Abstract
The promise of personalized medicine is highly dependent on the identification of biomarkers that inform diagnostic decisions and treatment options, as well as on the accurate, rapid and cost-effective detection and interpretation of these biomarkers. miRNAs, which are short noncoding regulatory RNAs, are rapidly emerging as a novel class of biomarkers with a unique set of biological and chemical properties that makes them very appealing candidates for theranostic applications in cancer. Since the utility of some protein-encoding gene biomarkers is already exploited in routine clinical practice, it will be important to identify areas in which miRNAs provide complementary or superior information to these existing (and other translational) biomarkers to enhance the diagnostic, prognostic and predictive power of molecular characterization of tumors. In this article, the challenges and opportunities for integration of miRNA-based assays in the clinical toolkit to improve care and management of patients afflicted with solid tumors will be discussed.
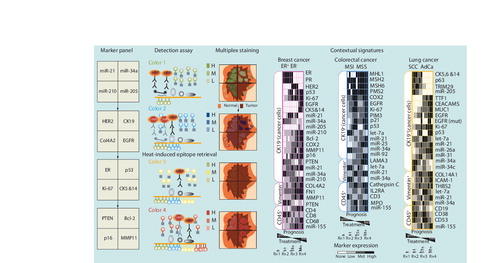
On the left is a flow chart of a conceptual high-density marker detection platform. Physical compartmentalization yields four contiguous but insulated compartments for the parallel detection of independent biomarkers from the tumor core to adjacent normal tissue. Specific locked nucleid acid-modified DNA probes are dispensed in each compartment (marker panel) and tissue slides are subjected to in situ hybridization assay. miRNA signal is revealed by tyramide signal amplification (TSA) reactions with green fluorochrome-tyramine substrate (color 1; detection assay). After hydrogen peroxide incubation to inactivate HRP from the preceding TSA reaction, protein expression is revealed by a new round of TSA reactions with a different fluorochrome–tyramine substrate (color 2). After heat-induced epitope retrieval, expression of additional proteins is revealed by sequential TSA reactions with other fluorochrome–tyramine substrates (colors 3 and 4). In this example, the colorized tissue cartoon provides a virtual rendition of the expected staining pattern for each marker on an ER+PR+HER2- breast cancer specimen (multiplex staining). On the right are tentative and speculative multimarker panels based on biomarkers already incorporated in routine clinical practice and promising translational biomarkers. Computer-assisted image analysis will be used to quantitate the expression levels of each marker. This information will be used to generate contextual signatures that reflect molecular changes within the cancer cells (CK19+) or other cellular compartments of the tumor microenvironment, such as reactive fibroblast (vimentin+) and immune cells (CD45+), to inform treatment selection and intensity in prevalent solid tumors.
AdCa: Adenocarcinoma; EGFR: EGF receptor; ER: Estrogen receptor; H: High; HER2: Human EGF receptor 2; HRP: Horseradish peroxidase; L: Low; M: Medium; MMP: Matrix metalloproteinase; MSI: Microsatellite instable: MSS: Microsatellite stable; PR: Progesterone receptor; SCC: Squamous cell carcinoma.
Cancer causes approximately 23% of all deaths in the USA Citation[1]. Standard clinical parameters such as tumor size, grade, lymph node involvement and tumor–node–metastasis (TNM) staging Citation[201] correlate with outcome and serve to stratify patients with respect to (neo)adjuvant chemotherapy and/or radiotherapy regimens. Incorporation of molecular markers in clinical practice has defined tumor subtypes that are more likely to respond to targeted therapy, as is the case for the response of breast cancers overexpressing estrogen receptor (ER) to hormone therapy (e.g., tamoxifen and aromatase inhibitors). However, stage-matched tumors grouped by histological or molecular subtypes respond differently to the same treatment regimen. This indicates that additional key genetic and epigenetic alterations exist, with important etiological contributions. A more detailed understanding of the molecular mechanisms and regulatory pathways at work in cancer cells and the tumor microenvironment (TME) could dramatically improve the design of novel anti-tumor drugs and inform the selection of optimal therapeutic strategies. Thus, there is an urgent need to develop and implement diagnostic, prognostic and therapeutic biomarkers to characterize the biology of each tumor and to assist clinicians in making important decisions with regard to individual patient care and treatment Citation[2,3].
In the last 5–10 years, high-throughput expression analysis platforms have been widely used to study tumors at a molecular level in order to further refine patient subgroups. While the general strategy and approach of these technologies are applicable and have been applied to the molecular characterization of hematologic (leukemias and lymphomas) and solid (carcinomas, sarcomas and melanomas) tumors, this article will focus on recent biomarker discoveries in breast, colorectal and lung cancer as examples of prevalent and representative solid tumors of epithelial origin (carcinomas). I will place a particular emphasis on recent clinical findings on miRNAs and will discuss how inherently distinct biological properties of miRNA and protein-encoding genes are important determinants of the clinical utility and performance of miRNAs as novel biomarkers.
miRNAs as emerging clinical biomarkers
MicroRNAS are evolutionary conserved short noncoding RNA genes that were discovered just 10 years ago Citation[4–6]. While their discovery occurred in studies of the nematode Caenorhabditis elegansCitation[7–9], we now know that miRNAs are an important and pervasive regulatory layer of gene expression that acts at the post-transcriptional level in all animals, including humans Citation[10,11]. miRNAs have been implicated as pivotal players in prevalent human diseases, including cancer and neurodegenerative conditions Citation[12–16]. The mature and biologically active miRNA is released from a much longer RNA molecule after sequential enzymatic cleavage. A primary 5´-capped and 3´-polyadenylated transcript is cleaved in the nucleus by the RNAse type III enzyme Drosha and associated proteins of the microprocessor, and yields a 70-nucleotide precursor RNA hairpin, which is exported via the Expo5 pathway into the cytoplasm Citation[17]. Once in the cytoplasm, the approximately 21–23 nucleotide-long mature miRNA is released after cleavage by another RNAse type III, Dicer, and loaded in an Argonaute-containing multiprotein miRNA-induced silencing complex Citation[17]. By binding to partially complementary sites in the 3´-untranslated region of mRNAs, the miRNA brings the miRNA-induced silencing complex in close proximity to target mRNA, which triggers increased mRNA degradation and/or inhibition of protein synthesis Citation[18].
The human genome is estimated to contain more than 1000 miRNA genes and a third of protein-encoding genes are predicted to be under miRNA-mediated regulation Citation[19–23]. This suggests that miRNAs constitute a novel and global mechanism to modulate gene expression, comparable to the genome-wide epigenetic and transcriptional changes associated with cancer. Indeed, the recent explosion in miRNA research has brought conceptual and technological advances and innovations that are expanding our understanding in the fields of cancer biology and molecular pathology Citation[12–14,24]. Unique chemical and biological characteristics make miRNAs very attractive candidates for theranostic applications in cancer as the expression and function of miRNAs are tightly associated. A single miRNA can regulate and modulate levels of hundreds of targets, some of which are components of the same signaling pathway and/or biological process Citation[21,22]. Accordingly, functional manipulation of a single miRNA could concertedly affect multiple target mRNAs (one drug, multiple hits), unlike siRNA-, antibody- or small molecule inhibitor-based therapies (one drug, one hit). Synthetic compounds exist to eliminate or decrease miRNA activity (e.g., antagomirs, tiny antisense locked nucleic acids [LNAs] and oligonucleotides) and to replenish or increase miRNA activity (e.g., siRNA-like compounds). Proof-of-concept experiments have demonstrated effective and safe in vivo delivery of these compounds in rodents and nonhuman primates Citation[25–30]. Therefore, the utility of key miRNAs as biomarkers for interrogating tumor biology could also be exploited to discover pharmacological miRNA targets that affect tumor biology.
Assay platforms & approaches for biomarker detection
Molecular characterization of tumors based on the expression of genes and gene products can be divided into two broad and distinct categories: RNA-based and tissue slide-based assays. RNA-based assays follow similar procedures for miRNA and mRNA detection. Typically, total RNA is extracted from whole tissue biopsies (fresh, frozen or fixed) to determine mRNA and miRNA levels. High-throughput platforms, such as microarray chips and deep sequencing have enabled transcriptome- and miRNome-wide tumor tissue characterizations. Tissue slide-based morphology-driven assays use archived formalin-fixed paraffin-embedded (FFPE) specimens to determine gene copy number by fluorescence in situ hybridization, mRNA or miRNA levels by in situ hybridization (ISH) and protein levels by immunohistochemistry (IHC). IHC is the most common assay for expression analysis of protein-encoding genes in routine clinical practice, as is ISH for expression of miRNAs in translational research.
The use of miRNA biomarkers have a distinct set of advantages over protein-encoding genes:
• miRNAs are more stable and are better preserved than mRNAs in biological specimens, including archived FFPE tissue blocks and blood samples. Thus, poor RNA quality and RNA degradation may compromise the information content and accuracy of mRNA signatures much more than that of miRNA signatures;
• The human miRNome is relatively simple, with approximately 1000 genes, enabling detection of all miRNAs in a single-tube reaction and quick generation of miRNA signatures, which are less bioinformatically demanding than mRNA expression signatures;
• RNA-based and tissue slide-based assays measure the same bioanalyte for miRNA genes – namely, the RNA levels of mature miRNA – whereas RNA-based assays measure mRNA levels (intermediary carrier of genetic information) and tissue slide-based assays measure protein levels (gene product) for protein-encoding genes;
• More straightforward interpretations of miRNA function can be drawn based on expression changes of protein function, as mature miRNA levels generally correlate with miRNA activity, whereas post-transcriptional regulation and post-translational modifications render a more complex correlation between protein activity and expression levels of mRNAs or proteins.
RNA-based assays
Expression profiling in whole tumor tissue samples has provided sets of differentially expressed mRNAs and miRNAs (expression signatures), with potential links to the etiology of the disease. However, this type of assay does not distinguish between altered RNA expression within the cancer cell compartment or other elements of the TME, including reactive stroma and infiltrating immune cells. This level of cellular complexity is probably captured in expression signatures obtained from whole-tissue RNA analysis, and thus it is not straightforward to interpret what these signatures measure or indicate biologically. Changes in RNA levels between normal and tumor tissues and among tumors from different patients can, in some cases, simply reflect a different ratio of cell type representation and tissue heterogeneity, whereas, in other instances, it can reflect specific molecular aberrations in the cancer cell or other cellular compartments of the TME that affect cancer cell behavior and aggressiveness. For protein-encoding genes, gene ontology analysis provides annotated information on expression and function that facilitates ascribing changes in mRNA levels to a specific cell type Citation[202]. For miRNA genes, there is only a fragmentary knowledge of cell type and tissue-specific expression and function in these different cellular contexts under physiological and pathological conditions. Upregulation of a miRNA within cancer cells or immune cells may have both etiological relevance and clinical significance, but obviously indicates different biological processes and has different diagnostic and therapeutic implications.
While one should be mindful of these caveats and limitations, high-throughput discovery tools have identified expression signatures with potential clinical utility for the diagnosis and prognosis of cancer that can be measured in single-tube highly sensitive multiplex quantitative reverse-transcriptase PCR assays. Some of these refined mRNA signatures have already received or are seeking US FDA approval for clinical use (see later). A miRNA signature for the diagnosis of pancreatic adenocarcinoma (miRInform® Pancreas) is currently offered by Asuragen, Inc. (TX, USA) as laboratory-developed test in a Clinical Laboratory Improvement Amendments-certified environment Citation[31].
Tissue slide-based assays
Tissue slide-based morphology-driven assays are the workhorse of clinical pathology. These include morphological and histological characterization of tumor lesions with specialized stains and dyes (e.g., hematoxylin and eosin), as well as molecular assays such as IHC (e.g., ER, MSH2 and EGF receptor [EGFR]) and fluorescence ISH (e.g, human EGF receptor 2 [HER2]). Routine clinical IHC assays detect one or two markers by chromogenic staining Citation[32]. The use of fluorescence-based staining can increase the multiplexing capability of this assay to three or more independent markers Citation[33,34]. To meet this increasing demand for digital pathology, several companies already offer multispectral whole slide scanning instruments and companion software packages for computer-assisted image analysis that enable fast and quantitative scoring of markers in specific cellular compartments Citation[33–36].
Protein molecules can accumulate in cells at several orders of magnitude (102–104) higher than their corresponding mRNAs Citation[37], whereas the maximum number of mature miRNA molecules equals the number of primary miRNA transcripts. Thus, sensitivity has been a challenge for detection of miRNAs by ISH on FFPE clinical specimens. The introduction of LNAs, high-affinity bycyclic RNA analogs Citation[38], for the synthesis of chimeric LNA/DNA probes has overcome the technical limitations of achieving specific and avid hybridization to the short RNA sequence of mature miRNAs. This has allowed several groups to characterize miRNA expression at single-cell resolution in different types of cancer Citation[39–53]. The ISH assay is an important validation tool to correctly interpret the biological basis and clinical relevance of altered expression of cancer-associated miRNAs, as determined by RNA-based assays on whole tissue samples Citation[52,53]. A highly sensitive fluorescence-based method for the codetection of multiple RNA and protein markers on the same tissue section has recently been reported Citation[52]. Sequential rounds of tyramide signal amplification reactions Citation[54] with in-house-synthesized fluorochromes enables the detection of additional RNA markers, such as abundant noncoding RNAs (e.g., U6 snRNA and 18S rRNA) that can be used to assess RNA quality and integrity, and protein biomarkers that can be used: to highlight a specific cellular compartment (e.g., CK19 for epithelial/cancer cells, CD45 for leukocytes) and to assess the cell source of miRNA expression; to characterize signaling pathways and cellular processes (e.g., Ki-67 for proliferation, p53 for compromised DNA damage and apoptotic responses); and to interrogate miRNA–target interaction networks (e.g., PTEN and PDCD4 for miR-21) Citation[52]. This ISH/IHC assay was deployed to characterize the spatial expression of cancer-associated miRNAs, including miR-21 and -155, two of the most frequently upregulated miRNAs in cancer, which are thought to be highly oncogenic Citation[12–14,24]. In a panel of breast, colorectal, lung, pancreatic and prostate carcinomas, the authors identified a subpopulation of immune cells to be the predominant source of miR-155 expression in these prevalent solid tumors, whereas miR-21 expression was upregulated within cancer cells and reactive fibroblasts to different extents depending on the organ site Citation[52]. By contrast, miR-34a expression was detected at lower levels in cancer whereas the surrounding stromal cells retained high levels of miR-34a in breast and lung cancer specimens, suggesting dynamic and independent regulation of this miRNA in different cellular compartments Citation[52]. In an effort to bring this type of miRNA-based assay closer to the clinic, the authors have already performed a fully automated protocol for the described IHC/ISH assay in FDA-approved automated staining stations in a laboratory environment that is compliant with gold-standard practice of current clinical IHC assays Citation[52,55].
Molecular assays in breast cancer
Breast cancer (BC) is the most prevalent type of cancer and is the second most common cause of cancer-related death of women in the USA Citation[56]. BC is not a single disease, but rather a complex and heterogeneous group of diseases with different molecular alterations Citation[57,58]. Current management of BC is mainly based on the activation status of ER, progesterone receptor (PR) and HER2. ER/PR/HER2 classification is used for diagnosis, prognosis and treatment selection, as targeted drugs have been developed to block these signaling pathways Citation[3]. Global mRNA expression studies have identified four major biological subtypes of BC, which differ with respect to their biology, prognosis and susceptibility to specific treatments Citation[59,60]. These molecular intrinsic subtypes closely correspond to ER/PR/HER2 classification Citation[61]: the immense majority of luminal A cases are ER+ and/or PR+(ER/PR+)HER2-; luminal B cases are ER/PR+HER2- (with a higher proliferation rate) or ER/PR+HER2+; HER2-overexpressing cases are ER/PR-HER2+; and basal cases are ER-PR-HER2-. These gene-expression studies also indicate the involvement of distinct subpopulations of epithelial cells among tumor subtypes; cells with myoepithelial/basal characteristics contribute almost exclusively to the aggressive basal subtype. This underscores the importance of ER and HER2 signaling in tumor initiation and evolution.
Given the technical and financial difficulties of conducting global mRNA expression analysis in all clinical samples, multiprotein marker panels and refined mRNA signatures have been identified for diagnosis and subtyping, prognosis and treatment selection Citation[62,63]. An eight-marker IHC assay has been proposed as a refinement of ER/PR/HER2 classification and as a better surrogate marker of intrinsic subtypes Citation[61,64,65]. In addition to ER, PR and HER2, this eight-marker IHC assay also includes CK5, CK14, EGFR, Ki-67 and p53. CK5 and CK14 are cytokeratins expressed by basal/myoepithelial cells and are used to identify ER-PR-HER2- breast cancer cases with a basal phenotype Citation[61,64]. Human EGFR is overexpressed in a large number of ER-PR-HER2- cases with a basal phenotype and is a potential druggable target. The Ki-67 proliferation index is used for grading and prognostics in BC and other cancer types. High proliferation index and elevated p53 expression (indicative of a mutant and dysfunctional p53) can be used to distinguish luminal A from luminal B tumors, which have a slightly less favorable outcome owing to reduced response to hormone therapy and chemotherapy Citation[65]. Refined mRNA signatures such as the 21-gene Oncotype DX® (Genomic Health Inc., CA, USA) Citation[66], 70-gene MammaPrint®Citation[67] and 50-gene PAM50 Citation[68] can predict the risk of recurrence and thus inform the course and intensity of treatment. For ER+ cases with a low risk of recurrence as assessed by these tests, adjuvant chemotherapy is not recommended since the adverse side effects and morbidity would outweigh any potential clinical benefit. Oncotype Dx and MammaPrint are the only RNA-based assays so far that have received approval or clearance by the FDA for use in the clinic Citation[62]. Remarkably, only one gene is shared in common between the Oncotype Dx and MammaPrint mRNA signatures, suggesting that these genes, besides the obvious exception of ER and HER2, probably serve more as readout of biological processes rather than the identification of key driver alterations in the cancer cells. For example, CD68, one of the genes in the Oncotype Dx assay, is a cell type-specific marker of monocytes and macrophages; mRNA levels of CD68 probably reflect the composition of immune cell infiltrates and by inference the overall nature of immune programs elicited against (tumoricidal) or recruited by (tumorigenic) the cancer cells. Indeed, three-marker IHC-based immune cell signature that measures the ratio of CD4+ T cells, CD8+ T cells and CD68+ monocytes/macrophages was recently shown to be a prognostic indicator of recurrence-free and overall survival in BC Citation[69]. The authors suggest that this deleterious effect is a result of decreased treatment response to chemotherapy in patients with a high number of infiltrating CD68+ cells Citation[69]. Similarly, RNA-based assays using microdissected stromal tissue as the input source have also revealed the influence of noncancer cell compartments in the course of the disease Citation[70–75]. Stromal signatures can stratify patients based on outcome independent of standard clinical prognostic factors and global mRNA expression signatures such as MammaPrint Citation[75]. These stromal signatures reflect a combination of biological processes (angiogenic, hypoxic and immune responses) in the different constituent cellular compartments such as reactive fibroblasts, vasculature and leukocytes.
Global miRNA expression studies have revealed consistent changes between normal and tumor tissues. miR-125b and miR-145 are frequently detected at lower levels, and miR-21, miR-155 and miR-210 at higher levels in tumor tissues Citation[40,76–79]. Unsupervised hierarchical clustering of cases based on global miRNA signatures differs from that of global mRNA signatures and well-established IHC surrogate markers, suggesting that miRNAs could provide additional diagnostic information Citation[80,81]. Other miRNAs have been shown to closely correlate with ER, PR and HER2 status, and these miRNAs could be useful to detect alterations in these signaling pathways and inform treatment response Citation[82–86]. Yet, there is little overlap of differentially expressed miRNAs among studies, and additional investigations will be needed to select the most informative and relevant miRNAs Citation[78,87]. Other clinical applications have been proposed for single miRNAs and refined miRNA signatures in specific intrinsic subtypes or subgroups of patients . miR-210 is a robust and independently validated prognostic biomarker in BC. Higher miR-210 levels have been associated with poor outcome in ER+ cases with negative lymph node involvement Citation[80,88,89] and in ER- cases with negative lymph node involvement Citation[88,90,91]. It is noteworthy that miR-210-based quantitative reverse-transcriptase PCR assay performed similarly to the 21-mRNA Oncotype Dx assay to predict outcome in ER+ cases that received adjuvant tamoxifen treatment for 5 years Citation[80]. miR-210 expression is induced by hypoxic conditions and has been shown to mediate anti-apoptotic, migratory and proinvasive programs in ER+ and ER- BC cell lines Citation[80,92]. Thus, miR-210 expression could serve as a hypoxic sensor and indicator of an aggressive phenotype in clinical samples.
The overwhelming majority of miRNA expression studies utilize RNA-based rather than tissue slide-based assays. Tissue slide-based assays provide an independent platform and a different vantage point to validate and refine expression profiling results. An ISH-based study found a subset of BC-associated miRNAs whose expression was altered within distinct subpopulations of mammary epithelial cells Citation[40]. Expression of let-7a and miR-141 was detected at varying levels, predominantly within luminal epithelial cells in normal tissue Citation[40]. Conversely, miR-145 and miR-205 expression was restricted to myoepithelial cells in normal epithelial structures, whereas their expression was reduced or completely eliminated in matching tumor specimens Citation[40]. The authors proposed miR-145 as a novel biomarker for early detection of malignancy and disease progression from noninvasive to preinvasive lesions based on the early manifestation of altered miR-145 expression in carcinoma in situ lesions adjacent to invasive carcinoma and thus presumed preinvasive Citation[40]. The authors also observed a positive correlation between miR-205 expression and favorable clinical outcome in ER-PR-HER2- cases and proposed miR-205 as a prognostic indicator for the aggressive ER-PR-HER2- (basal) subtype Citation[40].
Molecular assays in colorectal cancer
Colorectal cancer (CRC) is the third leading cause of cancer-related death in both men and women in the USA Citation[56]. CRC can be divided into two molecular subtypes: microsatellite stable (MSS) and microsatellite instable (MSI). The MSI phenotype is observed in approximately 15% of cases, is associated with a better prognosis and appears to have a different chemoresponse to therapeutic agents such as 5-fluorouracil (more resistant) and irinotecan (more sensitive) Citation[93,94]. MSI was first described in connection to Lynch syndrome or hereditary nonpolyposis CRC Citation[95]. The MSI phenotype can be determined by PCR-based genomic DNA analysis of mono- and di-nucleotide repeats of representative microsatellite sequences or by IHC-based assay of DNA mismatch repair proteins (MHL1, MSH2, MSH6 and PMS2) Citation[93,94]. The latter is a preferred screening assay in routine clinical practice; absence of one or more of these proteins is indicative of the MSI phenotype. This may be followed by microsatellite analysis to confirm diagnosis and/or DNA sequencing to determine hereditary or sporadic origin of the tumor Citation[94].
Surgical resection of the primary tumor is a successful curative treatment for approximately 80% of stage I and II CRC cases. Prognostic mRNA signatures have been identified to stratify cases into low risk and high-recurrence groups to minimize patient overtreatment with adjuvant chemotherapy Citation[96]. As noted earlier for BC, there is little overlap in the gene lists that compose these expression signatures Citation[96]. A recent study with large sample sizes for both training and validating sets of patients developed an 18 mRNA prognostic signature (ColoPrint®; Agendia, CA, USA) for stage II and III cases that has better predictive value than mutation status of BRAF, KRAS and/or PI3KCA genes or standard clinical parameters Citation[97]. Unsupervised hierarchical clustering analysis uncovered three main molecular subtypes in the training set. Overall, 86% of the MSI cases were included in the low-risk recurrence subtype in the training set, and 90% of the MSI cases in the validation set Citation[97]. This indicates that ColoPrint captures the already known good prognosis of MSI cases. Importantly, ColoPrint also serves to stratify MSS cases with a more mixed prognosis a prioriCitation[97]. The current standard of care for stage III cases, which are more often MSS tumors, is adjuvant chemotherapy. Results from these RNA-based assays may prompt a revision of this practice, at least for a subgroup of stage III cases with a low-risk recurrence signature.
Global miRNA expression studies have revealed consistent changes between normal and tumor tissues. miR-125a, miR-143 and miR-145 are frequently detected at lower levels and miR-31, miR-106a and miR-203 at higher levels in tumor tissues Citation[77,98,99]. An eight-miRNA signature was developed to classify CRC tissues as the MSS or MSI subtype Citation[100]. Two of these miRNAs, miR-25 and miR-92, are consistently detected at higher levels in MSS tissue samples and detection of these miRNAs in CRC could complement current diagnostic assays Citation[100]. Other studies have investigated the prognostic value of single miRNAs or refined miRNA signatures . Independent reports have associated high miR-21 expression with shorter recurrence-free and overall survival Citation[51,101–103]. One of these studies determined miR-21 expression by ISH analysis in a cohort of 197 stage II CRC cases Citation[51]. Upregulation of miR-21 expression was predominantly detected in the stromal cell compartment, mainly in tumor-associated fibroblasts Citation[51]. Interestingly, higher expression of miR-21 in reactive fibroblasts was associated with a poor outcome in colon cancer (n = 130), but not in rectal cancer (n = 67) specimens Citation[51].
Epidermal growth factor receptor is overexpressed in the majority of metastatic CRC (mCRC) cases Citation[104]. Treatment of mCRC with monoclonal blocking anti-EGFR antibodies (i.e., cetuximab or panitumumab) alone, or in combination with, chemotherapy has been shown to increase progression-free and overall survival in several randomized clinical trials Citation[104–106]. While EGFR overexpression correlates with treatment response to cetuximab Citation[104], wild-type status of the KRAS gene is currently a more accurate predictor of treatment response to cetuximab- and panitumumab-containing therapies Citation[105,106]. Since patients whose tumors harbor mutated KRAS gene as a group are unlikely to benefit from anti-EGFR therapies, these may exclude individual patients with mutated KRAS tumors for whom this treatment could be efficacious and include some patients with wild-type KRAS for whom this treatment would not be efficacious. RNA-based studies in CRC cell lines have reported mRNA and miRNA signatures that could improve prediction of treatment response over KRAS mutation status alone and thus inform patient eligibility to anti-EGFR-based treatments Citation[107,108]. Using cetuximab-sensitive and -resistant CRC cell lines to profile miRNA expression and mining miRNA expression data from previous studies in clinical samples, a three-miRNA signature was proposed to predict treatment response to cetuximab Citation[108]. Two of these miRNAs, let-7b and let-7e, are negative regulators of KRAS expression. The importance of let-7’s role is reinforced by molecular epidemiologic studies that indicate a poor outcome of cetuximab-treated mCRC patients whose tumors harbor a single-nucleotide polymorphism variant in the 3´-untranslated region of KRAS mRNA that disrupts let-7 binding Citation[109,110].
Molecular assays in lung cancer
Lung cancer (LC) is the leading cause of cancer-related death for men and women in the USA Citation[56]. LC can be divided into two major histological subtypes: small-cell LC and non-small-cell LC (NSCLC). Small cell LC has an endocrine cell of origin and an overall even worse prognosis than NSCLC, partly owing to its late presentation Citation[111]. NSCLC represent approximately 80% of all LC cases and can be divided into three histological groups: large cell carcinoma, squamous cell carcinoma (SCC) and adenocarcinoma (AdCa) Citation[111]. AdCa and SCC are the two most common subtypes and have traditionally received the same treatment regimens with equally disappointing results.
To gain further insight into the biological differences between these histological subtypes, global mRNA profiling experiments have been conducted with normal adjacent and tumor tissues. Initial studies identified a close correlation between histological subtypes and expression signatures, with the exception of AdCa cases, which were separated into three to four major groups with different outcomes Citation[112–114]. Two-marker (p63 and TFF-1) and six-marker (TRIM29, CEACAM5, SLC7A5, MUC1, CK5 and CK6) IHC assays can also accurately distinguish AdCa and SCC subtypes Citation[115]. Consensus expression signatures from multiple studies that compared normal tissues with AdCa tissues Citation[112,113,116–118] and normal tissues with SCC tissues Citation[112–114] contain some, but not all, of the genes that these IHC assays are based on. Numerous studies have described prognostic signatures for early-stage NSCLC cases; some studies combined expression data of AdCA and SCC cases while other studies focused on a specific histological subtype Citation[119,120]. As noted for BC and CRC, the overlap of genes in these signatures was low Citation[119]. Gene ontology analysis indicates that these signatures capture different alterations in both cancer cells and other cellular elements of the TME. For example, a 72-gene expression prognostic NSCLC classifier was noticed to be highly enriched for immune cell-expressed genes, suggesting the influence of differential immune cell responses in different tumors Citation[121].
Global miRNA expression studies have revealed consistent changes between normal and tumor tissues. Let-7a, miR-34a, -126 and -145 are frequently detected at lower levels, and miR-21, -155 and -221 at higher levels in tumor tissues Citation[77,122–124]. A 34-miRNA signature was developed to classify NSCLC tissues in the AdCa and SCC subtypes Citation[125]. Surprisingly, miR-205 was not part of this diagnostic signature. Several independent studies reported that high expression levels of miR-205 are associated with SCC cases and can be used to accurately separate AdCa and SCC histologies, even in samples with little tissue material Citation[126–130]. However, these studies did not test whether differential expression levels of miR-205 could have a prognostic value to stratify SCC cases into low- and high-risk groups. Other studies have investigated the prognostic value of single miRNAs or refined miRNA signatures . In one of the first miRNA studies in LC, low expression of let-7a and high expression of miR-155 correlated with poor outcome in mixed staged AdCa cases Citation[131]. High levels of miR-155 expression were also correlated with poor prognosis in 41 stage I AdCa cases Citation[131]. These findings on miR-155 expression have been difficult to validate in independent studies Citation[47,132,133].
The advent of systemic targeted therapies against EGFR with cetuximab or with tyrosine kinase inhibitors (i.e., gefitinib and erlotinib) and against processes with anti-VEGF-blocking antibody (i.e., bevacizumab) has exposed the need for molecular characterization to preselect patients who will benefit from these treatments Citation[134]. Unlike in mCRC, cetuximab does not appear to be efficacious in advanced NSCLC patients and the KRAS mutation status is not predictive of treatment response Citation[134,135]. Gefitinib and erlotinib are only efficacious in tumors with a mutated EGFR gene. These tumors mainly present with an AdCa histology and are more frequent among nonsmoker female patients Citation[134]. Approximately 15% of AdCa cases harbor a mutated EGFR gene and are eligible for gefitinib or erlotinib treatment; this excludes 85% of AdCa and virtually all SCC cases. Similarly, bevacizumab treatment is contraindicated due to an increased risk of severe bleeding events in SCC cases Citation[134]. Thus, the great majority of cases currently lack a targeted treatment. mRNA and miRNA expression analysis could provide a means to identify subgroups that may benefit from other therapies. let-7a-mediated regulation of KRAS expression was first described in LC Citation[136]. A SNP variant in the 3´-untranslated region of KRAS mRNA that disrupts let-7 binding is associated with an increased risk of developing NSCLC in moderate smokers Citation[137]. An independent study in AdCa cases did not find an association of this SNP variant with mutation status of KRAS gene or overall survival Citation[138]. However, expression levels of let-7 family members are much lower in SCC than in AdCa cases, and expression levels of let-7 family members only have prognostic value in SCC cases Citation[125]. SCC cases have virtually no incidence of mutated KRAS gene, whereas approximately 30% of AdCa tumors harbor activating KRAS mutation. The authors speculate that compromised let-7-mediated regulation in SCC cases increases levels of wild-type KRAS expression and consequently tumor aggressiveness and resistance to treatment, whereas activating KRAS mutations in AdCa cases renders these tumors less susceptible to let-7-mediated regulation Citation[125]. Other miRNAs, including miR-7 and miR-128b, have been shown to regulate EGFR expression in NSCLC cell lines Citation[139–141]. Loss of miR-128b expression by chromosomal deletion (loss of heterogeneity) correlated with a better clinical response and improved overall survival in a group of 58 NSCLC patients treated with gefitinib-containing therapy Citation[139]. The authors suggest that attenuation of miR-128b-mediated regulation of EGFR expression was responsible for this effect, as tumor expressing higher levels of EGFR may be more responsive to gefitinib Citation[139]. Thus, miR-128b and other miRNAs that modulate EGFR expression could be used to inform treatment response to anti-EGFR therapies.
Expert commentary
Discrepancies between studies and independent validation of most expression signatures remain a tall challenge. Heterogeneity of patient cohorts (subtypes, stages and age), heterogeneity of tissue specimens and RNA quality, different experimental designs, different detection platforms, different statistical analysis and data mining algorithms are all contributing and confounding factors to the generation of different expression signatures for the ‘same’ clinical questions Citation[142,143]. Nonetheless, gene-expression signatures defining intrinsic BC subtypes have been reproduced using different microarray platforms Citation[144] and salient miRNA examples have been independently validated . This suggests that information about robust and frequent biological processes can be obtained with these high-throughput discovery platforms, but as such further validation and understanding of what these signatures reflect is required.
Cellular complexity of tumor tissues is often overlooked in the design, analysis and interpretation of RNA-based assays. Each tumor lesion has a different ratio of cancer cells to reactive stroma and infiltrating immune cells; a biomarker can be expressed at different levels in different cell types. Therefore, the most appropriate molecular assay and biomarkers should be deployed to extract the contextual information embedded within different cell types to indicate aberrant and dysfunctional pathways in cancer cells or other cellular compartments of the TME and to avoid confounding effects of tissue heterogeneity (see later). This will be an especially important consideration to validate reported miRNA signatures in large cohorts of patients with similar clinical characteristics and to identify the cellular compartment of altered miRNA expression. A miRNA biomarker may only be informative if its overall levels can be specifically assessed in a particular cellular compartment or if a differential expression pattern between cancer cell compartment versus another compartment can be measured.
miR-21, miR-34a and miR-210 are among the few salient examples of miRNA biomarkers that are ready to be tested in a clinical setting. A critical head-to-head comparison of the performance of these miRNA biomarkers with existing and translational protein-encoding genes will determine whether miRNAs provide superior or complementary information to increase the molecular characterization of tumors and enhance the diagnostic power of current clinical assays. Pioneered global miRNA profiling indicated that miRNA signatures outperformed mRNA signatures to group together tumors derived from the GI tract from other carcinomas and to classify poorly differentiated tumors into the appropriate organ site Citation[145]. Other studies have also shown the utility of miRNA signatures to identify the organ site of carcinomas of unknown primary origin Citation[146,147]. This information can be useful to improve clinical outcome by matching treatment of the carcinomas of an unknown primary tumor with that recommended for the organ site. A miR-210-based assay yields comparable information to the 21-mRNA Oncotype Dx assay to predict response to tamoxifen in BC patients Citation[80]. It is likely that both assays reflect the coordinated effects of different biological processes, but a single miRNA assay will be more amenable to clinical implementation and readout interpretation.
While this is beyond the scope of this article, miRNA-based assays in blood samples is a rapidly expanding field with the potential to improve disease management Citation[148–151]. A blood test is a noninvasive procedure in which samples from the same patient can be collected repeatedly in a longitudinal manner. miRNA-based blood assays could be useful for early disease detection and for monitoring disease progression, especially after treatment. However, the exact mechanism and cell source(s) for altered miRNA levels in blood are not currently known. Do they mainly represent the content of lysed cancer cells or other cellular components of the TME? Do they mainly reflect an anti-tumor immune response or protumoral inflammation? Do cancer cells communicate systemically by secreting specific miRNAs in exosomes, lipoprotein complexes or other organized macrostructures?
Five-year view
As the clinical utility of more miRNA- and protein-encoding biomarkers is validated by independent prospective and longitudinal studies, it will be important to translate and integrate these findings into routine clinical practice. Innovative technological and conceptual approaches that combine the unique capabilities of RNA-based (multiple marker detection) and tissue slide-based (cell type-specific information) assays will enhance the diagnostic power and accuracy of molecular analysis. Enrichment of specific cell types by laser capture microdissection or similar techniques prior to RNA quantification is a feasible option to provide RNA-based assays with contextual information Citation[152,153]. However, this type of assay will be technically demanding and will require further processing steps. This contextual RNA-based assay will be amenable to implementation in a research setting, but it will probably be difficult to incorporate into routine clinical practice unless further technological advances are introduced, such as procedural automatization. Instead, I propose a transformative technological platform that shares the same technical principles and instruments as current tissue slide-based clinical assays and is designed to be seamlessly adaptable with minimal time loss or procedural modifications in high-volume clinical pathology laboratories. In order to enable codetection of an unprecedented large number of protein, DNA and RNA biomarkers on a single tissue slide, this impending platform will couple tissue compartmentalization for parallel marker detection with sequential rounds of permanent stains for cumulative marker detection. In 2010, Kim et al. engineered a four-chambered microfluidic device that was pressed down on top of BC tissue slides to physically separate independent compartments and dispense different solutions and antibodies to each compartment Citation[154]. Kim et al. demonstrated codetection of ER, PR, HER2 and Ki-67 on contiguous but insulated (there is no antibody cross-contamination) compartments on a single BC tissue section using chromogenic staining Citation[154]. Also in 2010, Sempere et al. demonstrated cumulative codetection of up to five independent RNA and protein markers on the same tissue section using multicolor fluorescent staining Citation[52]. illustrates the concept of this tissue slide-based high-density marker detection assay and how it could be deployed to maximize the informational content of a single tissue section using translational and clinically established biomarkers in BC, CRC and LC. Briefly, the whole tissue section is physically compartmentalized in large insulated fields. Each field contains a similar representation of tumor tissue from the core to the periphery of the tumor mass and adjacent normal tissue . Marker expression in cancer, stromal and immune cells in these different tissue regions can then be analyzed side by side. The use of cell type-specific protein markers to highlight and enumerate cancer cells (CK19+), reactive fibroblasts (vimentin+), and immune cells (CD45+) will enable the generation of contextual signatures in which the expression changes of each biomarker will be determined in the appropriate cellular compartment(s). In the next 5 years, as the necessary technologies continue to evolve, it will be feasible to acquire contextual expression signatures of 60 or more miRNA and/or protein biomarkers (within the range of current gene-expression signatures) on a single tissue slide in a fully automated manner from multiplex marker detection to computer-assisted image analysis. This exquisitely detailed molecular characterization of tumors will be conducive to personalized cancer treatment.
Table 1. Diagnostic and prognostic applications of miRNA signatures in solid tumors.
Table 2. Diagnostic and prognostic applications of single miRNAs in solid tumors.
Key issues
• miRNAs are short noncoding regulatory RNAs. Chemical and biological features make miRNAs attractive candidates as novel clinical biomarkers.
• Expression profiling experiments have identified diagnostic and prognostic mRNA and miRNA signatures that refine the classification and treatment of tumors based on current pathological and clinical criteria.
• Discrepancies among different studies of individual genes that compose these signatures present a challenge for the selection and clinical implementation of the most robust biomarkers.
• Specific miRNAs have demonstrated clinical utility to improve current diagnostic and prognostic assays. Single miRNAs provide comparable information to multigene mRNA signatures.
• Head-to-head comparisons of performance for miRNA and protein-encoding gene biomarkers will determine redundancy or synergy of combining miRNAs with other translational and clinically established biomarkers.
• Tissue heterogeneity and cell type complexity are confounding and contributing factors to accurate interpretation of expression signatures.
• Molecular assays that extract contextual information from tissue samples should improve the diagnostic power and accuracy of expression analysis.
• RNA-based and tissue slide-based assays can be developed to extract this contextual information. However, tissue slide-based assays will be easier to incorporate in high-volume clinical pathology laboratories.
Acknowledgements
The author is grateful to Elena Bryleva, Ethan Dmitrovsky and Wendy Wells for their insightful comments and suggestions on this manuscript.
Financial & competing interests disclosure
This work was supported, in part, by NIH and National Cancer Institute grants R03 CA141564 and R21 CA141017. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Kochanek KD, Xu J, Murphy SL, Miniño AM, Kung H-S. Deaths: preliminary data for 2009. National Vital Statistics Reports 59 No. 4, Division of Vital Statistics, National Center for Health Statistics, MD, USA, 1–51 (2011).
- Haber DA, Gray NS, Baselga J. The evolving war on cancer. Cell145, 19–24 (2011).
- Caskey CT. Using genetic diagnosis to determine individual therapeutic utility. Annu. Rev. Med.61, 1–15 (2010).
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science294, 858–862 (2001).
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science294, 853–858 (2001).
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science294, 862–864 (2001).
- Reinhart BJ, Slack FJ, Basson M et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature403, 901–906 (2000).
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell75, 843–854 (1993).
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell75, 855–862 (1993).
- Ambros V. The functions of animal microRNAs. Nature431, 350–355 (2004).
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet.5, 396–400 (2004).
- Sempere LF, Kauppinen S. Translational implications of microRNAs in clinical diagnostics and therapeutics. In: Handbook of Cell Signaling. Bradshaw RA, Dennis EA (Eds). Academic Press, Oxford, UK, 2965–2981 (2009).
- Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell136, 586–591 (2009).
- Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov.9, 775–789 (2010).
- De SE, Ferretti E, Gulino A. MicroRNAs as biomarkers for CNS cancer and other disorders. Brain Res.1338, 100–111 (2010).
- Saba R, Schratt GM. MicroRNAs in neuronal development, function and dysfunction. Brain Res.1338, 3–13 (2010).
- Ketting RF. MicroRNA biogenesis and function: an overview. Adv. Exp. Med. Biol.700, 1–14 (2011).
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet.11, 597–610 (2010).
- Bentwich I, Avniel A, Karov Y et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet.37, 766–770 (2005).
- Lim LP, Lau NC, Garrett-Engele P et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature433, 769–773 (2005).
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature455, 58–63 (2008).
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature455, 64–71 (2008).
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res.39, D152–D157 (2011).
- Ferracin M, Veronese A, Negrini M. Micromarkers: miRNAs in cancer diagnosis and prognosis. Expert. Rev. Mol. Diagn.10(3), 297–308 (2010).
- Elmen J, Lindow M, Silahtaroglu A et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res.36, 1153–1162 (2008).
- Elmen J, Lindow M, Schutz S et al. LNA-mediated microRNA silencing in non-human primates. Nature452, 896–899 (2008).
- Trang P, Wiggins JF, Daige CL et al. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther.19(6), 1116–1122 (2011).
- Wiggins JF, Ruffino L, Kelnar K et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res.70, 5923–5930 (2010).
- Trang P, Medina PP, Wiggins JF et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene29, 1580–1587 (2010).
- Ma L, Reinhardt F, Pan E et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol.28, 341–347 (2010).
- Szafranska-Schwarzbach AE, Adai AT, Lee LS, Conwell DL, Andruss BF. Development of a miRNA-based diagnostic assay for pancreatic ductal adenocarcinoma. Expert. Rev. Mol. Diagn.11, 249–257 (2011).
- Matos LL, Trufelli DC, de Matos MG, da Silva Pinhal MA. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark. Insights5, 9–20 (2010).
- Brennan DJ, O’Connor DP, Rexhepaj E, Ponten F, Gallagher WM. Antibody-based proteomics: fast-tracking molecular diagnostics in oncology. Nat. Rev. Cancer10, 605–617 (2010).
- Tholouli E, Sweeney E, Barrow E, Clay V, Hoyland JA, Byers RJ. Quantum dots light up pathology. J. Pathol.216, 275–285 (2008).
- Al-Janabi S, Huisman A, Van Diest PJ. Digital pathology: current status and future perspectives. Histopathology DOI: 10.1111/j.1365-2559.2011.03814.x (2011) (Epub ahead of print).
- Chen X, Zheng B, Liu H. Optical and digital microscopic imaging techniques and applications in pathology. Anal. Cell Pathol.34, 5–18 (2011).
- Schwanhausser B, Busse D, Li N et al. Global quantification of mammalian gene expression control. Nature473, 337–342 (2011).
- Kauppinen S, Vester B, Wengel J. Locked nucleic acid: high-affinity targeting of complementary RNA for RNomics. Handb. Exp. Pharmacol.405–422 (2006).
- Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA12, 187–191 (2006).
- Sempere LF, Christensen M, Silahtaroglu A et al. Altered microRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res.67, 11612–11620 (2007).
- Yamamichi N, Shimomura R, Inada K et al. Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin. Cancer Res.15, 4009–4016 (2009).
- Liu X, Sempere LF, Ouyang H et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J. Clin. Invest.120, 1298–1309 (2010).
- Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J. Gastrointest. Surg.12, 2171–2176 (2008).
- Habbe N, Koorstra JB, Mendell JT et al. MicroRNA miR-155 is a biomarker of early pancreatic neoplasia. Cancer Biol. Ther.8, 340–346 (2009).
- Preis M, Gardner TB, Gordon SR et al. microRNA-10b expression correlates with response to neoadjuvant therapy and survival in pancreatic ductal adenocarcinoma. Clin. Cancer Res.17, 5812–5821 (2011).
- Schneider M, Andersen DC, Silahtaroglu A et al. Cell-specific detection of microRNA expression during cardiomyogenesis by combined in situ hybridization and immunohistochemistry. J. Mol. Histol.42, 289–299 (2011).
- Donnem T, Eklo K, Berg T et al. Prognostic impact of miR-155 in non-small cell lung cancer evaluated by in situ hybridization. J. Transl. Med.9, 6 (2011).
- Gupta A, Mo YY. Detection of microRNAs in cultured cells and paraffin-embedded tissue specimens by in situ hybridization. Methods Mol. Biol.676, 73–83 (2011).
- Nelson PT, Dimayuga J, Wilfred BR. MicroRNA in situ hybridization in the human entorhinal and transentorhinal cortex. Front. Hum. Neurosci.4, 7 (2010).
- Schepeler T, Reinert JT, Ostenfeld MS et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res.68, 6416–6424 (2008).
- Nielsen BS, Jorgensen S, Fog JU et al. High levels of microRNA-21 in the stroma of colorectal cancers predict short disease-free survival in stage II colon cancer patients. Clin. Exp. Metastasis28, 27–38 (2011).
- Sempere LF, Preis M, Yezefski T et al. Fluorescence-based codetection with protein markers reveals distinct cellular compartments for altered microRNA expression in solid tumors. Clin. Cancer Res.16, 4246–4255 (2010).
- Nelson PT, Wilfred BR. In situ hybridization is a necessary experimental complement to microRNA (miRNA) expression profiling in the human brain. Neurosci. Lett.466, 69–72 (2009).
- Speel EJ, Hopman AH, Komminoth P. Tyramide signal amplification for DNA and mRNA in situ hybridization. Methods Mol. Biol.326, 33–60 (2006).
- Sempere LF, Korc M. Fully-automated multi-color co-detection of microRNA and protein biomarkers on formalin-fixed paraffin-embedded specimens of solid tumors. Presented at: 36th Annual National Society for Histotechnology Symposium/Convention. Seattle, WA, USA, 24–29 September 2010.
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J. Clin.60, 277–300 (2010).
- Perou CM, Sorlie T, Eisen MB et al. Molecular portraits of human breast tumours. Nature406, 747–752 (2000).
- Sorlie T, Perou CM, Tibshirani R et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA98, 10869–10874 (2001).
- Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur. J. Cancer40, 2667–2675 (2004).
- Sims AH, Ong KR, Clarke RB, Howell A. High-throughput genomic technology in research and clinical management of breast cancer. Exploiting the potential of gene expression profiling: is it ready for the clinic? Breast Cancer Res.8, 214 (2006).
- Carey LA, Perou CM, Livasy CA et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA295, 2492–2502 (2006).
- Ross JS. Multigene classifiers, prognostic factors, and predictors of breast cancer clinical outcome. Adv. Anat. Pathol.16, 204–215 (2009).
- Overdevest JB, Theodorescu D, Lee JK. Utilizing the molecular gateway: the path to personalized cancer management. Clin. Chem.55, 684–697 (2009).
- Cheang MC, Voduc D, Bajdik C et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res.14, 1368–1376 (2008).
- Cheang MC, Chia SK, Voduc D et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst.101, 736–750 (2009).
- Paik S, Shak S, Tang G et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med.351, 2817–2826 (2004).
- Weigelt B, Hu Z, He X et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res.65, 9155–9158 (2005).
- Parker JS, Mullins M, Cheang MC et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol.27, 1160–1167 (2009).
- DeNardo DG, Brennan DJ, Rexhepaj E et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Dis. DOI: 10.1158/2159-8274.CD-10-0028 (2011) (Epub ahead of print).
- Planche A, Bacac M, Provero P et al. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PLoS ONE6, e18640 (2011).
- Marchini C, Montani M, Konstantinidou G et al. Mesenchymal/stromal gene expression signature relates to basal-like breast cancers, identifies bone metastasis and predicts resistance to therapies. PLoS ONE5, e14131 (2010).
- Sharma M, Beck AH, Webster JA et al. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res. Treat.123, 397–404 (2010).
- Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res.11, R7 (2009).
- Farmer P, Bonnefoi H, Anderle P et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med.15, 68–74 (2009).
- Finak G, Bertos N, Pepin F et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med.14, 518–527 (2008).
- Iorio MV, Ferracin M, Liu CG et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res.65, 7065–7070 (2005).
- Volinia S, Calin GA, Liu CG et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl Acad. Sci. USA103, 2257–2261 (2006).
- Andorfer CA, Necela BM, Thompson EA, Perez EA. MicroRNA signatures: clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol. Med.17, 313–319 (2011).
- Le QJ, Caldas C. Micro-RNAs and breast cancer. Mol. Oncol.4, 230–241 (2010).
- Rothe F, Ignatiadis M, Chaboteaux C et al. Global MicroRNA expression profiling identifies miR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS ONE6, e20980 (2011).
- Janssen EA, Slewa A, Gudlaugsson E et al. Biologic profiling of lymph node negative breast cancers by means of microRNA expression. Mod. Pathol.23, 1567–1576 (2010).
- Lowery AJ, Miller N, Devaney A et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res.11, R27 (2009).
- Blenkiron C, Goldstein LD, Thorne NP et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumour subtype. Genome Biol.8, R214 (2007).
- Mattie MD, Benz CC, Bowers J et al. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer5, 24 (2006).
- Castellano L, Giamas G, Jacob J et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl Acad. Sci. USA106, 15732–15737 (2009).
- Yoshimoto N, Toyama T, Takahashi S et al. Distinct expressions of microRNAs that directly target estrogen receptor α in human breast cancer. Breast Cancer Res. Treat.130(1), 331–339(2011).
- Fu SW, Chen L, Man YG. miRNA biomarkers in breast cancer detection and management. J. Cancer2, 116–122 (2011).
- Foekens JA, Sieuwerts AM, Smid M et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc. Natl Acad. Sci. USA105, 13021–13026 (2008).
- Camps C, Buffa FM, Colella S et al. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin. Cancer Res.14, 1340–1348 (2008).
- Buffa FM, Camps C, Winchester L et al. microRNA associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res.71(17), 5635–5645 (2011).
- Radojicic J, Zaravinos A, Vrekoussis T, Kafousi M, Spandidos DA, Stathopoulos EN. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle10, 507–517 (2011).
- Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res.69, 1221–1229 (2009).
- Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol.7, 153–162 (2010).
- Pino MS, Chung DC. Microsatellite instability in the management of colorectal cancer. Expert Rev. Gastroenterol. Hepatol.5, 385–399 (2011).
- Fearon ER. Molecular genetics of colorectal cancer. Annu. Rev. Pathol.6, 479–507 (2011).
- Mutch MG. Molecular profiling and risk stratification of adenocarcinoma of the colon. J. Surg. Oncol.96, 693–703 (2007).
- Salazar R, Roepman P, Capella G et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol.29, 17–24 (2011).
- Luo X, Burwinkel B, Tao S, Brenner H. MicroRNA signatures: novel biomarker for colorectal cancer? Cancer Epidemiol. Biomarkers Prev.20, 1272–1286 (2011).
- Ma Y, Zhang P, Yang J, Liu Z, Yang Z, Qin H. Candidate microRNA biomarkers in human colorectal cancer: systematic review profiling studies and experimental validation. Int. J. Cancer DOI: 10.1002/ijc.26232 (2011) (Epub ahead of print).
- Lanza G, Ferracin M, Gafa R et al. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer6, 54 (2007).
- Kulda V, Pesta M, Topolcan O et al. Relevance of miR-21 and miR-143 expression in tissue samples of colorectal carcinoma and its liver metastases. Cancer Genet. Cytogenet.200, 154–160 (2010).
- Schetter AJ, Leung SY, Sohn JJ et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA299, 425–436 (2008).
- Shibuya H, Iinuma H, Shimada R, Horiuchi A, Watanabe T. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology79, 313–320 (2010).
- Yen LC, Uen YH, Wu DC et al. Activating KRAS mutations and overexpression of epidermal growth factor receptor as independent predictors in metastatic colorectal cancer patients treated with cetuximab. Ann. Surg.251, 254–260 (2010).
- Tol J, Punt CJ. Monoclonal antibodies in the treatment of metastatic colorectal cancer: a review. Clin. Ther.32, 437–453 (2010).
- Blanke CD, Goldberg RM, Grothey A et al. KRAS and colorectal cancer: ethical and pragmatic issues in effecting real-time change in oncology clinical trials and practice. Oncologist16(8), 1061–1068 (2011).
- Solmi R, Lauriola M, Francesconi M et al. Displayed correlation between gene expression profiles and submicroscopic alterations in response to cetuximab, gefitinib and EGF in human colon cancer cell lines. BMC Cancer8, 227 (2008).
- Ragusa M, Majorana A, Statello L et al. Specific alterations of microRNA transcriptome and global network structure in colorectal carcinoma after cetuximab treatment. Mol. Cancer Ther.9, 3396–3409 (2010).
- Zhang W, Winder T, Ning Y et al. A let-7 microRNA-binding site polymorphism in 3´-untranslated region of KRAS gene predicts response in wild-type KRAS patients with metastatic colorectal cancer treated with cetuximab monotherapy. Ann. Oncol.22, 104–109 (2011).
- Graziano F, Canestrari E, Loupakis F et al. Genetic modulation of the let-7 microRNA binding to KRAS 3´-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab–irinotecan. Pharmacogenomics J.10, 458–464 (2010).
- Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu. Rev. Pathol.1, 331–348 (2006).
- Bhattacharjee A, Richards WG, Staunton J et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl Acad. Sci. USA98, 13790–13795 (2001).
- Garber ME, Troyanskaya OG, Schluens K et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl Acad. Sci. USA98, 13784–13789 (2001).
- Yamagata N, Shyr Y, Yanagisawa K et al. A training-testing approach to the molecular classification of resected non-small cell lung cancer. Clin. Cancer Res.9, 4695–4704 (2003).
- Ring BZ, Seitz RS, Beck RA et al. A novel five-antibody immunohistochemical test for subclassification of lung carcinoma. Mod. Pathol.22, 1032–1043 (2009).
- Beer DG, Kardia SL, Huang CC et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med.8, 816–824 (2002).
- Powell CA, Spira A, Derti A et al. Gene expression in lung adenocarcinomas of smokers and nonsmokers. Am. J. Respir. Cell Mol. Biol.29, 157–162 (2003).
- Spira A, Beane JE, Shah V et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med.13, 361–366 (2007).
- Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J. Natl. Cancer Inst.102, 464–474 (2010).
- Lacroix L, Commo F, Soria JC. Gene expression profiling of non-small-cell lung cancer. Expert Rev. Mol. Diagn.8, 167–178 (2008).
- Roepman P, Jassem J, Smit EF et al. An immune response enriched 72-gene prognostic profile for early-stage non-small-cell lung cancer. Clin. Cancer Res.15, 284–290 (2009).
- Du L, Pertsemlidis A. MicroRNAs and lung cancer: tumors and 22-mers. Cancer Metastasis Rev.29, 109–122 (2010).
- Lin PY, Yu SL, Yang PC. MicroRNA in lung cancer. Br. J. Cancer103, 1144–1148 (2010).
- Yendamuri S, Kratzke R. MicroRNA biomarkers in lung cancer: MiRacle or quagMiRe? Transl. Res.157, 209–215 (2011).
- Landi MT, Zhao Y, Rotunno M et al. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res.16, 430–441 (2010).
- Lebanony D, Benjamin H, Gilad S et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol.27, 2030–2037 (2009).
- Fassina A, Cappellesso R, Fassan M. Classification of non-small cell lung carcinoma in trans-thoracic needle specimens using microRNA expression profiling. Chest DOI: 10.1378/chest.11-0708 (2011) (Epub ahead of print).
- Del V, V, Cantaloni C, Cucino A et al. miR-205 expression levels in nonsmall cell lung cancer do not always distinguish adenocarcinomas from squamous cell carcinomas. Am. J. Surg. Pathol.35, 268–275 (2011).
- Xing L, Todd NW, Yu L, Fang H, Jiang F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod. Pathol.23, 1157–1164 (2010).
- Bishop JA, Benjamin H, Cholakh H, Chajut A, Clark DP, Westra WH. Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res.16, 610–619 (2010).
- Yanaihara N, Caplen N, Bowman E et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell.9, 189–198 (2006).
- Saito M, Schetter AJ, Mollerup S et al. The association of microRNA expression with prognosis and progression in early-stage, non-small cell lung adenocarcinoma: a retrospective analysis of three cohorts. Clin. Cancer Res.17, 1875–1882 (2011).
- Voortman J, Goto A, Mendiboure J et al. MicroRNA expression and clinical outcomes in patients treated with adjuvant chemotherapy after complete resection of non-small cell lung carcinoma. Cancer Res.70, 8288–8298 (2010).
- Fruh M. The search for improved systemic therapy of non-small cell lung cancer – what are today’s options? Lung Cancer72, 265–270 (2011).
- Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J. Clin. Oncol.28, 4769–4777 (2010).
- Read ML, Spice R, Parker AL, Mir S, Logan A. 12th Annual Congress of the European Society of Gene Therapy. Expert Opin. Biol. Ther.5, 137–141 (2005).
- Chin LJ, Ratner E, Leng S et al. A SNP in a let-7 microRNA complementary site in the KRAS 3´ untranslated region increases non-small cell lung cancer risk. Cancer Res.68, 8535–8540 (2008).
- Nelson HH, Christensen BC, Plaza SL, Wiencke JK, Marsit CJ, Kelsey KT. KRAS mutation, KRAS-LCS6 polymorphism, and non-small cell lung cancer. Lung Cancer69, 51–53 (2010).
- Weiss GJ, Bemis LT, Nakajima E et al. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann. Oncol.19, 1053–1059 (2008).
- Rai K, Takigawa N, Ito S et al. Liposomal delivery of microRNA-7-expressing plasmid overcomes epidermal growth factor receptor-tyrosine kinase inhibitor-resistance in lung cancer cells. Mol. Cancer Ther.10(9), 1720–1727 (2011).
- Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, Leedman PJ. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem.284, 5731–5741 (2009).
- Subramanian J, Simon R. What should physicians look for in evaluating prognostic gene-expression signatures? Nat. Rev. Clin. Oncol.7, 327–334 (2010).
- Xu JZ, Wong CW. Hunting for robust gene signature from cancer profiling data: sources of variability, different interpretations, and recent methodological developments. Cancer Lett.296, 9–16 (2010).
- Hu Z, Fan C, Oh DS et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics7, 96 (2006).
- Lu J, Getz G, Miska EA et al. MicroRNA expression profiles classify human cancers. Nature435, 834–838 (2005).
- Ferracin M, Pedriali M, Veronese A et al. MicroRNA profiling for the identification of cancers with unknown primary tissue-of-origin. J. Pathol.225(1), 43–45 (2011).
- Varadhachary GR, Spector Y, Abbruzzese JL et al. Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary. Clin. Cancer Res.17, 4063–4070 (2011).
- Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer11, 426–437 (2011).
- Mostert B, Sieuwerts AM, Martens JW, Sleijfer S. Diagnostic applications of cell-free and circulating tumor cell-associated miRNAs in cancer patients. Expert Rev. Mol. Diagn.11, 259–275 (2011).
- Brase JC, Wuttig D, Kuner R, Sultmann H. Serum microRNAs as non-invasive biomarkers for cancer. Mol. Cancer9, 306 (2010).
- Scholer N, Langer C, Dohner H, Buske C, Kuchenbauer F. Serum microRNAs as a novel class of biomarkers: a comprehensive review of the literature. Exp. Hematol.38, 1126–1130 (2010).
- Hipp J, Cheng J, Hanson JC et al. SIVQ-aided laser capture microdissection: a tool for high-throughput expression profiling. J. Pathol. Inform.2, 19 (2011).
- Hanson JC, Tangrea MA, Kim S et al. Expression microdissection adapted to commercial laser dissection instruments. Nat. Protoc.6, 457–467 (2011).
- Kim MS, Kim T, Kong SY et al. Breast cancer diagnosis using a microfluidic multiplexed immunohistochemistry platform. PLoS. ONE5, e10441 (2010).
- Rodriguez-Gonzalez FG, Sieuwerts AM, Smid M et al. MicroRNA-30c expression level is an independent predictor of clinical benefit of endocrine therapy in advanced estrogen receptor positive breast cancer. Breast Cancer Res. Treat.127, 43–51 (2011).
- Ota D, Mimori K, Yokobori T et al. Identification of recurrence-related microRNAs in the bone marrow of breast cancer patients. Int. J. Oncol.38, 955–962 (2011).
- Yu SL, Chen HY, Chang GC et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell13, 48–57 (2008).
- Raponi M, Dossey L, Jatkoe T et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res.69, 5776–5783 (2009).
- Gong C, Yao Y, Wang Y et al. Up-regulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer. J. Biol. Chem.286, 19127–19137 (2011).
- Qian B, Katsaros D, Lu L et al. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-β1. Breast Cancer Res. Treat.117, 131–140 (2009).
- Yan LX, Huang XF, Shao Q et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA14, 2348–2360 (2008).
- Drebber U, Lay M, Wedemeyer I et al. Altered levels of the onco-microRNA 21 and the tumor-supressor microRNAs 143 and 145 in advanced rectal cancer indicate successful neoadjuvant chemoradiotherapy. Int. J. Oncol.39, 409–415 (2011).
- Diaz R, Silva J, Garcia JM et al. Deregulated expression of miR-106a predicts survival in human colon cancer patients. Genes Chromosomes Cancer47, 794–802 (2008).
- Nishida N, Yokobori T, Mimori K et al. MicroRNA miR-125b is a prognostic marker in human colorectal cancer. Int. J. Oncol.38, 1437–1443 (2011).
- Takamizawa J, Konishi H, Yanagisawa K et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res.64, 3753–3756 (2004).
- Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin. Chem.54, 1696–1704 (2008).
- Gao W, Shen H, Liu L, Xu J, Xu J, Shu Y. miR-21 overexpression in human primary squamous cell lung carcinoma is associated with poor patient prognosis. J. Cancer Res. Clin. Oncol.137, 557–566 (2011).
- Gallardo E, Navarro A, Vinolas N et al.miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis30, 1903–1909 (2009).
Websites
- American Joint Committee on Cancer www.cancerstaging.org
- The Gene Ontology www.geneontology.org