase in the future owing to an aging population, and also because obesity, which is strongly associated with OSA, is increasing dramatically in both children and adults Citation[13]. The increasing prevalence rates and lack of symptoms in patients with OSA will undoubtedly present healthcare providers with significant challenges in an attempt to reduce morbidity and mortality from OSA. This situation is further compounded by difficulties with patient access to overnight polysomnography Citation[14], but the development of simpler and more cost-effective diagnostic tests should help overcome these challenges.
Pathophysiology
Normal sleep
The transition from wakefulness to non-rapid eye movement (NREM) sleep is accompanied by increases in parasympathetic nervous activity and reductions in sympathetic nervous activity and metabolic rate Citation[15–17], which are associated with reductions in heart rate (HR), blood pressure (BP), systemic vascular resistance, stroke volume, cardiac output and a consequent reduction in myocardial workload, with cardiac electrical stability being relatively increased. During the cyclical transition from NREM to REM sleep, the state of parasympathetic tone predominance is interrupted by one in which sympathetic tone predominates and HR, BP, systemic vascular resistance, cardiac output and arrhythmogenicity increase towards levels seen during wakefulness. Since adults spend 75–85% of their total sleep time in NREM sleep, sleep is generally considered a time of parasympathetic tone predominance and cardiovascular quiescence. Apneic episodes disrupt this state of hemodynamic and autonomic stability. Repetitive apneic episodes lead to hemodynamic, autonomic, neuroendocrine, inflammatory and metabolic disruptions. Given the fact that we spend, on average, a third of our time asleep, it is not surprising that, apart from the acute effects, repeated apneic episodes from OSA can lead to adverse long-term cardiovascular effects.
Acute effects
The acute adverse cardiovascular effects from repeated obstructive apneic episodes are considered to be the result of three main pathological changes: hypoxemia, the generation of excessive negative intrathoracic pressure against an occluded pharynx and its effects on right and left ventricular (LV) function, and repeated arousals from sleep. The main effects of these changes include sympathetic nervous system activation, leading to surges in HR and BP, increased atrial and ventricular wall stress, myocardial workload and myocardial oxygen demand (and possibly myocardial ischemia), as well as decreased myocardial contractility and cardiac output. All these changes may promote the development of arrhythmias .
Hypoxemia
Hypoxia, as a result of the obstructive apneic episodes, is a potent stimulant of the sympathetic nervous system via reflex mechanisms. This increase in the sympathetic nerve activity (SNA) by hypoxia is amplified by increases in CO2 levels caused by the apnea Citation[18]. The net effects of these changes are surges in both HR and BP that typically occur immediately after arousals in the postapneic ventilatory phase. The surges in BP and HR lead to increased myocardial workload and oxygen consumption, which are compounded by concomitant hypoxemia. In addition, hypoxia induces pulmonary vasoconstriction, leading to raised pulmonary arterial pressure and, consequently, increased myocardial workload Citation[19]. The net effect on the heart and circulation is increased myocardial strain and depressed myocardial contractility Citation[20].
Although hypoxia typically causes HR surges during arousal from sleep and the postapneic ventilatory phase, it has varying effects on HR depending on the balance between its sympathetic and parasympathetic stimulatory effects. During the apneic episode, the combination of hypoxia and an absence of airflow result in carotid body chemoreceptor stimulation, leading to reflex bradycardia via vagal afferents Citation[21,22]. However, in the presence of airflow (e.g., during the postapneic ventilatory phase), hypoxia causes tachycardia, because stretching of the lung during ventilation leads to inhibition of parasympathetic outflow and allows unopposed sympathetic outflow to the heart Citation[23]. Therefore, the effect of hypoxia on HR depends on the ventilatory phase and the balance between sympathetic and parasympathetic cardiac outflow. These oscillations in sympathetic and parasympathetic activity in OSA may predispose patients to the development of both tachy- and bradyarrhythmias.
Negative intrathoracic pressure
The negative intrathoracic pressure generated during inspiratory efforts against an occluded pharynx (Müller manoeuvre) can be significant, reaching as low as -80 cmH2O. The resultant reduction in stroke volume, and therefore cardiac output, is due to three main factors. First, the excessive negative intrathoracic pressure generated leads to acute increases in LV transmural pressure as a result of the widening difference between the extracardiac and intracardiac pressures, which has the effect of acutely increasing LV afterload and reducing stroke volume Citation[24–26]. Second, venous return to the right heart increases, leading to right ventricular distension and, therefore, a leftward shift of the interventricular septum Citation[27]. This results in an impairment of LV diastolic filling, and consequently stroke volume and cardiac output. Third, the excessive negative intrathoracic pressure can directly impair LV relaxation, leading to further impairments of LV diastolic filling. The net effect of increased LV afterload and decreased LV preload and diastolic filling is a reduction in stroke volume and cardiac output that is proportional to the magnitude of negative intrathoracic pressure generated Citation[28–32]. The adverse impact of these effects on afterload is further augmented by LV systolic dysfunction. Furthermore, the repeated acute increases in LV afterload, in combination with augmented SNA during sleep and wakefulness, will lead to LV hypertrophy in the long term, contributing further to LV diastolic dysfunction, increases in left atrial filling pressure and left atrial enlargement Citation[2,33–35]. All these changes serve to promote the development of cardiac arrhythmias.
Arousals from sleep
Arousals from sleep act as a protective mechanism for asphyxiation from extended apneic episodes; they are triggered by hypoxemia, hypercapnia and increased ventilatory effort, and lead to the activation of upper airway dilator muscles, thereby restoring airway patency Citation[36]. Arousals contribute to surges in HR and BP in the postapneic ventilatory phase Citation[37] in combination with increases in ventilation at termination of apnea Citation[38]. These surges in SNA provide a potent stimulus to the development and persistence of arrhythmias.
Chronic effects
The long-term effects of OSA are not well understood, although autonomic nervous system dysregulation with chronic sympathetic activation and the development of systemic hypertension have been implicated. This may lead to the development of left atrial enlargement, LV hypertrophy, systolic and diastolic heart failure, and cardiac arrhythmias Citation[2,33–35,39–42]. In addition, there is evidence that OSA may result in a procoagulant and proinflammatory state, leading to vascular dysfunction and an increased risk of thromboembolic cardiovascular events Citation[43–45].
Autonomic nervous system dysregulation
Apart from changes in autonomic nervous system regulation during sleep, OSA also results in autonomic dysregulation during wakefulness. This is characterized by increased SNA, reduced HR variability, increased BP variability and blunted baroreceptor sensitivity Citation[46–50]. Compared with healthy controls, patients with OSA have higher SNA during sleep and wakefulness Citation[46–48]. Although not completely understood, it is thought that repeated episodes of hypoxia lead to increased peripheral chemoreceptor sensitivity, thereby leading to increased cardiac sympathetic outflow even when patients with OSA are normoxic and awake. This increase in SNA is also thought to result in reduced HR variability. Moreover, this increased SNA may lead to the development of systemic hypertension in OSA patients, although endothelial dysfunction may also contribute Citation[51–55]. While under normal conditions, an increase in BP due to increased SNA leads to activation of the carotid sinus and aortic arch baroreceptors, resulting in reflex bradycardia via vagal afferents, the repetitive BP surges during sleep in OSA patients may lead to downregulation of baroreceptor sensitivity over time Citation[56], leading to reduced vagal-mediated inhibition and, therefore, increased SNA and BP variability. Treatment of OSA with continuous positive airway pressure (CPAP) therapy has been shown to improve BP in some but not all randomized trials, and normalize increased SNA by night and day Citation[46,57].
Vascular endothelial dysfunction
Increased SNA, BP surges from repeated hypoxic episodes, increased oxidative stress and insulin resistance may all lead to endothelial dysfunction in OSA Citation[43]. Increased endothelin-1 release and depressed circulating nitric oxide levels have been implicated in this and may contribute to the development of systemic hypertension in patients with OSA Citation[51–54]. In addition, patients with OSA have an increased prevalence of surrogate markers for atherosclerosis including increased carotid intima-media thickness, carotid calcification, coronary calcification and subsequent presentations with coronary events Citation[58–61]. Treatment with CPAP therapy in patients with OSA has also been shown to improve baseline endothelial nitric oxide release and systemic and pulmonary vascular endothelial function Citation[43,62–64].
Proinflammatory state
Obstructive sleep apnea appears to be associated with a proinflammatory state. Elevations in C-reactive protein, TNF-α, IL-6 and IL-8 plasma levels have been reported in OSA patients and may be reduced by CPAP therapy Citation[45,65,66]. The levels of expression of NF-kB and nitrotyrosine, which are markers of vascular inflammation, have also been shown to be elevated in OSA patients, and this elevation can be reversed with CPAP therapy Citation[63]. OSA also promotes the oxidation of lipoproteins, increases the expression of adhesion molecules and monocyte adherence to endothelial cells, and promotes vascular smooth muscle proliferation Citation[67–70]. Indeed, this proinflammatory and oxidative state may be an important contributor to the development of cardiovascular disease in OSA, and may be reversed with OSA treatment.
Procoagulant state
In contrast to healthy controls, patients with OSA have significant increases in platelet aggregability overnight in association with elevated nocturnal catecholamine levels Citation[44,71–73]. Treatment with CPAP therapy has been shown to reduce both platelet aggregability and nocturnal catecholamine levels Citation[44,72,74]. In addition, OSA patients have increased hematocrit levels (probably due to nocturnal hypoxemia), fibrinogen levels and blood viscosity Citation[75–77]. This procoagulant state in patients with OSA may predispose to cardiovascular events, and account for the increased rate of cardiovascular events during sleep in OSA patients Citation[7].
OSA & the metabolic syndrome
It is well known that obesity is associated with an increased risk of cardiovascular events, independent of traditional risk factors Citation[78–80]. This may be explained in part by its association with a proinflammatory, oxidative, prothrombotic and proatherogenic state Citation[78,81,82]. The clustering of other risk factors with obesity, including hypertension, insulin resistance and proatherogenic dyslipidemia, has been termed the metabolic syndrome or ‘syndrome X’. This risk factor cluster is significant as these individual risk factors may potentiate the effects of each other. However, an important confounding factor that interacts with central obesity and the metabolic syndrome is its strong links with OSA.
Obstructive sleep apnea may lead to the development of both insulin resistance and to individual components of the metabolic syndrome, independent of the effects of obesity Citation[83,84]. This close linkage between OSA and the metabolic syndrome led us to propose the term ‘syndrome Z’ to draw attention to this relationship (Box 1)Citation[85,86]. The metabolic syndrome is a proatherogenic, proinflammatory and, potentially, proarrhythmic state that is adversely impacted by concurrent OSA.
Potential mechanisms linking OSA to arrhythmias
The acute and chronic autonomic and hemodynamic changes, myocardial ischemia, as well as changes in atrial and ventricular structure and function, are all potent contributors to the development and persistence of cardiac arrhythmias .
Acute & chronic changes in the autonomic nervous system
The repetitive oscillations between sympathetic and parasympathetic predominance in OSA during sleep, depending on the ventilatory phase of the apneic episode and the phase of sleep, provide the perfect milieu for the development of cardiac arrhythmias. When parasympathetic tone predominates, bradyarrhythmias may occur; when sympathetic tone predominates, both atrial and ventricular tachyarrhythmias may occur. In addition, atrial fibrillation (AF) may be triggered by both surges in vagal and adrenergic tone Citation[87]; hence, acute autonomic fluctuations in OSA may also predispose to the development of AF. Apart from the surges in SNA during these acute autonomic fluctuations, OSA patients also have chronically raised SNA when awake Citation[46,48]. Both these factors may favor the development of ventricular arrhythmias and sudden cardiac death (SCD) during sleep and wakefulness.
Myocardial ischemia
The combination of increased SNA and hypoxemia in OSA leads to increased myocardial oxygen consumption and decreased oxygen supply. This situation is compounded further by increases in HR, BP and afterload peaking during the postapneic phase Citation[88], and is also caused by the acutely and chronically raised SNA and fluctuations in intrathoracic pressure. The net effect of these changes may be myocardial ischemia, which will undoubtedly predispose to a variety of cardiac arrhythmias. Furthermore, the procoagulant and proinflammatory state in OSA further predisposes to arterial thrombosis, myocardial ischemia and infarction; it is therefore intuitive that this state of heightened adrenergic, inflammatory and procoagulant activity may lead to increased risk of ventricular arrhythmias and SCD.
Changes in cardiac structure
The combination of repetitive fluctuations in HR, BP and intrathoracic pressure during sleep with increased SNA during sleep and wakefulness leads to significant left and right ventricular wall stress as well as remodeling of the heart over time, with the development of both LV and right ventricular hypertrophy, and systolic and diastolic heart failure Citation[2,35,39–42]. This may lead to an increased risk of ventricular arrhythmias and SCD. The acute effects of OSA also directly affect the atria, leading to both acute and chronic atrial enlargement Citation[33,34,42]. The development of diastolic ventricular dysfunction in OSA may also contribute to this process. This notion is supported by reports of raised serum atrial natriuretic peptide levels and the symptom of nocturia in patients with OSA Citation[89]. Furthermore, it has been reported that OSA may increase left atrial size independently of obesity, hypertension and diastolic dysfunction Citation[34]. Such changes of atrial stretching and enlargement may lead to the development of atrial remodeling and fibrosis, and therefore to an increased risk of paroxysmal and persistent AF.
Arrhythmias in OSA
OSA & bradycardias
Studies on the prevalence of bradyarrhythmias in patients with OSA have yielded conflicting results, with some reporting no increase in rates Citation[90,91] and others reporting rates of between 7 and 50% Citation[92–94]. Some of the possible reasons for the conflicting results include differences in the severity of OSA in the study patients and differences in the duration of cardiac monitoring. One of the largest and earliest studies by Guilleminault et al. assessed 400 patients with severe OSA with polysomnography and 24-h Holter monitoring, and reported bradyarrhythmias occurring in 18% of the study patients Citation[93]. A subgroup of 50 patients with significant arrhythmias was restudied after tracheostomy and the previously documented arrhythmias were abolished with the exception of frequent ventricular ectopic beats.
More recently, Becker et al. assessed 239 consecutive patients with OSA with polysomnography and 24-h Holter monitoring Citation[92]. They reported that 7.5% of these patients had significant bradyarrhythmias (i.e., second- or third-degree atrioventricular [AV] block, or sinus pauses >2 s). In addition, they found that the prevalence of bradyarrhythmias correlated strongly with OSA severity and degree of nocturnal desaturation, such that all patients with significant bradyarrhythmias had an AHI 60/h or more (accounting for 20% of this subgroup of patients). By contrast, studies by Flemon et al.Citation[90] and, more recently, the Sleep Heart Health Study Citation[91], which compared 228 patients with severe OSA with 338 subjects without OSA matched for age, sex, race and BMI, showed no increased prevalence rates of bradyarrhythmia in OSA patients. In the latter study, cardiac monitoring was derived only from the overnight polysomnography Citation[91]. Detection of arrhythmias using such a short period of monitoring is well known to be an insensitive method compared with longer duration recording methods such as implanted pacemakers, defibrillators or loop recorders. Given the night-to-night variability in OSA severity, it is possible that significant bradyarrhythmias may have been missed in this study. This possibility is confirmed by a study by Simantirakis et al., who studied 23 patients with at least moderate OSA with an implantable loop recorder Citation[95]. They reported that 47% of these untreated patients had significant nocturnal bradyarrhythmias. The authors also used a 48-h Holter monitoring in this study and, in contrast, bradyarrhythmias were noted in only 13% of the patients. This suggests that the difference in the observed rate of bradyarrhythmias in OSA patients might be related to the extent of cardiac monitoring used in the various studies.
Obstructive sleep apnea as a cause of bradyarrhythmias in these patients is supported by studies showing that CPAP therapy abolishes the bradyarrhythmias in the majority of OSA patients Citation[95–99]. These results are in keeping with other studies reporting normal or mild abnormalities in sinus node function and AV conduction during electrophysiological studies in patients with OSA and significant nocturnal bradyarrhythmias Citation[94,100]. These mild conduction abnormalities were completely reversible with atropine administration in most patients, which further supports the concept of increased parasympathetic tone in the pathophysiology of bradyarrhythmias in OSA patients Citation[100,101].
In addition, Garrigue et al. reported that undiagnosed OSA was present in approximately half of patients with pacemakers implanted for symptomatic bradycardia, AV block or heart failure Citation[102]. There is therefore an argument that patients with bradyarrhythmias, especially those occurring predominantly during sleep, should also be screened for OSA. The notion that patients with OSA and bradycardia may be able to avoid pacemaker implantation is fallacious, because if a patient has a clinically important arrhythmia, CPAP or other treatments such as oral appliances, while highly effective, have inferior compliance rates compared with the compliance and efficacy rates for an implanted pacemaker. There are, however, other potential benefits from treatment of OSA in patients with bradyarrhythmias, which include reducing the amount of ventricular pacing required and the burden of tachyarrhythmias such as AF, atrial flutter or ventricular arrhythmias.
The notion that atrial overdrive pacing (AOP) may reduce apneic episodes is interesting, as it is thought that suppressing periods of SDB-related bradycardia with AOP may help restore the autonomic balance disrupted by the repeated apneic episodes, thereby reducing SDB severity. This theory was supported by Garrigue et al., who reported that AOP significantly reduced the number of episodes of central or OSA without reducing the total sleep time Citation[103]. However, subsequent studies have failed to find an improvement with AOP, or reported only marginal benefit of unknown clinical significance Citation[104,105]. A recent meta-analysis concluded that AOP may be effective in patients with central sleep apnea; however, the role of AOP in OSA remains unclear Citation[106].
OSA & atrial arrhythmias
The relationship between OSA and AF was observed over 25 years ago from an observational study of 400 adults with moderate-to-severe OSA who underwent ambulatory Holter monitoring Citation[93]. Nocturnal episodes of AF were observed in 3% of these patients, and all patients who received definitive treatment of OSA had complete resolution of AF up to 6 months later.
More recently, several studies have confirmed an increased prevalence of AF in OSA patients. Gami et al. prospectively studied 151 consecutive patients undergoing electrical cardioversion for AF and 312 consecutive patients without past or current AF referred to a general cardiology practice Citation[107]. Both groups had similar age, gender, BMI, and prevalence of diabetes, hypertension and heart failure. They reported that the prevalence of OSA was significantly higher in the AF group than in the general cardiology group (49 vs 32%), with an adjusted odds ratio for the association between AF and OSA of 2.2.
The Sleep Heart Health Study compared 228 patients with severe OSA with 338 patients without OSA matched for age, sex, race and BMI Citation[91]. Based on cardiac monitoring from overnight polysomnography, the authors found that 4.8% of patients with severe OSA had AF, as compared with only 0.9% of patients without OSA. Even after adjusting for age, sex, BMI and known coronary artery disease, the risk of AF in individuals with OSA was increased fourfold . These results are further strengthened by reports from Stevenson et al.Citation[108]. In this study, the authors compared 90 patients with AF with 45 control patients without a history of AF. All patients had a structurally normal heart. They reported that the proportion of patients with at least moderate OSA was significantly higher in the AF group than in the control group (62 vs 38%). In addition, the study also revealed a relationship between AF burden (in terms of frequency and duration of AF) and OSA, such that the proportion of patients with at least moderate OSA was significantly higher in those with high-frequency AF (i.e., >six episodes/year) as opposed to those with low-frequency AF (i.e., ≤six episodes/year), being 75 versus 43%, respectively Citation[108].
Apart from these cross-sectional studies, several longitudinal studies have reported that the presence of OSA predicts the risk of future AF Citation[109,110]. Kanagala et al. assessed the risk of recurrent AF after electrical cardioversion in inadequately treated or untreated OSA Citation[109]. In this study, 39 patients with a diagnosis of OSA from a previous sleep study were compared with 79 patients who did not undergo a previous sleep study. A total of 27 out of the 39 patients were identified as being untreated or receiving inadequate treatment of OSA. They reported that the recurrence of AF at 12 months in these 27 patients was significantly higher compared with either the treated OSA group (n = 12) or control group (n = 79; 82 vs 42 and 53%, respectively). Moreover, among the patients whose OSA was untreated, the nocturnal desaturation was greater in those who had recurrence of AF compared with those without. It is also possible that the reported modest efficacy of antiarrhythmic medications in the treatment of AF in many of the published trials Citation[111–113] may in fact be confounded by the presence of undiagnosed and untreated OSA.
In a study that examined postoperative AF after heart surgery, Mooe et al. found that OSA predicted postoperative AF requiring therapy prior to hospital discharge, with a 32% incidence of AF compared with 18% in the control group without OSA. They also found that the severity of oxygen desaturation predicted AF independently of other factors Citation[114].
More recently, Gami et al. prospectively followed 3542 patients who underwent a diagnostic overnight polysomnography without a past or current history of AF over a mean duration of 4.7 years Citation[110]. They reported that, in patients under 65 years of age, the presence of OSA conferred a 2.2-fold increased risk of development of AF over the study follow-up period. In addition, both obesity and OSA were independent predictors of incident AF in subjects under 65 years of age.
A recent study by Monahan et al. reported that, although the absolute risk of arrhythmia was low, the relative risk of paroxysmal AF during sleep was markedly increased (odds ratio: 18) shortly after a respiratory disturbance Citation[115]. Schulz et al. also reported a case of paroxysmal AF occurring after a prolonged apneic episode with marked oxygen desaturation Citation[116]. These studies illustrate a direct temporal link between OSA events and the development of AF. They also suggest that the duration of apnea and degree of desaturation, rather than the ‘severity’ of OSA as defined by the AHI, may play a more important role in the development of clinically important adverse cardiac effects, and that a single prolonged apneic episode may be enough to trigger an arrhythmia. Moreover, the ‘severity’ of OSA may vary from night-to-night and according to sleep stage, and even apparently ‘mild’ OSA can have clinically significant cardiovascular consequences.
The aforementioned concept is further supported by a study involving pregnant women with preeclampsia and upper airway resistance syndrome (heavy snoring without OSA or significant nocturnal desaturation), showing that CPAP therapy resulted in a reduction in sleep-induced BP increments Citation[117]. Another study found that even individuals with ‘mild’ SDB (AHI: 5–15/h) had a greater than twofold increased risk of stroke Citation[5]. These findings suggest that even ‘milder’ forms of SDB may have adverse cardiac effects.
The association between OSA and ischemic stroke is also significant, with a reported adjusted hazard ratio of at least 2 in all the major studies Citation[5,118,119]. The Sleep Heart Health study, the largest to date assessing OSA and stroke risk, prospectively followed 5422 subjects with untreated OSA over a median of 8.7 years, and reported a strong adjusted association between ischemic stroke and AHI Citation[119]. They also found that even a low-to-moderate AHI was associated with an increased stroke risk. Some of the possible mechanisms for this association include apnea-induced hypertension, hypercoagulability and proinflammatory state, all of which may lead to endothelial dysfunction, progression of atherosclerosis, carotid plaque rupture and thrombosis, as well as nocturnal cerebral hypoxemia.
A much more plausible explanation for the increased risk of stroke in OSA is the increased prevalence of AF, conferring an increased thromboembolic risk. This possibility has not been specifically addressed in the published studies on OSA and stroke risk Citation[5,118,119]. Furthermore, LV dysfunction, even to a mild degree, is associated with a nearly fourfold increased risk of stroke Citation[120]. It is therefore also possible that LV dysfunction and heart failure caused by OSA can, in itself, promote the development of AF, thereby leading to an increased risk of stroke. Thus, OSA, AF and LV dysfunction are likely to be an adverse combination. Moreover, AF occurring in OSA patients is often paroxysmal, and can therefore be missed on cardiac monitoring. In fact, any patient presenting with an unexplained thromboembolic event should probably be assessed for possible OSA.
The impact of AF triggered by OSA is likely to be affected substantially by the presence and type of underlying heart disease. Hypertrophic cardiomyopathy (HCM), the most common inherited cardiac condition, was recently reported to be associated with a high prevalence of OSA Citation[121]. The adverse hemodynamic changes in LV preload, afterload and diastolic function in OSA can all lead to increases in LV outflow tract obstruction and symptomatic decline in patients with HCM. Apart from the importance of treating OSA in these patients to possibly prevent further disease progression through worsening hypertrophy and diastolic dysfunction, preventing AF in these patients is also important as it is often poorly tolerated in HCM, and the onset of persistent or permanent AF often heralds a relentless functional decline in these patients.
Although OSA is associated with increased risk of AF, it is currently not known whether treatment of OSA will lead to a reduced risk of AF. Several observational studies suggest that risk of AF can be reduced by treatment of OSA Citation[93,109]. Treatment of OSA by tracheostomy was shown to abolish nocturnal AF in 12 patients with severe OSA and nocturnal paroxysmal AF Citation[93]. While tracheostomy has not been the first-line therapy for OSA since CPAP was developed in the early 1980s Citation[122], the significance of this mode of treatment is its high compliance and efficacy for the treatment of OSA when not associated with cardiac or respiratory failure. The recurrence of AF 12 months after electrical cardioversion was also compared between OSA patients treated and untreated with CPAP, and the study found that recurrence rates were drastically reduced with CPAP use, being 42 and 82%, respectively Citation[109]. A more recent study confirmed that CPAP therapy in OSA patients drastically reduced the occurrence of paroxysmal AF Citation[123]. Randomized clinical trials are currently underway to answer this question more definitively.
While CPAP is effective in nearly all patients with OSA, compliance is typically between 50 and 70% in usual clinical practice. This may be related, in part, to the fact the many patients with OSA are asymptomatic and not sleepy. As in other settings, assessment of the impact of therapy requires that compliance and efficacy are measured, as treatment effects can be underestimated. Modern CPAP devices are often capable of recording these parameters.
OSA & SCD
Seppala et al. first reported that a history of ‘habitual snoring’ obtained from relatives of the deceased subjects was associated with a significantly increased risk of early morning cardiovascular death compared with the general population in Finnish men, with an odds ratio of 4 Citation[4]. More recently, another study assessed 112 patients who had undergone a previous polysomnography and who died of SCD. In contrast to the general population, where the predilection of SCD peaks from 6 am to noon and has a nadir from midnight to 6 am Citation[124], this study found a nocturnal predilection of SCD in patients with OSA Citation[7]. From midnight to 6 am, the authors found that SCD occurred in 46% of subjects with OSA, as compared with only 21% of subjects without OSA, 16% of the general population, and the 25% expected by chance. In subjects with OSA, the relative risk of SCD from midnight to 6 am was 2.6, compared with the relative risk of 0.8 in non-OSA subjects. This pattern is reversed for the time interval from 6 am to noon, with the relative risk of SCD being 0.8 and 2.1 for OSA and non-OSA subjects, respectively . In addition, subjects who died from SCD between midnight and 6 am had a more severe OSA than those dying during other time intervals, and the severity of OSA correlated directly with the relative risk of SCD from midnight to 6 am.
The results from these studies suggest that OSA may be associated with increased risk of SCD, especially at night. There are three possible mechanistic explanations for this. First, hypoxemia, increased SNA, and the proinflammatory and procoagulant state caused by the apneic episodes may result in myocardial ischemia, thereby predisposing to nocturnal cardiac death. Indeed, there are several studies supporting this, describing an association between OSA and occurrence of nocturnal myocardial ischemia Citation[125–127]. A recent study compared 21 symptomatic OSA patients with 12 snorers (without hypersomnolence) and 15 healthy controls, and found that OSA patients had a higher frequency of ST-segment depression episodes than snoring and control subjects Citation[128]. Moreover, ST-segment changes were found to be related to sympathetic tone and sleep fragmentation. Second, sinus arrest or AV block occurring while asleep during periods of parasympathetic tone predominance in OSA can also lead to nocturnal sudden death. Third, an increased risk of ventricular arrhythmias, either as a result of nocturnal myocardial ischemia or increased SNA, may account for the increased risk of nocturnal SCD in OSA patients. Several studies support these findings, and report an increased risk of ventricular ectopy or nonsustained ventricular tachycardia (NSVT) in OSA patients Citation[91,115].
The Sleep Heart Health Study reported that OSA patients had a significantly higher rate of ventricular arrhythmias compared with non-OSA patients, being 5.3 versus 1.2%, respectively, for NSVT, and 25 versus 14.5%, respectively, for complex ventricular ectopy Citation[91]. Compared with those without OSA and even after adjusting for age, sex, BMI and known coronary artery disease, individuals with OSA had 3.4-times the odds of NSVT and almost twice the odds of complex ventricular ectopy . Another more recent study reported that the odds of NSVT after an apneic episode was drastically increased, with an odds ratio of 17.4 compared with that after normal breathing during sleep Citation[115]. The findings from this study provided direct evidence of a relationship between apneic events and the development of ventricular arrhythmias, likely but not proven to be a casual one.
There are several studies reporting the benefit of OSA treatment and reduction of nocturnal myocardial ischemia and ventricular arrhythmias. Franklin et al. reported an abolishment of nocturnal angina after treatment with CPAP therapy in all but one OSA patient Citation[125]. These findings were confirmed by another study reporting that treatment with CPAP significantly decreased the nocturnal ST-segment depression time from 78 to 33 min Citation[129]. Ryan et al. conducted a randomized trial on 18 heart failure patients with OSA, and found that CPAP therapy, compared with placebo, was associated with a 58% reduction in premature ventricular ectopy, as well as a reduction in daytime systolic BP and increased LV ejection fraction and overnight urinary norepinephrine Citation[130]. This is confirmed by a more recent study showing a reduction in premature ventricular ectopy in OSA patients treated with CPAP therapy Citation[123]. It is possible, based on these studies, that the net benefit from CPAP therapy may result in a reduced risk of nocturnal SCD in OSA patients. However, a recent small randomized study disputes these earlier findings, and suggests no difference in the rates of ventricular arrhythmias in unselected OSA patients treated with therapeutic CPAP therapy compared with subtherapeutic therapy Citation[131]. Further randomized clinical trials with clinically relevant end points are needed to address this issue.
Conclusion
Obstructive sleep apnea is a prevalent condition that is increasingly recognized to be associated with cardiovascular disease, including arrhythmias. The hemodynamic and autonomic changes caused by OSA promote a range of arrhythmias, including bradyarrhythmias, AF and ventricular arrhythmias, and possibly confer an increased risk of SCD. Further trials are needed to establish the role of OSA treatment in reducing arrhythmic risk. Finally, increasing the awareness of the association between OSA and cardiovascular disease among cardiologists is vital to reduce cardiovascular morbidity and mortality.
Expert commentary & five-year view
Obstructive sleep apnea is a prevalent condition that remains underdiagnosed. Given the wide spectrum of potential cardiovascular complications of OSA, it is likely that diagnosis and treatment of OSA will become an integral part of the assessment and treatment of patients with cardiovascular disease. Randomized controlled trials are also currently underway to determine the cardiovascular benefits of treating OSA. Given the fact that a considerable proportion of patients with severe OSA may be asymptomatic, it may be necessary for the diagnosis and treatment of SDB to be incorporated into pre-existing cardiovascular guidelines to change practice in this emerging area, especially in patients presenting with conditions such as refractory hypertension, premature coronary disease, arrhythmias (especially AF), heart failure, stroke and unexplained thromboembolic events.
Furthermore, SDB is a potential confounder of the results in the major published cardiovascular clinical trials. An increasing awareness of this problem by clinical trialists is vital, and adjusting for the presence or absence of SDB during data analyses in these trials should be undertaken to allow for more accurate interpretation of study conclusions. One of the challenges involves patient access to a diagnostic sleep study, but this will hopefully be improved with the development and use of simpler, more cost-effective diagnostic tests and treatment algorithms. Finally, the definition of the ‘severity’ of SDB may need to be redefined according to cardiovascular rather than respiratory events.
Table 1. Arrhythmia risk during sleep in severe sleep apnea (apnea–hypopnea index >30/h) in the Sleep Heart Health Study.
Box 1. Components of ‘syndrome Z’.
• Obstructive sleep apnea
• Hypertension
• Central (visceral) obesity
• Increased sympathetic nerve activity
• Insulin resistance/Type 2 diabetes
• Proatherogenic dyslipidemia
• Proinflammatory state
• Procoagulant state
Key issues
• Obstructive sleep apnea (OSA) is prevalent and is frequently undiagnosed.
• Given the increasing epidemic of obesity and the aging population, the prevalence of OSA is expected to increase.
• Although presenting with predominantly respiratory symptoms, the most serious associations of OSA are cardiovascular.
• The ‘severity’ of OSA may be better defined according to its cardiovascular effects rather than respiratory events.
• There is an increasing body of evidence that OSA is associated with a wide spectrum of cardiac arrhythmias including bradyarrhythmias, atrial fibrillation and ventricular arrhythmias.
• The association between OSA and stroke may, in part, be due to the increased incidence of atrial fibrillation.
• OSA may be associated with increased risk of nocturnal sudden cardiac death.
• Treatment of OSA has the potential to reduce or eradicate arrhythmias, and complement drug- and device-based therapy.
Financial & competing interests disclosure
Ian Wilcox is a clinical consultant to ResMed Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
Modified from Citation[85].
References
- Somers V, White D, Amin R et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. J. Am. Coll. Cardiol.52, 686–717 (2008).
- Chami H, Devereux R, Gottdiener J et al. Left ventricular morphology and systolic function in sleep-disordered breathing. The Sleep Heart Health Study. Circulation117, 2599–2607 (2008).
- Lattimore J, Celermajer D, Wilcox I. Sleep apnea and cardiovascular disease. J. Am. Coll. Cardiol.41, 1429–1437 (2003).
- Seppälä T, Partinen M, Penttilä A, Aspholm R, Tiainen E, Kaukianen A. Sudden death and sleeping history among Finnish men. J. Int. Med.229, 23–28 (1991).
- Valham F, Mooe T, Rabben T, Stenlund H, Wiklund U, Franklin K. Increased risk of stroke in patients with coronary artery disease and sleep apnea. A 10 year follow-up. Circulation118, 955–960 (2008).
- Wilcox I, Semsarian C. Obstructive sleep apnea. A respiratory syndrome with protean cardiovascular manifestations. J. Am. Coll. Cardiol.54, 1810–1812 (2009).
- Gami A, Howard D, Olson E, Somers V. Day–night pattern of sudden death in obstructive sleep apnea. N. Engl. J. Med.352, 1206–1214 (2005).
- Dement W, Kleitman N. Cyclic variations in EEG during sleep and their relation to eye movements, body motility, and dreaming. Electroencephalogr. Clin. Neurophysiol.9, 673–690 (1957).
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N. Engl. J. Med.328, 1230–1235 (1993).
- Bixler E, Vgontzas A, Have TT, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am. J. Respir. Crit. Care Med.157, 144–148 (1998).
- Duran J, Esnaola S, Rubio R, Iztueta A. Obstructive sleep apneahypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am. J. Respir. Crit. Care Med.163, 685–689 (2001).
- Young T, Evans L, Finn L, Palta M. Estimation of the clinically diagnosed proportion of sleep apnea syndrome in middle-aged men and women. Sleep20, 705–706 (1997).
- Young T, Peppard P, Taheri S. Excess weight and sleep-disordered breathing. J. Appl. Physiol.99, 1592–1599 (2005).
- Flemon W, Douglas N, Kuna S, Rodenstein D, Wheatley J. Access to diagnosis and treatment of patients with suspected sleep apnea. Am. J. Respir. Crit. Care Med.169, 668–672 (2004).
- White D, Weil J, Zwillich C. Metabolic rate and breathing during sleep. J. Appl. Physiol.59, 384–391 (1985).
- Mancia G. Autonomic modulation of the cardiovascular system during sleep. N. Engl. J. Med.328, 347–349 (1993).
- Hornyak M, Cejnar M, Elam M, Matousek M, Wallin B. Sympathetic muscle nerve activity during sleep in man. Brain114, 1281–1295 (1991).
- Somers V, Mark A, Zavala D, Abboud F. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J. Appl. Physiol.67, 2101–2106 (1989).
- Stoohs R, Guilleminault C. Cardiovascular changes associated with obstructive sleep apnea syndrome. J. Appl. Physiol.72, 583–589 (1992).
- Kusuoka H, Weisfeldt M, Zweier J, Jacobus W, Marban E. Mechanism of early contractile failure during hypoxia in intact ferret heart: evidence for modulation of maximal Ca2+-activated force by inorganic phosphate. Circ. Res.59, 270–282 (1986).
- Daly M, Scott M. The cardiovascular responses to stimulation of the carotid chemoreceptors in the dog. J. Physiol.165, 179–197 (1963).
- Daly M, Scott M. The effects of stimulation of the carotid body chemoreceptors on heart rate in the dog. J. Physiol.144, 148–166 (1958).
- Leung R, Bradley T. Sleep apnea and cardiovascular disease. Am. J. Respir. Crit. Care Med.164, 2147–2165 (2001).
- White S, Fletcher E, Miller C. Acute systemic blood pressure elevation in obstructive and nonobstructive breath hold in primates. J. Appl. Physiol.79, 324–330 (1995).
- O’Donnell C, Ayuse T, King E, Schwartz A, Smith P, Robotham J. Airway obstruction during sleep increases blood pressure without arousal. J. Appl. Physiol.80, 773–781 (1996).
- Chen L, Scharf S. Comparative hemodynamic effects of periodic obstructive and simulated central apneas in sedated pigs. J. Appl. Physiol.83, 485–494 (1997).
- Brinker J, Weiss J, Lappe D et al. Leftward septal displacement during right ventricular loading in man. Circulation61, 626–633 (1980).
- Bradley T. Right and left ventricular functional impairment and sleep apnea. Clin. Chest Med.13, 459–479 (1992).
- Hall M, Ando S, Floras J, Bradley T. Magnitude and time course of hemodynamic responses to Mueller maneuvers in patients with congestive heart failure. J. Appl. Physiol.85, 1476–1484 (1998).
- Guilleminault C, Motta J, Mihm F, Melvin K. Obstructive sleep apnea and cardiac index. Chest89, 331–334 (1986).
- Tolle F, Judy W, Yu P, Markand O. Reduced stroke volume related to pleural pressure in obstructive sleep apnea. J. Appl. Physiol.55, 1718–1724 (1983).
- Stoohs R, Guilleminault C. Cardiovascular changes associated with obstructive sleep apnea syndrome. J. Appl. Physiol.72, 583–589 (1992).
- Oliveira W, Campos O, Lira-Filho EB et al. Left atrial volume and function in patients with obstructive sleep apnea assessed by real-time three-dimensional echocardiography. J. Am. Soc. Echocardiogr.21, 1355–1361 (2008).
- Otto M, Belohlavek M, Romero-Corral A et al. Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am. J. Cardiol.99, 1298–1302 (2007).
- Arias MA, García-Río F, Alonso-Fernández A, Mediano O, Martínez I, Villamor J. Obstructive sleep apnea syndrome affects left ventricular diastolic function: effects of nasal continuous positive airway pressure in men. Circulation112, 375–383 (2005).
- Phillipson E, Bowes G. Control of breathing during sleep. In: Handbook of Physiology (Volume 2). Cherniack N, Widdicombe J (Eds). Williams & Wilkins, Bethesda, MD, USA, 649–689 (1986).
- Horner R, Brooks D, Kozar L, Tse S, Phillipson E. Immediate effects of arousal from sleep on cardiac autonomic outflow in the absence of breathing in dogs. J. Appl. Physiol.79, 151–162 (1995).
- Guyenet P. Lower brainstem mechanisms of respiratory integration. In: Sleep Apnea: Implications in Cardiovascular and Cerebrovascular Disease. Bradley T, Floras J (Eds). Marcel Dekker, NY, USA, 61–98 (2000).
- Usui K, Parker J, Newton G, Floras J, Ryan C, Bradley T. Left ventricular structural adaptations to obstructive sleep apnea in dilated cardiomyopathy. Am. J. Respir. Crit. Care Med.173, 1170–1175 (2006).
- Laaban J, Pascal-Sebaoun S, Bloch E, Orvoen-Frija E, Oppert J, Huchon G. Left ventricular systolic dysfunction in patients with obstructive sleep apnea syndrome. Chest122, 1133–1138 (2002).
- Fung J, Li T, Choy D et al. Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest121, 422–429 (2002).
- Romero-Corral A, Somers V, Pellikka P et al. Decreased right and left ventricular myocardial performance in obstructive sleep apnea. Chest132, 1863–1870 (2007).
- Ip M, Tse H, Lam B, Tsang K, Lam W. Endothelial function in obstructive sleep apnea and response to treatment. Am. J. Respir. Crit. Care Med.169, 348–353 (2004).
- Sanner B, Konermann M, Tepel M, Groetz J, Mummenhoff C, Zidek W. Platelet function in patients with obstructive sleep apnoea syndrome. Eur. Respir. J.16, 648–652 (2000).
- Yokoe T, Minoguchi K, Matsuo H et al. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continous positive airway pressure. Circulation107, 1129–1134 (2003).
- Somers V, Dyken M, Clary M, Abboud F. Sympathetic neural mechanisms in obstructive sleep apnea. J. Clin. Invest.96, 1897–1904 (1995).
- Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin G. Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J. Hypertens. Suppl.6, S529–S531 (1988).
- Carlson J, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin B. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest103, 1763–1768 (1993).
- Narkiewicz K, Montano N, Cogliati C, van de Borne P, Dyken M, Somers V. Altered cardiovascular variability in obstructive sleep apnea. Circulation98, 1071–1077 (1998).
- Narkiewicz K, van de Borne P, Cooley R, Dyken M, Somers V. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation98, 772–776 (1998).
- Carlson J, Rangemark C, Hedner J. Attenuated endothelium dependent vascular relaxation in patients with sleep apnoea. J. Hypertens.14, 577–584 (1996).
- Kato M, Roberts-Thomson P, Phillips B et al. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation102, 2607–2610 (2000).
- Saarelainen S, Seppala E, Laasonen K, Hasan J. Circulating endothelin-1 in obstructive sleep apnea. Endothelium5, 115–118 (1997).
- Grimpen F, Kanne P, Schulz E, Hagenah G, Hasenfuss G, Andreas S. Endothelin-1 plasma levels are not elevated in patients with obstructive sleep apnoea. Eur. Respir. J.15, 320–325 (2000).
- Kraiczi H, Hedner J, Peker Y, Carlson J. Increased vasoconstrictor sensitivity in obstructive sleep apnea. J. Appl. Physiol.89, 493–498 (2000).
- Parati G, Rienzo MD, Bonsignore M et al. Autonomic cardiac regulation in obstructive sleep apnea syndrome: evidence from spontaneous baroreflex analysis during sleep. J. Hypertens.15, 1621–1626 (1997).
- Becker H, Jerrentrup A, Ploch T et al. Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation107, 68–73 (2003).
- Dean R, Wilcox I. Possible atherogenic effects of hypoxia during obstructive sleep apnea. Sleep16(Suppl. 8), S15–S21 (1993).
- Drage L, Bortolotto L, Krieger E, Lorenzi-Filho G. Additive effects of obstructive sleep apnea and hypertension on early markers of atherosclerosis. Hypertension53, 64–69 (2009).
- Sorajja D, Gami A, Somers V, Behrenbeck T, Garcia-Touchard A, Lopez-Jimenez F. Independent association between obstructive sleep apnea and subclinical coronary artery disease. Chest133, 927–933 (2008).
- Peker Y, Carlson J, Hedner J. Increased incidence of coronary artery disease in sleep apnoea: a long-term follow-up. Eur. Respir. J.28, 596–602 (2006).
- Lattimore J, Wilcox I, Skilton M, Langenfeld M, Celermajer D. Treatment of obstructive sleep apnoea leads to improved microvascular endothelial function in the systemic circulation. Thorax61, 491–495 (2006).
- Jelic S, Lederer D, Adams T et al. Vascular inflammation in obesity and sleep apnea. Circulation121, 1014–1021 (2010).
- Lattimore J, Wilcox I, Adams M, Kilian J, Celermajer D. Treatment of obstructive sleep apnoea leads to enhanced pulmonary vascular nitric oxide release. Int. J. Cardiol.126, 229–233 (2008).
- Ryan S, Taylor C, McNicholas W. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation112, 2660–2667 (2005).
- Ryan S, Taylor C, McNicholas W. Predictors of elevated nuclear factor-κB-dependent genes in obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med.174, 824–830 (2006).
- Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J. Respir. Crit. Care Med.165, 934–939 (2002).
- Bedwell S, Dean R, Jessup W. The action of defined oxygen-centred free radicals on human low-density lipoprotein. Biochem. J.262, 707–712 (1989).
- Faller D. Endothelial cell responses to hypoxic stress. Clin. Exp. Pharmacol. Physiol.26, 74–84 (1999).
- El-Solh A, Mador M, Sikka P, Dhillon R, Amsterdam D, Grant B. Adhesion molecules in patients with coronary artery disease and moderate-to-severe obstructive sleep apnea. Chest121, 1541–1547 (2002).
- Larsson P, Wallen N, Hjemdahl P. Norepinephrine-induced human platelet activation in vivo is only partly counteracted by aspirin. Circulation89, 1951–1957 (1994).
- Bokinsky G, Miller M, Ault K, Husband P, Mitchell J. Spontaneous platelet activation and aggregation during obstructive sleep apnea and its response to therapy with nasal continuous positive airway pressure. A preliminary investigation. Chest108, 625–630 (1995).
- Dimsdale J, Coy T, Ziegler M, Ancoli-Israel S, Clausen J. The effect of sleep apnea on plasma and urinary catecholamines. Sleep18, 377–381 (1995).
- Eisensehr I, Ehrenberg B, Noachtar S et al. Platelet activation, epinephrine, and blood pressure in obstructive sleep apnea syndrome. Neurology51, 188–195 (1998).
- Hoffstein V, Herridge M, Mateika S, Redline S, Strohl K. Hematocrit levels in sleep apnea. Chest106, 787–791 (1994).
- Chin K, Ohi M, Kita H et al. Effects of NCPAP therapy on fibrinogen levels in obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med.153, 1972–1976 (1996).
- Nobili L, Schiavi G, Bozano E, Carli FD, Ferrillo F, Nobili F. Morning increase of whole blood viscosity in obstructive sleep apnea syndrome. Clin. Hemorheol. Microcirc.22, 21–27 (2000).
- Lakka H, Lakka T, Tuomilehto J, Salonen J. Abdominal obesity is associated with increased risk of acute coronary events in men. Eur. Heart J.23, 706–713 (2002).
- Hubert H, Feinleib M, McNamara P, Castelli W. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation67, 968–977 (1983).
- Lee I, Manson J, Hennekens C, Paffenbarger R Jr. Body weight and mortality: a 27-year follow-up of middle-aged men. JAMA270, 2823–2828 (1993).
- Rosito G, D’Agostino R, Massaro J et al. Association between obesity and a prothrombotic state: the Framingham Offspring Study. Thromb. Haemost.91, 683–689 (2004).
- Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation110, 1564–1571 (2004).
- Ip M, Lam B, Ng M, Lam W, Tsang K, Lam K. Obstructive sleep apnea is independently associated with insulin resistance. Am. J. Respir. Crit. Care Med.165, 670–676 (2002).
- Punjabi N, Sorkin J, Katzel L, Goldberg A, Schwartz A, Smith P. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am. J. Respir. Crit. Care Med.165, 677–682 (2002).
- Wilcox I, McNamara S, Collins F, Grunstein R, Sullivan C. ‘Syndrome Z’: the interaction of sleep apnoea, vascular risk factors and heart disease. Thorax53(Suppl. 3), S25–S28 (1998).
- Nock N, Li L, Larkin E, Patel S, Redline S. Empirical evidence for ‘Syndrome Z’: a hierarchical 5-factor model of the metabolic syndrome incorporating sleep disturbance measures. Sleep32, 615–622 (2009).
- Coumel P. Cardiac arrhythmias and the autonomic nervous system. J. Cardiovasc. Physiol.4, 338–355 (1993).
- Peled N, Abinader E, Pillar G, Sharif D, Lavie P. Nocturnal ischemic events in patients with obstructive sleep apnea syndrome and ischemic heart disease J. Am. Coll. Cardiol.34, 1744–1749 (1999).
- Krieger J, Laks L, Wilcox I et al. Atrial natriuretic peptide release during sleep in patients with obstructive sleep apnoea before and during treatment with nasal continuous positive airway pressure. Clin. Sci.77, 407–411 (1989).
- Flemons W, Remmers J, Gillis A. Sleep apnea and cardiac arrhythmias. Is there a relationship? Am. Rev. Respir. Dis.148, 618–621 (1993).
- Mehra R, Benjamin E, Shahar E et al. Association of nocturnal arrhythmias with sleep-disordered breathing. The Sleep Heart Health Study. Am. J. Respir. Crit. Care Med.173, 910–916 (2006).
- Becker H, Koehler U, Stammnitz A, Peter J. Heart block in patients with sleep apnoea. Thorax53, 29–32 (1998).
- Guilleminault C, Connolly S, Winkle R. Cardiac arrhythmia and conduction disturbances during sleep in 400 patients with sleep apnea syndrome. Am J. Cardiol.52, 490–494 (1983).
- Tilkian A, Guilleminault C, Schroeder J, Lehrman K, Simmons F, Dement W. Sleep-induced apnea syndrome. Prevalence of cardiac arrhythmias and their reversal after tracheostomy. Am. J. Med.63, 348–358 (1977).
- Simantirakis E, Schiza S, Marketou M et al. Severe bradyarrhythmias in patients with sleep apnoea: the effect of continuous positive airway pressure treatment: a long-term evaluation using an insertable loop recorder. Eur. Heart J.25, 1070–1076 (2004).
- Harbison J, O’Reilly P, McNicholas W. Cardiac rhythm disturbances in the obstructive sleep apnea syndrome: effects of nasal continuous positive airway pressure therapy. Chest118, 591–595 (2000).
- Grimm W, Koehler U, Fus E et al. Outcome of patients with sleep apnea-associated severe bradyarrhythmias after continuous positive airway pressure therapy. Am. J. Cardiol.86, 688–692, A9 (2000).
- Koehler U, Fus E, Grimm W et al. Heart block in patients with obstructive sleep apnoea: pathogenetic factors and effects of treatment. Eur. Respir. J.11, 434–439 (1998).
- Stegman S, Burroughs J, Henthorn R. Asymptomatic bradyarrhythmias as a marker for sleep apnea: appropriate recognition and treatment may reduce the need for pacemaker therapy. Pacing Clin. Electrophysiol.19, 899–904 (1996).
- Grimm W, Hoffmann J, Menz V et al. Electrophysiologic evaluation of sinus node function and atrioventricular conduction in patients with prolonged ventricular asystole during obstructive sleep apnea. Am. J. Cardiol.77, 1310–1314 (1996).
- Zwillich C, Devlin T, White D, Douglas N, Weill J, Martin R. Bradycardia during sleep apnea: characteristics and mechanism. J. Clin. Invest.69, 1286–1292 (1982).
- Garrigue S, Pepin J, Defaye P et al. High prevalence of sleep apnea syndrome in patients with long-term pacing. The European multicenter polysomnographic study. Circulation115, 1703–1709 (2007).
- Garrigue S, Bordier P, Jaïs P et al. Benefit of atrial pacing in sleep apnea syndrome. N. Engl. J. Med.346, 404–412 (2002).
- Pepin J, Defaye P, Garrigue S, Poezevara Y, Levy P. Overdrive atrial pacing does not improve obstructive sleep apnoea syndrome. Eur. Respir. J.25, 343–347 (2005).
- Sharafkhaneh A, Sharafkhaneh H, Bredikus A, Guilleminault C, Bozkurt B, Hirshkowitz M. Effect of atrial overdrive pacing on obstructive sleep apnea in patients with systolic heart failure. Sleep Med.8, 31–36 (2007).
- Weng C, Chen Q, Ma Y, He Q. A meta-analysis of the effects of atrial overdrive pacing on sleep apnea syndrome. Pacing Clin. Electrophysiol.32, 1434–1443 (2009).
- Gami A, Pressman G, Caples S et al. Association of atrial fibrillation and obstructive sleep apnea. Circulation110, 364–367 (2004).
- Stevenson I, Teichtahl H, Cunnington D, Ciavarella S, Gordon I, Kalman J. Prevalence of sleep disordered breathing in paroxysmal and persistent atrial fibrillation patients with normal left ventricular function. Eur. Heart J.29, 1662–1669 (2008).
- Kanagala R, Murali N, Friedman P et al. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation107, 2589–2594 (2003).
- Gami A, Hodge D, Herges R et al. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J. Am. Coll. Cardiol.49, 565–571 (2007).
- Wyse D, Waldo A, DiMarco J et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N. Engl. J. Med.347, 1825–1833 (2002).
- Hagens V, Ranchor A, Sonderen EV et al. Effect of rate or rhythm control on quality of life in persistent atrial fibrillation: results from the Rate Control Versus Electrical Cardioversion (RACE) Study. J. Am. Coll. Cardiol.43, 241–247 (2004).
- Carlsson J, Miketic S, Windeler J et al. Randomized trial of rate-control versus rhythm-control in persistent atrial fibrillation: the Strategies of Treatment of Atrial Fibrillation (STAF) study. J. Am. Coll. Cardiol.41, 1690–1696 (2003).
- Mooe T, Gullsby S, Rabben T, Eriksson P. Sleep-disordered breathing: a novel predictor of atrial fibrillation after coronary artery bypass surgery. Coron. Artery Dis.7, 475–478 (1996).
- Monahan K, Storfer-Isser A, Mehra R et al. Triggering of nocturnal arrhythmias by sleep-disordered breathing events. J. Am. Coll. Cardiol.54, 1797–1804 (2009).
- Schulz R, Eisele H, Seeger W. Nocturnal atrial fibrillation in a patient with obstructive sleep apnoea. Thorax60, 174 (2005).
- Edwards N, Blyton D, Kirjavainen T, Kesby G, Sullivan C. Nasal continuous positive airway pressure reduces sleep-induced blood pressure increments in preeclampsia. Am. J. Respir. Crit. Care Med.162, 252–257 (2000).
- Yaggi H, Concato J, Kernan W, Lichtman J, Brass L, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N. Engl. J. Med.353, 2034–2041 (2005).
- Redline S, Yenokyan G, Gottlieb D et al. Obstructive sleep apnea hypopnea and incident stroke: the Sleep Heart Health Study. Am. J. Respir. Crit. Care Med. DOI: 10.1164/rccm.200911–1746OC (2010) (Epub ahead of print).
- Hays A, Sacco R, Rundek T et al. Left ventricular systolic dysfunction and the risk of ischemic stroke in a multiethnic population. Stroke37, 1715–1719 (2006).
- Eleid M, Konecny T, Orban M et al. High prevalence of abnormal nocturnal oximetry in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol.54, 1805–1809 (2009).
- Sullivan C, Issa F, Berthon-Jones M, Eves L. Reversal of obstructive sleep apnoea by continuous positive airway pressure applied through the nares. Lancet1(8225), 862–865 (1981).
- Abe H, Takahashi M, Yaegashi H et al. Efficacy of continuous positive airway pressure on arrhythmias in obstructive sleep apnea patients. Heart Vessels25, 63–69 (2010).
- Cohen M, Rohtla K, Lavery C, Muller J, Mittleman M. Meta-analysis of the morning excess of acute myocardial infarction and sudden cardiac death. Am. J. Cardiol.79, 1512–1516 (1997).
- Franklin K, Nisson J, Sahlin C, Naslund U. Sleep apnea and nocturnal angina. Lancet345, 1085–1087 (1995).
- Schafer H, Koehler U, Ploch T, Peter J. Sleep related myocardial ischemia and sleep structure in patients with obstructive sleep apnea and coronary heart disease. Chest111, 387–393 (1997).
- Hanly P, Sasson Z, Zuberi N, Lunn K. ST segment depression during sleep in obstructive sleep apnea. Am. J. Cardiol.71, 1341–1345 (1993).
- Alonso-Fernández A, García-Río F, Racionero M et al. Cardiac rhythm disturbances and ST-segment depression episodes in patients with obstructive apnea-hypopnea syndrome and its mechanisms. Chest127, 15–22 (2005).
- Peled N, Abinader E, Pillar G, Sharif D, Lavie P. Nocturnal ischemic events in patients with obstructive sleep apnea syndrome and ischemic heart disease. Effects of continuous positive air pressure treatment. J. Am. Coll. Cardiol.34, 1744–1749 (1999).
- Ryan C, Usui K, Floras J, Bradley T. Effect of continuous positive airway pressure on ventricular ectopy in heart failure patients with obstructive sleep apnoea. Thorax60, 781–785 (2005).
- Craig S, Pepperell J, Kohler M, Crosthwaite N, Davies R, Stradling J. Continuous positive airway pressure treatment for obstructive sleep apnoea reduces resting heart rate but does not affect dysrhythmias: a randomised controlled trial. J. Sleep Res.18, 329–336 (2009).