Abstract
There is heterogeneity in patient responses to current asthma medications. Significant progress has been made identifying genetic polymorphisms that influence the efficacy and potential for adverse effects to asthma drugs, including; β2-adrenergic receptor agonists, corticosteroids and leukotriene modifiers. Pharmacogenetics holds great promise to maximise clinical outcomes and minimize adverse effects. Asthma is heterogeneous with respect to clinical presentation and inflammatory mechanisms underlying the disease, which is likely to contribute to variable results in clinical trials targeting specific inflammatory mediators. Genome-wide association studies have begun to identify genes underlying asthma (e.g., IL1RL1), which represent future therapeutic targets. In this article, we review and update the pharmacogenetics of current asthma therapies and discuss the genetics underlying selected Phase II and future targets.
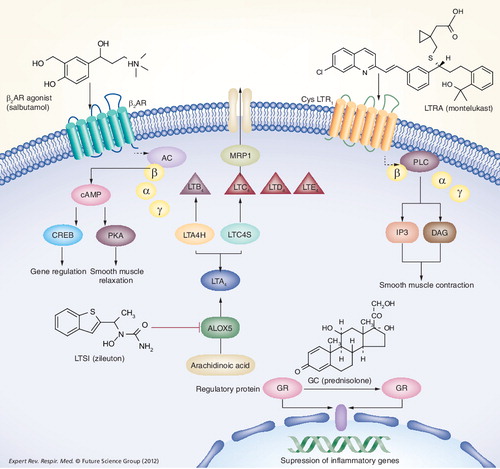
Corticosteroids cross the cell membrane and bind to receptors that are stabilized by regulatory proteins. The glucocorticoid receptor (GR) then crosses the nuclear membrane, binds to a response element and modulates transcription. β2-adrenergic receptor agonists bind to the β2-adrenergic receptor and mediate smooth muscle relaxation via PKA and regulate various genes via the transcription factor CREB. The cysteinyl leukotrienes in LTC4, LTD4 and LTE4,and dihydroxy leukotriene LTB4 are synthesized from arachidonic acid in a series of steps via an enzymatic cascade as depicted. Following translocation into the extracellular space, cysteinyl leukotrienes activate their cognate GPCRs (including CYSLTR1) and mediate a variety of effects such as smooth muscle contraction.
AC: Adenylcyclase; ALOX5: Arachidonate 5-lipoxygenase; β2AR: β2-adrenergic receptor; cAMP: Cyclic adenosine monophosphate; cysLTR: Cysteinyl Leukotriene receptor; CREB: Cyclic AMP response element-binding; DAG: Diacyl glycerol; GC: Glucocorticoid; GPCR: G-protein-coupled receptor; GR: Glucocorticoid receptor; IP3: Inositol-triphosphate; LTA4: Leukotriene A4; LTB4: Leukotriene B4; LTC4: Leukotriene C4; LTD4: Leukotriene D4; LTE4: Leukotriene E4; LTC4S: Leukotriene C4 synthase; LTA4H: Leukotriene A4 hydrolase; LTSI: Leukotriene synthesis inhibitor; LTRA: Leukotriene receptor antagonist; PKA: Cyclic-AMP dependent protein kinase; PKC: Protein kinase C; PLC: Phospholipase C.
Asthma is characterised by reversible airway obstruction, increased bronchial hyper-responsiveness (BHR) and chronic inflammation. However, it is recognized that asthma is heterogeneous regarding the underlying clinical and inflammatory phenotypes present. In the UK, 5.4 million people are currently receiving treatment for asthma, which represents a major burden with respect to mortality, morbidity and National Health Service costs – estimated to be GB£1 billion per year. Current asthma medications are effective in most but not all patients, with a subgroup of patients (<5%) that have limited disease control, persistent symptoms and exacerbations despite medication use Citation[1]. There is a need for alternative therapies for these refractory patients and a need to target existing drugs towards those subjects most likely to gain clinical benefit.
Asthma is considered a complex genetic disease involving the interaction of multiple genes with each other (gene–gene interactions) and the environment (gene–environment interactions), including; infection (viral, bacterial), allergen exposure, pollution and smoking Citation[2,3]. There has been good progress in recent years in asthma-susceptibility gene discovery, driven mainly by genome-wide association (GWA) approaches that investigate up to 1 million single-nucleotide polymorphisms (SNPs) in large populations composed of thousands of individuals. Asthma susceptibility genes identified using this approach include IL1RL1, IL18R, TSLP, PDE4D, MHC II, DQ, IL-33, LRRC32, SMAD3, ORMDL3, GSDMB and IL2RBCitation[4–9].
Pharmacogenetics in asthma
Pharmacogenetics, the investigation of the effect of genetic variability on treatment response or risk of serious side effects, is the first step in developing personalized prescribing. Thus far, asthma studies have mainly been limited to small numbers of subjects (<100 subjects) and restricted to candidate gene/pathway SNPs using retrospective analyses. These studies have focused on pharmacodynamic aspects – for example, changes in forced expiratory volume in 1 s (FEV1) or symptom scores postmedication stratified based on genotype .
Improving our understanding of asthma pharmacogenetics for the selected targeting of the right therapy to the right patient aims to reduce hospital admissions due to poorly controlled asthma as well as reducing asthma fatalities. Current asthma medications have few apparent severe adverse drug effects; however, some detrimental effects have been observed – for example arrhythmias with β2-adrenergic receptor agonists and osteoporosis with steroids. Similarly, Churg–Strauss syndrome has been linked to leukotriene modifier therapy. Pharmacogenetic knowledge, leading to targeted therapy, has the potential to reduce exposure to these effects. Asthma pharmacogenetics also has a role to play in the development of new therapeutics – for example improving the accuracy and safety of Phase II trials. In the current review we provide:
• A comprehensive update on recent pharmacogenetic developments in the major drug classes used to treat asthma – that is, β2-adrenergic receptor agonists, corticosteroids and leukotriene modifiers;
• Preliminary pharmacogenetic data for newer drugs in Phase II development;
• Discuss the potential pharmacogenetic considerations for newer targets identified in GWA studies in asthma.
Pharmacogenetics of current asthma therapy: an update
There are currently three major classes of asthma pharmacotherapy:
• Inhaled β2-adrenergic receptor agonists for the relief of airway obstruction, which are further divided into short-acting β2-adrenergic receptor agonists (e.g., salbutamol) that are used for the short-term relief of bronchospasm and long-acting β2-adrenergic receptor agonists (i.e., salmeterol) that are used for the long-term treatment of bronchospasm;
• Inhaled and systemic corticosteroids (e.g., beclomethasone and prednisolone);
• Leukotriene receptor antagonists (LTRAs) (e.g., montelukast and zafirlukast), and leukotriene synthesis inhibitors (LTSIs; e.g., zileuton, although LTSIs have not been licensed for use in the UK.
These medications are used in a stepwise approach (British Thoracic Society guidelines Citation[10]), changing as the patient’s outlook improves or worsens. The fact that patient response to medication is highly heterogeneous warrants approaches to develop a more personalized approach to asthma treatment. In the next sections we provide an update on the pharmacogenetic aspects of current asthma medication and focus on more recent findings. For a comprehensive review of earlier studies of the pharmacogenetics of these drug classes see reviews Citation[11–15].
β2-adrenergic receptor agonists
β2-adrenergic receptor agonists act by binding to the β2-adrenergic receptor, a 413-amino-acid G-protein-coupled receptor encoded by an intronless gene (ADRB2) located on chromosome 5q31.32. Receptor binding results in the activation of adenylcyclase through stimulatory Gαs proteins that activate protein kinase A. The latter phosphorylates several target proteins, resulting in a decrease in intracellular calcium, importantly causing smooth muscle relaxation in the airways.
ADRB2 is highly polymorphic with 49 SNPs and two insertion/deletion variants validated to date Citation[16]. ADRB2 has been extensively studied for its pharmacogenetic effects on β2-adrenergic receptor agonist responses. Most studies have focused on the role of nonsynonymous coding-region polymorphisms Arg16Gly, Gln27Glu, Val34Met and Thr164Ile Citation[17]. The frequency of the position 16 and 27 polymorphisms were found to be 59% at Arg16Glu and29% at Gln27Glu in the Caucasian population Citation[17]. The Val34Met and Thr164Ile polymorphisms are rare, with approximate frequencies of <0.001 and 0.05%, respectively. Although the Arg16 variant has been associated with an enhanced acute response to β2-adrenergic receptor agonist a decline of asthma control following prolonged use and a subsensitivity of response for bronchoprotection. Several studies have failed to reproduce these effects. Therefore, a common consensus on the contribution of Arg16 has yet to be reached. At least 12 different haplotypes (a specific combination of SNPs across the gene) have been described in the ADRB2 gene Citation[18], which may explain the lack of consensus examining genotype effects in isolation Citation[11]. Interestingly, recent GWA studies have failed to identify a signal to ADRB2 suggesting this gene is not a major asthma susceptibility gene Citation[4–9].
In vitro work using cell lines has identified functional effects of these β2-adrenergic receptor coding region polymorphisms, including the Thr164Ile variant receptor that caused impairment in agonist binding and reduced adenylcyclase activity. Enhanced agonist-mediated receptor downregulation of the Gly16 variant and resistance to downregulation of the Glu27 variant were also observed (reviewed in Citation[11,12,14,15]). A recent study did not identify a significant effect of the common ADRB2 coding region polymorphisms on responses to a range of β2-adrenergic receptor agonists, including indacaterol in vitroCitation[19]. However, this study did confirm that the Thr164Ile variant receptor produced impairment agonist-mediated cAMP production in these recombinant cell lines Citation[19].
More recent clinical studies have again been hindered by small sample sizes and conflicting results; for example, Lee and colleagues identified an association between the Gln27Glu and terbutaline nebulizer response (percentage change in peak expiratory flow rate [ΔPEFR (%)]) Gln/Gln = 8.2 ± 9.4%, Glu/Glu = 23.4 ± 14.4; p < 0.05) and an absence of effect for the Arg16Gly polymorphism in children. However, it should be noted that these outcomes were only based on 27 patients Citation[20]. In one of the largest studies to date, Basu and colleagues recently identified an increased risk of exacerbations per copy of Arg16 in 1182 young (3–22 years) asthmatic patients (odds ratio [OR]: 1.30; 95% CI: 1.09–1.55; p = 0.003) on daily exposure to β2-adrenergic receptor agonists (regular inhaled corticosteroids plus salbutamol on demand and regular inhaled corticosteroids plus salmeterol and salbutamol on demand groups) Citation[21]. Importantly, this effect was driven by asthma patients that used salbutamol and/or salmeterol daily Citation[21]. While an interesting finding, the authors highlight the limitations of the study, namely that all participants were using β2-adrenergic receptor agonists as relievers and a study arm of non-β2-adrenergic receptor agonist reliever use would have provided clearer interpretation for the detrimental effects of salbutamol/salmeterol in the Arg16 subjects. While this large study suggested a pharmacogenetic effect of the Arg16 variant on adverse effects of β2-adrenergic receptor agonists, it is important to note that previous large studies have failed to observe a clinically relevant effect of these polymorphisms. In a study of 2250 asthma patients randomly assigned to budesonide plus formoterol maintenance and reliever therapy; fixed dose budesonide plus formoterol; or fixed dose fluticasone plus salmeterol for 6 months, no overall effect of the Gly16Arg genotype was found on clinical outcomes including; exacerbations and secondary end point FEV1Citation[22].
The different study designs may underlie these differential outcomes, for example, in a study of 2250 subjects, patients were not stratified based on as-needed reliever use (increase or decrease daily). Interestingly, salmeterol has been further investigated in a prospective study exploring the potential pharmacogenetic effects of 11 ADRB2 SNPs in patients taking fluicasone/salmeterol (n = 268) or salmeterol alone (n = 266) Citation[23]. The main conclusion from this study was that there was a lack of pharmacogenetic effect for Arg16Gly on key outcomes, including morning peak flow and symptom-free days; however, modest associations were observed for other SNPs in ADRB2 regulatory regions, although it is unlikely these would survive correction for multiple testing Citation[23]. Another key observation from this study was the lack of any significant adverse effects following active treatment withdrawal that have previously been described post β2-adrenergic receptor agonist (salbutamol) withdrawal Citation[24]. An absence of effect of the Arg16 genotype on fluicasone/salmeterol clinical outcomes, including lung function and symptoms, was also observed in a smaller, multicenter, randomized, double-blind, placebo-controlled trial in adult asthma subjects over a 18-week period Citation[25]. Importantly, in this prospective study the Arg/Arg and Gly/Gly subjects were matched (morning peak expiratory flow, FEV1, symptom scores) prior to the trial to maximize genotype-specific effects Citation[25].
While the majority of studies have focused on Arg16Gly as outlined above, ADRB2 is highly polymorphic, with potential polymorphisms in the regulatory regions. Recent in vitro approaches using ‘whole-gene’ transfection have identified differential effects on receptor expression and downregulation that are haplotype driven, by studying eight common haplotypes based on 26 SNPs Citation[26]. Interestingly this study identified four common haplotypes with elevated receptor expression and two haplotypes with enhanced receptor downregulation. The clinical significance of these findings remains to be resolved; however, previous haplotype-based data have identified significant clinical effects, suggesting that multiple polymorphisms may be clinically relevant Citation[11]. Recent interest has focused on 3´UTR investigations that have identified a variable-length poly-C tract polymorphism that altered ADRB2 expression and receptor downregulation in vitroCitation[27], but was not found to associate with clinical responses to salmeterol when investigated in an asthma population Citation[28].
Overall, recent larger prospective studies have failed to identify a pharmacogenetic effect of the common ADRB2 Arg16Gly and Gln27Glu polymorphisms, questioning the initial findings from smaller studies but also the translation of in vitro functional work to a clinical setting where single SNPs are anticipated to have modest effect size on clinical outcome measures. However, there still remains evidence of clinically relevant effects – that is, in carefully selected individuals that are using daily β2-adrenergic receptor agonist relievers, as outlined by Basu and colleagues Citation[21].
Novel regulators of β2-adrenergic receptor agonist responses
The mechanism of action of β2-adrenergic receptor agonists is complex and multifactorial, and a large number of gene polymorphisms may be expected to influence the efficacy of the drug. While cAMP is considered a major second messenger in the beneficial effects of this drug class – that is, smooth-muscle relaxation, several other mechanisms may be of relevance. Using a novel algorithm implemented in a family-based association test, Litonjua et al. screened the association of 844 genotyped SNPs in 111 candidate genes (42 involved in β2-adrenergic receptor signalling/regulation, 28 genes involved in glucocorticoid regulation, 41 genes from prior asthma association studies) in 209 children and their parents participating in the Childhood Asthma Management Program for their association with acute response to inhaled β2-adrenergic receptor agonists. This study identified the Arginase 1 (ARG1) SNP rs2781659 as being significantly associated with bronchodilator response (p = 0.047) Citation[29]. In agreement, polymorphisms spanning ARG1 have been identified that influence patient response to salbutamol in a recent candidate gene study involving 221 asthma subjects Citation[30]. The ARG1 polymorphisms identified in both studies were in linkage disequilibrium (inherited together), suggesting a common causative mechanism involving potential transcriptional regulation as these polymorphisms were predominantly 5´ to the gene. This alteration in transcription has now been confirmed in promoter-reporter studies, with the key ARG haplotype associated with improved bronchodilator response driving the highest level of ARG1 promoter activity Citation[31]. Interestingly, in the study by Vonk and colleagues, ARG2 SNPs were also associated with patient responses to salbutamol Citation[30]. Data from guinea pig models of allergic airway disease suggest that arginase enzymes deplete stores of L-arginine, a nitric oxide synthase substrate, in the airways leading to decreased nitric oxide levels, which normally acts to relax smooth muscle, resulting in airway hyper-responsiveness Citation[32,33].
Recently, GSNOR SNP (rs1154400, promoter region) was associated with a decreased response to salbutamol in 107 African–American children Citation[34]. Within the same study, a post-hoc multilocus analysis identified that a combination of rs1154400 with ADRB2 Arg16Gly, Gly27Glu and the carbamoyl phosphate synthetase-1 (CPS1) SNP rs2230739 gave a 70% predictive value for lack of response to therapy Citation[34]. This implies that pharmacogenetic regulation of β2-adrenergic receptor agonist therapy may depend on several loci acting together via gene–gene interactions. In confirmation, four out of five SNPs tested within GSNOR were associated with asthma patient responses to salbutamol in 168 Puerto Rican asthma patients Citation[35]. These SNPs were also associated with asthma susceptibility and the key risk haplotype was associated with increased transcriptional activity based on promoter-reporter studies Citation[35]. GSNOR is an alcohol dehydrogenase that breaks down S-nitrosoglutathione, an endogenous bronchodilator Citation[36]. In addition, S-nitrosoglutathione regulates nitrosylation of proteins leading to alterations in function, including G protein-coupled receptor kinase 2, which phosphorylates and desensitises the β2-adrenergic receptor Citation[37].
The identification of those patients at risk from potential adverse effects of β2-adrenergic receptor agonists remains a critical clinical question, as does the targeting of this class of drug to those patients most likely to gain benefit. While there has been clear progress with respect to study design for example, examining haplotypes instead of genotypes in isolation and adequately powered studies using thousands of individuals – there is still a need for large prospective studies of asthma patients with matched phenotypes and carefully controlled covariates including environmental influences. Similarly, these studies would benefit from GWA approaches and the identification of gene–gene interactions.
Leukotriene modifiers
Cysteinyl leukotrienes contribute to the inflammatory process in asthma and are synthesized from arachidonic acid via the 5-lipoxygenase pathway . In the synthesis reaction, arachidonic acid is converted to 5-hydroperoxyeicosatetraenoic acid and leukotriene A4 (LTA4) by membrane-bound 5-lipoxygenase (5LO/ALOX5) and 5-lipoxygenase activating protein (FLAP/ALOX5AP). LTA4 is then converted to leukotriene B4 (LTB4) by LTA4 hydrolase (LTA4H) or is conjugated with reduced glutathione by leukotriene C4 (LTC4) synthase to form LTC4. LTC4 is then transported to the extracellular space via the multidrug resistance protein 1 (MRP1) . Leukotriene modifiers include LTSIs, which act by targeting the leukotriene pathway, blocking cysteinyl leukotriene production from arachidonic acid, while LTRAs block cysteinyl leukotrienes from binding to their primary receptor, CYSLTR1.
There are excellent data demonstrating heterogeneity in patient responses to LTRAs – that is, a key study by Malmstrom and colleagues investigating the efficacy of montelukast (10 mg once daily) in chronic asthma (12 weeks; n = 895) potentially suggesting genetics may influence response Citation[38]. Similarly, there have been extensive candidate gene studies investigating the effect of SNPs on LTSI and LTRA responses, with pharmacogenetic associations observed for polymorphisms within ALOX5, ALOX5AP, LTC4S, CYSLTR1, CYSLTR2 and MRP1. However, few of these associations have been replicated Citation[11–13]. More recent studies have again focused on enzyme/receptor genes of the leukotriene pathway; however, novel pharmacogenetic effects have also been found – that is, OATP2B1 and montelukast absorption Citation[39].
5-lipoxygenase (ALOX5)
5-lipoxygenase catalyzes the conversion of arachidonic acid to LTA4. The ALOX5 gene has been mapped to chromosome 10q11.2, which spans approximately 82 kb and is composed of 14 exons and 13 introns. Extensive evidence suggests that polymorphisms spanning the ALOX5 gene influence clinical responses to LTRAs and LTSIs; most notably, a functional Sp1 repeat polymorphism in the promoter region of the gene associated with altered gene transcription was associated with response to the LTSI, ABT-761 Citation[13,40]. More recently, studies have included multiple polymorphisms and larger cohorts looking at both LTRA and LTSI responses. Tantisira and colleagues identified an association between ALOX5 intronic SNPs; rs892690, rs2029253 and rs2115819, and change in FEV1 postzileuton treatment, in a cohort of 577 asthma patients Citation[41]. Importantly, the rs2115819 SNP was also a predictor of response to montelukast in a previous study of 252 asthma subjects using percentage change in FEV1 as the primary outcome Citation[42]. In both cases the GG versus GA or AA had the greatest improvement in FEV1 – for example, change in FEV1 % predicted postmontelukast GG = 30%, GA = 4.4% and AA = 2.0% (p = 0.017) Citation[42]. The potential lack of signal for the rs892690 and rs2029253 SNPs in the montelukast study may be due to the reduced power of this study compared with the zileuton study. The functional significance of these intronic SNPs remains to be resolved. Klotsman and colleagues identified two ALOX5 SNPs (rs4987105 [synonymous Thr120Thr] and rs4986832 (5´-region)) that were associated with improvements in peak expiratory flow on treatment with montelukast in a 12 week study in 174 asthma patients – for example, rs4987105: CC = 33.7 (n = 110), CT = 55.3 (n = 42) and TT = 94.8 (n = 10) Citation[43].
Leukotriene C4 synthase (LTC4S)
Leukotriene C4 synthase conjugates glutathione with LTA4 to form LTC4, the first cysteinyl leukotriene in the 5-lipoxygenase pathway. The human LTC4 synthase gene (LTC4S) consists of five exons, ranging between 71 and 257 bp, spans 2.51 kb, and has been mapped to chromosome 5q35. A gene promoter polymorphism (A-444C, rs730012) has been previously associated with increased production and expression of LTC4 by blood eosinophils (C allele) Citation[44]. Five of the ten published pharmacogenetic studies on the LTC4S -444 A>C polymorphism showed an improved response to LTRAs (FEV1) from patients carrying the C allele Citation[42,44–47], although results were not always statistically significant Citation[44,46,47]. The other five published studies could not reproduce these findings Citation[43,48–51]. More recently, a study of zileuton responses (FEV1 change over time) in 577 asthma subjects failed to identify a pharmacogenetic effect of the LTC4S-444 polymorphism. However, an alternative LTC4S polymorphism (rs272431, intron 1) was associated with improved responses (mean FEV1 AA = 2.30 l; AC = 3.11 l; p = 0.006) Citation[41]. As these results were obtained in different populations and using different end points, the relative contribution of LTC4S polymorphism to LTRA or LTSI therapeutic responses in asthma are still unclear. We therefore suggest that further studies are required in multiple ethnic background populations in order to evaluate these multiple polymorphisms spanning the LTC4S gene.
CYSLTR1 & CYSLTR2
The target of LTRAs such as montelukast, pranlukast and zafirlukast, is the CYSLTR1 receptor. CYSLTR1 is 337 amino acids long, 38.5 kDa and encoded by an intronless gene on Xq13-21. CYSLTR2 resides on chromosome 13q14. The gene is 2548 bp long, encoding 346 amino acids. Recently, York and colleagues identified that polymorphisms within CYSLTR1 (in combination with ALOX5 SNPs) are associated with responses to montelukast Citation[52]. However, in isolation CYSLTR1 SNPs tested to date do not influence responses to montelukast Citation[42] or zileuton Citation[40]. Interestingly, an association between CYSLTR2 (rs91227 and rs912278 [3´-UTR]) and morning PEF (e.g., rs91227 TT = 29.1 [n = 60]; TC = 41.3 [n = 75]; CC = 71.6 [n = 28; p = 0.008]) was identified in a recent study of 174 asthma subjects taking montelukast for 12 weeks Citation[43].
Novel regulators of leukotriene modifier drugs
Mougey and colleagues recently identified that solute carrier organic anion transporter family member 2B1 (SLCO2B1, alternative name; organic anion transported SB1; OATP2B1) mediates montelukast permeability using a Madin–Darby canine kidney II cell line overexpressing recombinant OATP2B1 Citation[39]. The authors went on to identify that a key nonsynonymous SNP in OATP2B1, rs12422149, Arg312Gln was associated with reduced morning plasma concentrations of montelukast following an evening dose during 1- or 6-month treatment in 80 asthma subjects of mixed ethnic background. The GA (Arg/Gln) genotype had approximately 20% lower montelukast concentration than the GG (Arg) group at 1 month and an approximate 30% lower concentration at 6 months Citation[39]. Importantly, carriers of the variant A allele did not benefit from montelukast administration, as determined by a symptom utility score Citation[39]. These authors went on to replicate the effect of OATP2B1, rs12422149 and Arg312Gln on montelukast absorption in a further study Citation[53]. This study highlights the importance of genetic variation on both pharmacokinetics and the pharmacodynamics in asthma medication. Similarly, SNPs within another transporter protein, MRP1 (alternative name ABCC1), that is, rs119774 have been associated with montelukast response (change in FEV1 % predicted; CC = 2.2%; CT = 24%; p = 0.004) Citation[54]. More recently, multiple MRP1 SNPs have been associated with responses to zileuton, including the rs119774 SNP, confirming the potential pharmacogenetic significance of this MRP1 SNP marker Citation[41]. This SNP is intronic and the underlying mechanism explaining these pharmacogenetic effects remains to be resolved.
Overall, there has been good progress in the pharmacogenetics of leukotriene modifier therapy in asthma, with SNPs in key leukotriene synthesizing enzymes (e.g., ALOX5) showing association with both LTRA and LTSI responses. The molecular mechanisms underlying these effects require further study. Importantly, studies have begun to identify the importance of genetic variation on both pharmacokinetics and pharmacodynamics, including the role of polymorphisms spanning multiple transporter proteins. However, these studies require replication in larger cohorts to adequately determine the clinical implications of these findings.
Corticosteroids
Corticosteroids are the most effective and commonly (inhaled dosage form) used drugs for the treatment of chronic asthma, but may result in serious adverse effects such as osteoporosis and adrenal suppression. Inhaled, oral and intravenous corticosteroids – for example, prednisolone and beclomethasone – have been central in the management of asthma for the last 30 years. There is substantial interindividual variability in the response to corticosteroids Citation[38], which is highly reproducible, properties that support a genetic basis for the response to corticosteroids in asthma.
Initial pharmacogenetic investigations studied the glucocorticoid receptor gene (GR; NR3C1), which maps to chromosomal region 5q31, a region associated with multiple asthma phenotypes. Several polymorphisms have been described in the GR gene with functional consequences, that is, a Val641Asp polymorphism influences the binding affinity for dexamethasone; however, several of the polymorphisms are rare and their functional significance questionable Citation[11]. Haplotype analyses identified an association with a lower response to dexamethasone using a suppression test in 216 UK Caucasians Citation[55].
SNPs in the corticotrophin-releasing hormone receptor 1 (CRHR1) have been found to be associated with response to inhaled corticosteroid treatment in three asthmatic cohorts using a candidate gene approach (131 SNPs in 14 genes; n = 1117 asthma subjects in total; end point % change in FEV1 following 8 weeks treatment) Citation[56]. For example CRHR1 SNP rs242941 (intronic) showed genotype-specific responses; FEV1 % predicted change; AA = 13.28 ± 3.11%, GG = 5.49 ± 1.40; p = 0.025 (n = 470 adult asthma subjects). CRHR1 is thought to be involved in the regulation of endogenous levels of corticosteroid and therefore may be predicted to influence responses to exogenously administered corticosteroid. This study was the first study to show a pharmacogenetic effect for steroid efficacy in an asthmatic cohort.
Using BHR (4-year change) as an outcome in asthma subjects, TBX21 nonsynonymous SNP (rs2240017C/G, His33Gln) was shown to be a predictor of improvements in BHR post-corticosteroid treatment in children (G allele gave greatest improvements), although the allele frequency of this SNP is low in Caucasians (MAF ∼0.04) and thus few subjects (n = 5) contributed to this observation Citation[56]. TBX21 is a transcription factor involved in T-cell differentiation, particularly Th1/Th2, lineages and the authors demonstrated that dexamethasone can suppress TBX21 levels in human T cells Citation[56]. Previous data have suggested that TBX21 is involved in the development of BHR, with TBX21-/- mice spontaneously developing BHR Citation[57]. Subsequently, Ye and colleagues demonstrated that the TBX21 (His33Gln) polymorphism was significantly associated with improved asthma control in the presence of corticosteroids, although the alternative allele to that described previously was associated with greater control in 53 Korean asthma patients during 5–12 weeks treatment Citation[58]. In the same study, Ye and colleagues identified that Neurokinin 2 receptor (NK2R) SNP (rs77038916G/A, Gly231Glu) was associated with improved asthma control in the presence of corticosteroid, with the G allele (Gly) associated with greatest improvement Citation[58]. Neurokinin A induces bronchoconstriction and inflammation, therefore modulation of the neurokinin receptor may influence the magnitude of these responses. Amino acid 231 is in the third intracellular loop of the receptor, which is involved in agonist-mediated signaling, providing a putative mechanism.
Hawkins and colleagues have recently reported that multiple SNPs in stress-induced phosphoprotein 1 (STIP1) were associated with variable FEV1 responses to treatment with inhaled flunisolide in 382 asthma subjects Citation[59]. This study interrogated SNPs spanning eight candidate genes and identified STIP1 SNPs rs4980524 (intron 1), rs6591838 (intron 1) and rs2236647 (intron 5) affecting percent change in FEV1 in response to flunisolide at 4 and 8 weeks. For example, rs4980524; AA = 5.10 ± 17.16, AC = 5.40 ± 19.00, CC = 11.03 ± 24.03; p = 0.044 at 4 weeks Citation[59]. STIP1 is an adaptor protein that coordinates functions with HSP70 and may be involved in formation of the glucocorticosteroid receptor heterocomplex.
Dual-specificity phosphatase 1 (DUSP1) has dual specificity for tyrosine and threonine and inactivates p38 MAPK. Recently, several SNPs, including a 5´-region SNP rs881152, were associated with bronchodilator response and asthma control in the presence of corticosteroid using a cohort of asthma patients (n = 430) Citation[60].
The mechanism underlying these effects is unclear; however, it was suggested that corticosteroids induce DUSP1 expression (which may be altered in carriers of the rs881152 5´-region SNP), which influences the ability of DUSP1 to target the p38 MAPK signaling pathway.
A SNP within FCER2 (the low-affinity IgE receptor) has been shown to be associated with asthma exacerbations during budesonide therapy in 311 asthmatic children Citation[61]. In particular, SNP T2206C (rs28364072, intronic) was identified (relative risk of severe exacerbations while taking budesonide: 3.62; 95% CI: 2.02–6.49 for homozygous [CC] versus all other genotype groups) Citation[61]. Importantly, FcεRII expression on lymphoblastoid cells was shown to be dependent on T2206C genotype, with the C allele conferring lower expression. The C allele was associated with elevated IgE levels in the 311 asthma patients Citation[61]. This pharmacogenetic association has now been replicated in two further asthma cohorts (n = 386 and 939, respectively), with the 2206C allele being associated with increased hospital visits (OR: 1.91; 95% CI: 1.08–3.40) and uncontrolled asthma (OR: 2.64; 95% CI: 1.00–6.98) in patients receiving corticosteroid Citation[62]. It is potentially not surprising that this polymorphic variation in an IgE receptor influences receptor expression, which alters IgE levels and is associated with more severe forms of asthma and, in turn these subjects cannot be adequately controlled by corticosteroid treatment.
During the writing of this review, the first GWA study in asthma pharmacogenetics was published identifying GLCC1 SNPs as determinants of glucocorticoid responses in 935 asthma subjects Citation[63]. This study examined 534,290 SNPs using the HumanHap 550v3 BeadChip in an initial cohort of 403 parent and child trios, with the primary outcome being change in FEV1 and identified 100 SNPs of highest power for follow on. One SNP, GLCC1 rs37972C/T, was associated with change in FEV1 post-corticosteroid in three out of four cohorts tested; change in FEV1 4 weeks post-treatment; cohort 1: CC = 11.7 ± 2.6, TT = 2.7 ± 3.8; cohort 2: CC = 29.5 ± 1.7, TT = 3.1 ± 1.7; cohort 3: CC = 11.8 ± 1.8, TT = 3.1 ± 2.7; and cohort 4: CC = 9.8 ± 1.6, TT = 4.5 ± 2.8 Citation[63]. rs37972 is located in the promoter region of GLCC1 and is in linkage disequilibrium with another SNP, rs37973, which together were shown to lead to allele-specific transcription using reporter-based approaches in several cell lines; the rs37972C gave lower activity Citation[63]. The mechanism underlying this novel finding and clinical significance requires further clarification.
Pharmacogenetics of asthma drugs in Phase II development
Progress in asthma therapy has mainly been through new compounds/indications within existing drug classes – for example, improved duration of action of long-acting β2-adrenergic receptor agonists. Drug development has also focused on areas including:
• Cytokine inhibition; for example, anti-IL-5 (mepolizumab), anti-TNFα (etanercept, soluble TNFR), anti-IL-13 (lebrikizumab);
• Chemokine receptor antagonists; for example, chemokine (C-X-C motif) receptor 2 (CXCR2; SCH527123);
• Signalling pathway inhibitors with anti-inflammatory effects; for example, targeting p38 MAPK (SD-282), PDE4 (roflumilast) Citation[64].
The targeting of specific mediators is unlikely to provide a therapeutic option for asthma in a broad sense; however, targeting subpopulations has demonstrated clinical efficacy. In a recent clinical trial of anti-IL-5 (mepolizumab), careful selection of patients based on refractory asthma and sputum eosinophilia (>3%) counts demonstrated a robust clinical benefit; for example, reduced exacerbation rates, 2.0 versus 3.4 mean exacerbations per subject following 50 weeks treatment with mepolizumab or placebo respectively Citation[65]. Similarly, a recent study examining anti-IL-13 antibody therapy (lebrikizumab) efficacy in asthma used preselected individuals with IL-13/Th2-dominated asthma (as defined by total IgE, blood eosinophilia and serum periostin levels) Citation[66]. The asthma patients with a preselected IL-13/Th2 phenotype showed greatest improvements, including an increase in mean baseline FEV1 (% predicted) of 8.2% compared with the low periostin group – 1.6% following 12 weeks treatment Citation[66]. Both of these studies demonstrate that careful selection of asthma subjects, in these examples based on phenotype, is critical in asthma drug development. It is important to note that serum eosinophilia has a clear genetic contribution involving polymorphism within IL-5, IL-33, lL1RL1, WDR36 and MYB genes Citation[7] and that polymorphisms spanning the IL-13 locus show some of the most robust asthma associations to date including in recent GWA study Citation[5]. Therefore, it is highly likely that these drug-responsive subphenotypes have a pharmacogenetic basis.
Preliminary data supports genetic targeting in Phase II trials of newer compounds, as shown in a recent report of a IL-4/IL-13 dual antagonist (pitrakinra). Polymorphic variation in the target receptor for this antagonist, that is, IL4Rα, significantly influenced outcomes in allergic asthma subjects Citation[67]. This antagonist is a recombinant form of IL-4 with two amino acid residue changes (i.e., a mutein, R121D/Y124D) and was shown to reduce late-phase antigen responses (LAR) to inhaled antigen Citation[68]. The LAR was defined by changes in FEV1 over 4–10 h postantigen following a 4-week period of active treatment (twice daily, nebulized pitrakinra or placebo), with an increased LAR ratio correlating with a reduced FEV1. Stratification of subjects (pitrakinra n = 15; placebo n = 14) into genotype groups for nonsynonymous IL4RA SNP rs1801275 (Gln576Arg) identified that Arg/Arg carriers had an attenuated LAR (p < 0.0001) following pitrakinra treatment compared with Gln/Gln or Gln/Arg genotypes. Similarly, stratification based on rs1805011 (Glu400Ala) showed an attenuated LAR in the Glu/Ala group, not the Glu/Glu group Citation[67]. Interestingly, the Arg576 variant (when in combination with Val75) has been shown to be a risk factor for allergic asthma, and can lead to enhanced IL4Rα signalling post IL-4 stimulation Citation[69]. While preliminary, these data suggest that selecting a subgroup of patients with a particular genotype where it is anticipated the receptor/pathway may have a more dominant role in that individual’s asthma is critical to interpreting Phase II clinical trials.
Pharmacogenetic considerations for genes identified in recent asthma GWA studies
As our understanding of the genetic basis of asthma increases, new drug targets are becoming apparent. Recent GWA studies have identified a large number of genes that contribute to the susceptibility to develop asthma, including chromosome 17q21 (ORMDL3/GSDMB), IL33, IL1RL1/IL18R1, HLA-DQ, IL2RB, SMAD3, PDE4D, TSLP and DENND1B, LRRC32 and IL6R. Similarly, multiple genes have been identified using positional cloning, such as ADAM33, NPSR1, HLA-G, PCDH1 and PLAURCitation[2,3]. It remains largely unknown how these genes predispose individuals to specific clinical phenotypes that contribute to asthma. However, it is anticipated that polymorphic variation within these genes and/or relevant pathways will define subgroups and have pharmacogenetic effects on drugs designed to target these pathways. To illustrate these concepts, we will focus on IL33 and IL1RL1, two genes that show replicated association with asthma susceptibility and provide a therapeutic opportunity in asthma, and PDE4D a target of asthma drugs already in Phase II/III development.
IL-33 receptor antagonists
IL-33 belongs to the IL-1 cytokine family, which includes IL-1 and IL-18, and signals via its cognate receptor, ST2 (IL1RL1). ST2 is part of the IL-1 receptor/Toll-like receptor (TLR) family and is characterized by containing a TLR/IL1R (TIR) signaling domain. IL-33/ST2 ligation activates NF-κB and MAPK; p38, extracellular receptor kinase1/2 and JNK 1/2 pathways in immune cells. A soluble form of ST2, also exists which attenuates IL-33 function with respect to inflammatory cell recruitment Citation[70].
As proof-of-principle, treatment with anti-IL-33 significantly reduced induction of allergic airway disease in mice, as determined by reduced serum IgE and airway eosinophil and lymphocyte counts Citation[71]. Importantly, not only induction but also resolution of an ongoing allergic airway response was influenced by targeting this ligand–receptor interaction Citation[72].
The role of IL-33/ST2 in human asthma remains to be resolved; however, IL-33 has been shown to be elevated in airway structural cells, including bronchial epithelial cells and airway smooth muscle cells in asthma patients Citation[73]. Recent findings suggest that IL-33 is one of the earliest cytokines released by airway epithelial cells after contact with house dust-mite allergen or the bacterial product LPS via TLR4 Citation[74]. Similarly, levels of soluble ST2 in serum (the potential decoy receptor) are elevated during asthma exacerbation Citation[75]. These data strongly suggest that the IL-33/ST2 axis are dysregulated in asthma. The IL-33/ST2 axis is the focus of intense interest in inflammatory disease, with patents filed by Schering Corporation, Millennium Pharmaceuticals and Roche Diagnostics.
SNPs spanning IL33 and IL1RL1 have reproducibly been associated with asthma susceptibility in the main GWA studies to date Citation[5,7,76]. The key IL33 SNPs span approximately 90 kb and are located in the distal 5´ region (key SNP rs340908; 86.9 kb from transcription start site [TSS]), proximal promoter (rs3939286; 5.7 kb from TSS) or in the first intron (rs2066362) of IL33. It is possible that these risk alleles could alter IL-33 expression. The IL1RL1 SNPs associated with asthma are more variable between studies and map to the IL1RL1 5´ region, for example, rs11685480, which has been shown to influence expression levels of soluble form of ST2 in serum Citation[77]. In addition, coding region variation in the TIR domain, that is, rs10204137 (Glu501Arg), rs10192157 (Thr549Ile) and rs10206753 (Leu551Ser), have shown associations with asthma Citation[5], suggesting that protein structure/receptor signalling may be altered in carriers of these asthma risk genotypes.
Taken together, these data strongly suggest that any approach to target IL-33 induction and expression; and inhibit IL-33-ST2 interactions needs to take into account these potentially functionally relevant SNPs.
Phosphodiesterase inhibitors
There has been intense interest in PDE4 inhibition as an asthma and COPD treatment Citation[78]. Phosphodiesterase enzymes catalyze the breakdown of cAMP and cGMP to 5´ monophosphates. PDE4 inhibition is thought to lead to elevated levels of intracellular cAMP, which has many physiological implications; for example, suppression of inflammatory cell influx and function, inhibition of mucin production from airway epithelial cells and alterations in airway smooth muscle tone Citation[79,80]. The two main PDE4 inhibitors that have reached Phase III development are roflumilast (Altana) and cilomilast (GlaxoSmithKline), and both compounds have shown some clinical efficacy in asthma, with primary end points being; baseline FEV1, early and late response to allegen (FEV1), morning peak expiratory flow and asthma symptom scores Citation[81,82]. These compounds are second-generation PDE inhibitors, and have greater PDE4D selectivity and fewer side effects – for example, nausea.
The identification of PDE4D as an asthma susceptibility gene in 2009 further confirmed the importance of this enzyme in underlying mechanisms of relevance to asthma. The top SNPs identified involve predominantly intronic SNPs – that is, rs1588265 and rs1544791. However, these SNPs are in linkage disequilibrium with SNPs in the promoter region, therefore it is tempting to speculate that these SNPs may tag polymorphisms that influence mRNA expression and/or protein structure Citation[6]. These findings are extremely pertinent to the recent report that suggested both PDE4D expression and PDE activity are increased (approximately twofold) in airway smooth muscle cells isolated from asthma patients compared with control subjects Citation[83]. Importantly, this elevated PDE4D expression in the asthma airway smooth muscle was associated with approximately 50% lower cAMP production in response to β2-adrenergic receptor agonists, including salbutamol and formoterol (in the presence of IBMX, a pan PDE inhibitor) Citation[83]. Overall, these data suggest that SNPs within PDE4D are risk factors for the development of asthma and that alterations in PDE4D expression and activity are a feature of asthma patients airway smooth muscle influencing responses to β2-adrenergic receptor agonists. PDE4D SNPs may at least, in part, determine the clinical efficacy of PDE4D inhibitors, that is, these individuals have a subphenotype of asthma driven more by PDE4D dysregulation. Interestingly this class of drugs has several reported adverse effects including nausea, headache and diarrhea and thus, pharmacogenetics has the potential to avoid these effects in patients unlikely to respond to this specific therapy. We have identified SNPs spanning PDE4D as potential genetic determinants of FEV1 in a general population of 20,288 individuals, again suggesting a role for this enzyme beyond asthma Citation[84].
To date, no Phase II or III trials of PDE inhibitors have been reported or retrospectively analyzed to include pharmacogenetics; however, based on the PDE4D GWA study data this would appear to be an opportunity. Similarly, an investigation of the role of PDE4D genotypes influencing β2-adrenergic receptor agonists is warranted.
Conclusion & expert commentary
A greater understanding of the pharmacogenetics of current asthma medications has the potential to maximize clinical outcomes and minimize adverse effects, leading to improvement in the management of asthma. There has been good progress in the last 5 years with larger prospective studies that importantly interrogate multiple SNPs in haplotypes instead of SNPs in isolation being completed. While predominantly candidate gene/pathway approaches have been used, genes that influence leukotriene modifier response, for example, ALOX5 and genes that influence glucocorticoid response, for example, FCER2 have now been identified with confidence, showing independent replication for specific SNPs and direction of effect. Importantly, the magnitude of changes in clinical measures between genotypes including, for example, FEV1, can be considered clinically relevant. In addition, a large number of novel genes regulating responses to current asthma medications have been identified; however, these need further replication and validation. Interestingly, these pharmacogenetic effects account for a only a small proportion of the variability in response to these compounds and have been confounded by factors including SNP/haplotype analyses, gene–gene interactions and the relative contribution of SNPs in different ethnic backgrounds. While our knowledge has dramatically increased, there is a need for prospective trials of these current compounds involving large numbers of subjects and hypothesis-free approaches, including GWA studies.
To date, only one GWA study has been completed in asthma with respect to patient responses to current medication. This study identified a novel gene, GLCC1 as a determinant of corticosteroid responses using a hypothesis-free approach Citation[63]. However, it is important to note that while the use of this hypothesis free approach is a strength of the study, the design of the study preselected the 100 most powered SNPs, meaning the vast majority of SNPs were not tested across the cohorts. Large, adequately powered GWA studies interrogating all available SNP data are required to truly identify novel markers of patient responses to existing asthma medications. Only then will the relative contribution of common genetic polymorphisms to drug responses to existing asthma medications be defined, leading to translation to clinical relevance. This is clearly a challenge for existing pharmacogenetic datasets as the majority of studies have been limited in sample size.
Many new compounds in development to treat asthma target specific mediators or pathways and it is unlikely these approaches will show utility in all asthma subjects. Preliminary data suggest careful patient selection for Phase II trials will be essential to adequately evaluate these new compounds for clinical efficacy, as exemplified by anti-IL-5 and anti-IL-13 trials in asthma. It is highly likely that these patient subphenotypes in asthma have a genetic basis. Supporting this, recent Phase II trials of IL-4/IL-13 dual antagonist (pitrakinra) suggest patient selection is critical to evaluating the clinical efficacy of this compound with specific IL4Rα genotypes identifying responder groups. More pharmacogenetic integration is required for Phase II trials in asthma.
As the genetic basis of asthma is further defined by positional cloning, and more recently GWA studies new therapeutic opportunities are becoming apparent in specific pathways – for example, targeting the IL-33/ST2 interaction. In the evaluation of these new approaches, particularly Phase II clinical trials, it will be essential that the most appropriate patients are targeted based on genotype within these studies.
The impact of our pharmacogenetic knowledge on asthma management and prescribing practice is limited at this time as there is a need to further define the complex genetic basis of responders/nonresponders or patients with/without adverse effects in large prospective studies for all drug classes. As technological developments move at a dramatic pace; for example, GWA study platforms comprising of a million common SNPs or several million rare SNPs, gene-expression studies and whole-exome or targeted resequencing, these approaches will allow unprecedented detailed analyses of the human genome in these patient subgroups. This drug-specific genetic profile likely involving many genes (e.g., receptors, signalling intermediates, transcription factors) may provide a step towards personalized medicine in asthma, with associated benefits including avoidance of adverse side effects and the adequate control of the patients’ asthma, avoiding exacerbation and hospitalization due to lack of control or potential fatal asthma. As the cost of these genetic platforms comes down, the potential of a relatively simple test reducing asthma treatment costs becomes a real possibility. However, the introduction of routine genetic testing into clinical practice will require a clear demonstration of health benefits and/or cost–effectiveness that outperforms the current stepwise approach to the management of asthma.
Five-year view
We anticipate that pharmacogenetic investigations will continue to focus on known targets for current asthma drugs and genes will be identified with confidence through replication. The greatest progress will be made when the full utility of GWA approaches is applied to large cohorts involving thousands of individuals. We anticipate many novel common variants/genes will be identified underlying responses to current asthma medication. This information will be critical for profiling responders/nonresponders to existing asthma medications using multiple SNP markers across multiple genes, but importantly, may provide new insight into the mechanisms underlying the efficacy of existing drugs, leading to new therapeutic opportunities. If this information is going to impact clinical practice, there will be need for a simple, reproducible diagnostic test based on SNP combinations with adequately high specificity and sensitivity where responders and nonresponders are distinguished. This requirement is also a prerequisite for adequate evaluation of novel therapies in asthma – that is, conducting trials in the most appropriate populations likely to gain clinical benefit. Future challenges will be the handling of vast amounts of genomics information – GWA studies, whole-exomere-sequencing, expression profiling data and so on, and the standardization of clinical outcomes while accounting for nongenetic factors including the environment.
Table 1. Recently identified pharmacogenetic effects on current asthma therapies.
Table 2. Asthma susceptibility genes recently identified by genome-wide association studies.
Key issues
• To date, the majority of pharmacogenetic studies have been limited in size and the number of polymorphic variants studied in one or multiple genes small, leading to inconclusive findings for current asthma medication.
• In general, genotypes (single-nucleotide polymorphisms [SNPs]) instead of haplotypes (combinations of SNPs spanning the gene of interest) have been examined, potentially leading to conflicting data.
• Larger prospective studies have now been completed and conclusions regarding specific variants are becoming clearer, although there has still been a lack of continuity of the clinical outcomes studied.
• Where pharmacogenetic effects have been observed, these have been of a magnitude that can be considered clinically relevant; however, larger prospective studies are required to accurately determine effect sizes.
• To date, pharmacogenetic effects identified account for only a small proportion of the genetic variability in response to asthma medication (e.g., CRHR1 <3% of corticosteroid response), suggesting that asthma medications cannot be successfully personalized unless we resolve a larger degree of genetic variability and/or environmental influences.
• Most pharmacogenetic studies in asthma have focused on pharmacodynamic aspects of drug responses; however, there is a need to carry out more pharmacokinetic investigations of current asthma medications, particularly for systemically administered drugs.
• Technological advances allow the assessment of over 1 million SNPs in parallel (up to 6 million using imputation). These approaches are needed to provide an unprecedented evaluation of the contribution of common genetic factors to drug responses in asthma. Additional approaches, including expression profiling analyses and resequencing to identify rare variants, are required to further define underlying genetic mechanisms.
• Asthma is a heterogeneous disease and newer therapies targeting specific mediators are unlikely to provide benefit in all subjects; therefore, there is an immediate need for the integration of pharmacogenetics (driving specific phenotypes) in Phase II trials of newer asthma drugs to adequately evaluate these drugs for clinical efficacy.
Financial & competing interests disclosure
Research in the authors’ laboratory is funded by the MRC, BMA and Asthma UK. M Portelli is funded by a STEPS (Malta) studentship. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- No authors listed. Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society. Am. J. Respir. Crit. Care Med.162(6), 2341–2351 (2000).
- Ober C, Vercelli D. Gene–environment interactions in human disease: nuisance or opportunity? Trends Genet.27(3), 107–115 (2011).
- Holloway JW, Arshad SH, Holgate ST. Using genetics to predict the natural history of asthma? J. Allergy Clin. Immunol.126(2), 200–209; quiz 210–201 (2010).
- Moffatt MF, Kabesch M, Liang L et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature448(7152), 470–473 (2007).
- Moffatt MF, Gut IG, Demenais F et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med.363(13), 1211–1221 (2010).
- Himes BE, Hunninghake GM, Baurley JW et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am. J. Hum. Genet.84(5), 581–593 (2009).
- Gudbjartsson DF, Bjornsdottir US, Halapi E et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet.41(3), 342–347 (2009).
- Sleiman PM, Flory J, Imielinski M et al. Variants of DENND1B associated with asthma in children. N. Engl. J. Med.362(1), 36–44 (2010).
- Ferreira MA, Mcrae AF, Medland SE et al. Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21 with asthma risk in Australia. Eur. J. Hum. Genet.19(4), 458–464 (2011).
- BTS. British Guideline on the Managment of Asthma. British Thoracic Society, London, UK (2009).
- Sayers I, Hall IP. Pharmacogenetic approaches in the treatment of asthma. Curr. Allergy Asthma Rep.5(2), 101–108 (2005).
- Hall IP, Sayers I. Pharmacogenetics and asthma: false hope or new dawn? Eur. Respir. J.29(6), 1239–1245 (2007).
- Duroudier NP, Tulah AS, Sayers I. Leukotriene pathway genetics and pharmacogenetics in allergy. Allergy64(6), 823–839 (2009).
- Pascual RM, Bleecker ER. Pharmacogenetics of asthma. Curr. Opin. Pharmacol.10(3), 226–235 (2010).
- Tse SM, Tantisira K, Weiss ST. The pharmacogenetics and pharmacogenomics of asthma therapy. Pharmacogenomics J.11(6), 383–392 (2011).
- Hawkins GA, Tantisira K, Meyers DA et al. Sequence, haplotype, and association analysis of ADRβ2 in a multiethnic asthma case–control study. Am. J. Respir. Crit. Care Med.174(10), 1101–1109 (2006).
- Reihsaus E, Innis M, Macintyre N, Liggett SB. Mutations in the gene encoding for the β2-adrenergic receptor in normal and asthmatic subjects. Am. J. Respir. Cell Mol. Biol.8(3), 334–339 (1993).
- Drysdale CM, Mcgraw DW, Stack CB et al. Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc. Natl Acad. Sci. USA97(19), 10483–10488 (2000).
- Sayers I, Hawley J, Stewart CE et al. Pharmacogenetic characterization of indacaterol, a novel β2-adrenoceptor agonist. Br. J. Pharmacol.158(1), 277–286 (2009).
- Lee MY, Cheng SN, Chen SJ, Huang HL, Wang CC, Fan HC. Polymorphisms of the β2-adrenergic receptor correlated to nocturnal asthma and the response of terbutaline nebulizer. Pediatr. Neonatol.52(1), 18–23 (2011).
- Basu K, Palmer CN, Tavendale R, Lipworth BJ, Mukhopadhyay S. Adrenergic β(2)-receptor genotype predisposes to exacerbations in steroid-treated asthmatic patients taking frequent albuterol or salmeterol. J. Allergy Clin. Immunol.124(6), 1188–1194 (2009).
- Bleecker ER, Postma DS, Lawrance RM, Meyers DA, Ambrose HJ, Goldman M. Effect of ADRB2 polymorphisms on response to longacting β2-agonist therapy: a pharmacogenetic analysis of two randomised studies. Lancet370(9605), 2118–2125 (2007).
- Bleecker ER, Nelson HS, Kraft M et al. β2-receptor polymorphisms in patients receiving salmeterol with or without fluticasone propionate. Am. J. Respir. Crit. Care Med.181(7), 676–687 (2010).
- Israel E, Drazen JM, Liggett SB et al. The effect of polymorphisms of the β(2)-adrenergic receptor on the response to regular use of albuterol in asthma. Am. J. Respir. Crit. Care Med.162(1), 75–80 (2000).
- Wechsler ME, Kunselman SJ, Chinchilli VM et al. Effect of β-adrenergic receptor polymorphism on response to longacting β2 agonist in asthma (LARGE trial): a genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet374(9703), 1754–1764 (2009).
- Panebra A, Wang WC, Malone MM et al. Common ADRB2 haplotypes derived from 26 polymorphic sites direct β2-adrenergic receptor expression and regulation phenotypes. PLoS One5(7), e11819 (2010).
- Panebra A, Schwarb MR, Swift SM et al. Variable-length poly-C tract polymorphisms of the β2-adrenergic receptor 3´-UTR alter expression and agonist regulation. Am. J. Physiol. Lung Cell Mol. Physiol.294(2), L190–L195 (2008).
- Bleecker ER, Emmett A, Crater G, Knobil K, Kalberg C. Lung function and symptom improvement with fluticasone propionate/salmeterol and ipratropium bromide/albuterol in COPD: response by β-agonist reversibility. Pulm. Pharmacol. Ther.21(4), 682–688 (2008).
- Litonjua AA, Lasky-Su J, Schneiter K et al.ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am. J. Respir. Crit. Care Med.178(7), 688–694 (2008).
- Vonk JM, Postma DS, Maarsingh H, Bruinenberg M, Koppelman GH, Meurs H. Arginase 1 and arginase 2 variations associate with asthma, asthma severity and β2 agonist and steroid response. Pharmacogenet. Genomics20(3), 179–186 (2010).
- Duan QL, Gaume BR, Hawkins GA et al. Regulatory haplotypes in ARG1 are associated with altered bronchodilator response. Am. J. Respir. Crit. Care Med.183(4), 449–454 (2011).
- Meurs H, Mckay S, Maarsingh H et al. Increased arginase activity underlies allergen-induced deficiency of cNOS-derived nitric oxide and airway hyperresponsiveness. Br. J. Pharmacol.136(3), 391–398 (2002).
- Maarsingh H, Zuidhof AB, Bos IS et al. Arginase inhibition protects against allergen-induced airway obstruction, hyperresponsiveness, and inflammation. Am. J. Respir. Crit. Care Med.178(6), 565–573 (2008).
- Moore PE, Ryckman KK, Williams SM, Patel N, Summar ML, Sheller JR. Genetic variants of GSNOR and ADRB2 influence response to albuterol in African–American children with severe asthma. Pediatr. Pulmonol.44(7), 649–654 (2009).
- Choudhry S, Que LG, Yang Z et al. GSNO reductase and β2-adrenergic receptor gene–gene interaction: bronchodilator responsiveness to albuterol. Pharmacogenet. Genomics20(6), 351–358 (2010).
- Gaston B, Reilly J, Drazen JM et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc. Natl Acad. Sci USA90(23), 10957–10961 (1993).
- Whalen EJ, Foster MW, Matsumoto A et al. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell129(3), 511–522 (2007).
- Malmstrom K, Rodriguez-Gomez G, Guerra J et al. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma. A randomized, controlled trial. Montelukast/Beclomethasone study group. Ann. Intern. Med.130(6), 487–495 (1999).
- Mougey EB, Feng H, Castro M, Irvin CG, Lima JJ. Absorption of montelukast is transporter mediated: a common variant of OATP2B1 is associated with reduced plasma concentrations and poor response. Pharmacogenet. Genomics19(2), 129–138 (2009).
- Drazen JM, Yandava CN, Dube L et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet.22(2), 168–170 (1999).
- Tantisira KG, Lima J, Sylvia J, Klanderman B, Weiss ST. 5-lipoxygenase pharmacogenetics in asthma: overlap with Cys-leukotriene receptor antagonist loci. Pharmacogenet. Genomics19(3), 244–247 (2009).
- Lima JJ, Zhang S, Grant A et al. Influence of leukotriene pathway polymorphisms on response to montelukast in asthma. Am. J. Respir. Crit. Care Med.173(4), 379–385 (2006).
- Klotsman M, York TP, Pillai SG et al. Pharmacogenetics of the 5-lipoxygenase biosynthetic pathway and variable clinical response to montelukast. Pharmacogenet. Genomics17(3), 189–196 (2007).
- Sampson AP, Siddiqui S, Buchanan D et al. Variant LTC(4) synthase allele modifies cysteinyl leukotriene synthesis in eosinophils and predicts clinical response to zafirlukast. Thorax55(Suppl. 2), S28–S31 (2000).
- Asano K, Shiomi T, Hasegawa N et al. Leukotriene C4 synthase gene A(-444)C polymorphism and clinical response to a CYS-LT(1) antagonist, pranlukast, in Japanese patients with moderate asthma. Pharmacogenetics12(7), 565–570 (2002).
- Mastalerz L, Nizankowska E, Sanak M et al. Clinical and genetic features underlying the response of patients with bronchial asthma to treatment with a leukotriene receptor antagonist. Eur. J. Clin. Invest.32(12), 949–955 (2002).
- Whelan GJ, Blake K, Kissoon N et al. Effect of montelukast on time-course of exhaled nitric oxide in asthma: influence of LTC4 synthase A(-444)C polymorphism. Pediatr. Pulmonol.36(5), 413–420 (2003).
- Currie GP, Lima JJ, Sylvester JE, Lee DK, Cockburn WJ, Lipworth BJ. Leukotriene C4 synthase polymorphisms and responsiveness to leukotriene antagonists in asthma. Br. J. Clin. Pharmacol.56(4), 422–426 (2003).
- Kim SH, Ye YM, Hur GY et al. CYSLTR1 promoter polymorphism and requirement for leukotriene receptor antagonist in aspirin-intolerant asthma patients. Pharmacogenomics8(9), 1143–1150 (2007).
- Lee SY, Kim HB, Kim JH et al. Responsiveness to montelukast is associated with bronchial hyperresponsiveness and total immunoglobulin E but not polymorphisms in the leukotriene C4 synthase and cysteinyl leukotriene receptor 1 genes in Korean children with exercise-induced asthma (EIA). Clin. Exp. Allergy37(10), 1487–1493 (2007).
- Kang MJ, Kwon JW, Kim BJ et al. Polymorphisms of the PTGDR and LTC4S influence responsiveness to leukotriene receptor antagonists in Korean children with asthma. J. Hum. Genet.56(4), 284–289 (2011).
- York TP, Vargas-Irwin C, Anderson WH, Van Den Oord EJ. Asthma pharmacogenetic study using finite mixture models to handle drug-response heterogeneity. Pharmacogenomics10(5), 753–767 (2009).
- Mougey EB, Lang JE, Wen X, Lima JJ. Effect of citrus juice and SLCO2B1 genotype on the pharmacokinetics of montelukast. J. Clin. Pharmacol.51(5), 751–760 (2011).
- Lima JJ. Treatment heterogeneity in asthma: genetics of response to leukotriene modifiers. Mol. Diagn. Ther.11(2), 97–104 (2007).
- Stevens A, Ray DW, Zeggini E et al. Glucocorticoid sensitivity is determined by a specific glucocorticoid receptor haplotype. J. Clin. Endocrinol. Metab.89(2), 892–897 (2004).
- Tantisira KG, Lake S, Silverman ES et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum. Mol. Genet.13(13), 1353–1359 (2004).
- Finotto S, Neurath MF, Glickman JN et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science295(5553), 336–338 (2002).
- Ye YM, Lee HY, Kim SH et al. Pharmacogenetic study of the effects of NK2R G231E G>A and TBX21 H33Q C>G polymorphisms on asthma control with inhaled corticosteroid treatment. J. Clin. Pharm. Ther.34(6), 693–701 (2009).
- Hawkins GA, Lazarus R, Smith RS et al. The glucocorticoid receptor heterocomplex gene STIP1 is associated with improved lung function in asthmatic subjects treated with inhaled corticosteroids. J. Allergy Clin. Immunol.123(6), 1376–1383 (2009).
- Jin Y, Hu D, Peterson EL et al. Dual-specificity phosphatase 1 as a pharmacogenetic modifier of inhaled steroid response among asthmatic patients. J. Allergy Clin. Immunol.126(3), 618–625.e1–2 (2010).
- Tantisira KG, Silverman ES, Mariani TJ et al. FCER2: a pharmacogenetic basis for severe exacerbations in children with asthma. J. Allergy Clin. Immunol.120(6), 1285–1291 (2007).
- Koster ES, Maitland-van der Zee AH, Tavendale R et al. FCER2 T2206C variant associated with chronic symptoms and exacerbations in steroid-treated asthmatic children. Allergy66(12), 1546–1552 (2011).
- Tantisira KG, Lasky-Su J, Harada M et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N. Engl. J. Med.365(13), 1173–1183 (2011).
- Barnes KC. Ancestry, ancestry-informative markers, asthma, and the quest for personalized medicine. J. Allergy Clin. Immunol.126(6), 1139–1140 (2010).
- Haldar P, Brightling CE, Hargadon B et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med.360(10), 973–984 (2009).
- Corren J, Lemanske RF, Hanania NA et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med.365(12), 1088–1098 (2011).
- Slager RE, Hawkins GA, Ampleford EJ et al. IL-4 receptor α polymorphisms are predictors of a pharmacogenetic response to a novel IL-4/IL-13 antagonist. J. Allergy Clin. Immunol.126(4), 875–878 (2010).
- Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two Phase 2a studies. Lancet370(9596), 1422–1431 (2007).
- Risma KA, Wang N, Andrews RP et al. V75R576 IL-4 receptor α is associated with allergic asthma and enhanced IL-4 receptor function. J. Immunol.169(3), 1604–1610 (2002).
- Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J. Biol. Chem.282(36), 26369–26380 (2007).
- Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem. Biophys. Res. Commun.386(1), 181–185 (2009).
- Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am. J. Respir. Crit. Care Med.179(9), 772–781 (2009).
- Prefontaine D, Lajoie-Kadoch S, Foley S et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J. Immunol.183(8), 5094–5103 (2009).
- Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med.15(4), 410–416 (2009).
- Oshikawa K, Kuroiwa K, Tago K et al. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am. J. Respir. Crit. Care Med.164(2), 277–281 (2001).
- Torgerson DG, Ampleford EJ, Chiu GY et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet.43(9), 887–892 (2011).
- Savenije OE, Kerkhof M, Reijmerink NE et al. Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. J. Allergy Clin. Immunol.127(3), 750–756, e751–e755 (2011).
- Spina D. PDE4 inhibitors: current status. Br. J. Pharmacol.155(3), 308–315 (2008).
- Gamble E, Grootendorst DC, Brightling CE et al. Antiinflammatory effects of the phosphodiesterase-4 inhibitor cilomilast (Ariflo) in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med.168(8), 976–982 (2003).
- Mata M, Sarria B, Buenestado A, Cortijo J, Cerda M, Morcillo EJ. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax60(2), 144–152 (2005).
- Van Schalkwyk E, Strydom K, Williams Z et al. Roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor, attenuates allergen-induced asthmatic reactions. J. Allergy Clin. Immunol.116(2), 292–298 (2005).
- Fan Chung K. Phosphodiesterase inhibitors in airways disease. Eur. J. Pharmacol.533(1–3), 110–117 (2006).
- Trian T, Burgess JK, Niimi K et al. β2-agonist induced cAMP is decreased in asthmatic airway smooth muscle due to increased PDE4D. PLoS ONE6(5), e20000 (2011).
- Obeidat M, Wain LV, Shrine N et al. A comprehensive evaluation of potential lung function associated genes in the SpiroMeta general population sample. PLoS ONE6(5), e19382 (2011).