Abstract
α-1-antitrypsin deficiency (A1ATD) is an under-recognized hereditary disorder associated with the premature onset of chronic obstructive pulmonary disease. There is considerable heterogeneity in the phenotypic expression of lung disease in A1ATD and the pathophysiology is complex, involving the interaction of multiple pathways. Other genetic factors that may contribute to emphysema risk in A1AT-deficient individuals are beginning to be identified. Methods of monitoring disease progression have evolved, including the use of computed tomography densitometry and biomarkers of disease activity. Progress in the development of novel treatment strategies continues, including the hope for a potential cure through the use of gene therapies. In this article, the authors review the recent advances in this field and outline potential future directions of research in A1ATD.
Medscape: Continuing Medical Education Online
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Expert Reviews Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/expertrespiratory; (4) view/print certificate.
Release date: 4 June 2013; Expiration date: 4 June 2014
Learning objectives
Upon completion of this activity, participants will be able to:
• Assess the pathophysiology of A1ATD
• Analyze the clinical presentation of patients with A1ATD
• Compare methods to predict the progression of emphysema among patients with A1ATD
• Evaluate treatment options for patients with A1ATD
Financial & competing interests disclosure
EDITOR
Elisa Manzotti
Publisher, Future Science Group, London, UK.
Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME AUTHOR
Charles P Vega, MD
Associate Professor and Residency Director, Department of Family Medicine, University of California, Irvine, CA, USA.
Disclosure: Charles P Vega, MD, has disclosed no relevant financial relationships.
AUTHORS AND CREDENTIALS
Judith A Brebner
The ADAPT Project, Lung Function and Sleep Department, University Hospital Birmingham, Birmingham, UK.
Disclosure: JA Brebner has disclosed the following relevant financial relationships: funded by an educational grant from Grifols.
Robert A Stockley
The ADAPT Project, Lung Function and Sleep Department, University Hospital Birmingham, Birmingham, UK.
Disclosure: RA Stockley has disclosed the following relevant financial relationships: unrestricted research grant from Grifols and acts in an advisory role for Baxter, CSL Behring and Kamada.
Reproduced with permission from Citation[153].
![Figure 1. a-1-antitrypsin structure.Reproduced with permission from Citation[153].](/cms/asset/722084d5-3b40-4f7c-b87e-18a7f3c88e02/ierx_a_11219368_f0001_b.jpg)
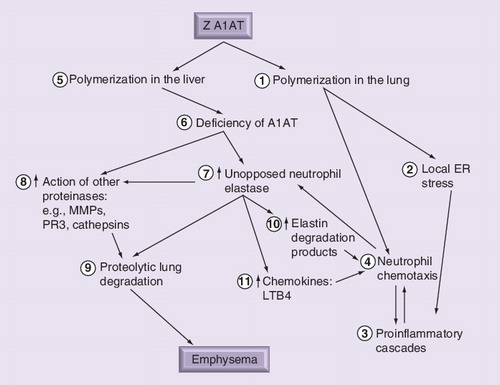
Polymerization of Z A1AT in the lung (1) leads to local ER stress (2) and establishment of a postinflammatory cascade (3) including increased neutrophil recruitment (4). Lung polymers are also chemoattractants, recruiting and localizing neutrophils further (4). Polymerization in the liver (5) results in serum and lung deficiency (6), causing unopposed neutrophil elastase activity (7) as a result of the recruited neutrophils (4), which in turn can activate other classes of enzymes (8) in addition to other uninhibited serine proteinases. The net result is proteolytic degradation of lung tissue (9), leading to emphysema. In addition, neutrophil elastase-derived peptide (10) and chemokines (11) can amplify the neutrophilic load and accelerate parenchymal damage through further release of proteinases.
A1AT: α-1-antitrypsin; ER: Endoplasmic reticulum; LTB4: Leukotriene B4;
MMP: Matrix metalloproteinase; PR3: Proteinase 3.
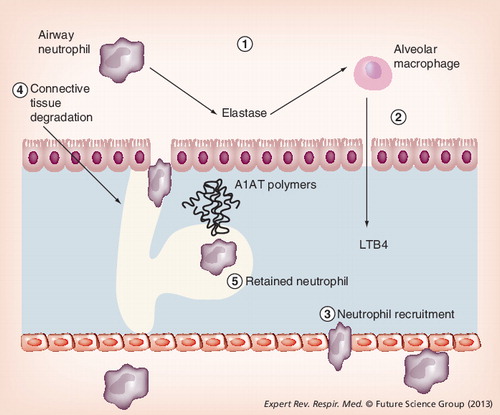
Local deficiency of A1AT leads to excess unopposed neutrophil elastase activity (1). Elastase binds to macrophages stimulating release of leukotriene-B4 (2), a potent neutrophil chemotactic mediator. This promotes neutrophil recruitment (3), causing further release of neutrophil elastase and tissue damage (4). In addition, mutant A1AT polymers colocalize with neutrophils, retaining them in the interstitium and promoting further connective tissue degradation (5).
FEV1: Forced expiratory volume in 1 s; KCO: Carbon monoxide transfer coefficient.
α-1-antitrypsin (A1AT) deficiency (A1ATD) is an under-recognized hereditary disorder first described by Laurell and Eriksson in 1963, after they linked the absence of an α1-globulin band on serum protein electrophoresis to the presence of premature emphysema Citation[1]. A1AT is a 52 kDa, single-chain glycoprotein with a 394 amino acid sequence Citation[2]. It is synthesized predominantly in the liver and functions as a serine proteinase inhibitor or serpin, providing essential protection to lung tissue against the actions of proteolytic enzymes such as neutrophil elastase (NE) and proteinase 3 (PR3).
The A1AT protein is encoded for by the SERPINA1 gene, which is composed of seven exons on the long arm of chromosome 14 (14q31–32.3) Citation[2]. There have now been over 500 single nucleotide polymorphisms reported at this gene locus Citation[201] (although many of these are yet to be validated) and inheritance occurs in an autosomal codominant manner. The traditional proteinase inhibitor (Pi) nomenclature uses alphabetical abbreviations to denote the speed of migration of the different allelic variants on gel electrophoresis Citation[3]. The M allele is the ‘normal’ variant and the commonest deficiency variants are S and Z, which migrate more slowly. The Pi phenotype refers to the type of circulating A1AT identified by serum electrophoresis, whereas genotyping relies on the use of specific probes to identify abnormal allelic sequences. The majority of individuals are homozygous for the normal M allele. Rarely, individuals can also inherit ‘null’ alleles, which do not produce any detectable A1AT protein by routine quantitative methods and hence cannot be phenotyped. The most common deficient phenotype is PiZ, which encompasses both PiZZ and PiZ null genotypes. Deficiency of circulating A1AT is associated with multiple conditions including emphysema, hepatic cirrhosis Citation[4], panniculitis Citation[5], bronchiectasis Citation[6] and vasculitis Citation[7].
In this review, the authors focus on lung disease in A1ATD with respect to its underlying pathophysiology and factors affecting variability in disease severity and clinical phenotypes. The authors also discuss the methods of monitoring disease progression, current treatment strategies and the recent advances in the development of novel therapeutic agents.
Genetic epidemiology of A1ATD
The majority of individuals with ‘severe pathophysiological deficiency’, classified by a serum A1AT level below the putative protective threshold (11 µM), have the PiZZ genotype Citation[8]. The highest prevalence of the Z variant is seen in northern Europe and it is thought to have arisen from southern Scandinavia Citation[9]. The S variant distribution differs, as the highest gene frequency is seen in southern Europe in the Iberian Peninsula Citation[10] and generally it leads to a mild deficiency not thought to be central to the pathophysiology of lung disease even when combined with a Z gene (SZ heterozygotes).
Recently, de Serres and Blanco used information from genetic epidemiological studies to develop estimates of the frequency of the two main deficiency alleles (PiS and PiZ) in 97 countries worldwide. More than 190 million individuals are estimated to have a deficient genotype, defined as having at least one of these alleles. In terms of severe deficiency, 0.1% of these individuals have the ZZ genotype with the majority living in Europe and North America Citation[11].
Less is known about the so-called ‘rare’ A1AT variants (non-Z and non-S deficient). It is likely that many of these cases have historically been misclassified as having the PiZ variant due to their absence or low levels and difficulty in their identification using isoelectric focusing phenotyping methods. Genotyping using PCR and DNA sequencing, in combination with measurement of serum A1AT levels, is becoming the new gold standard for diagnosing A1ATD and several defects have been identified, including major gene deletions, point mutations and frame shift mutations leading to the absence or truncated forms of the protein with intracellular polymerization or degradation Citation[12]. Our knowledge of the true prevalence of such rare variants and their associated clinical phenotypes is therefore likely to expand. A recent study of the prevalence of rare alleles in Spain highlighted the importance of standardization of methodology to obtain conclusive prevalence rates among different populations Citation[13]. In 2007, the Alpha One International Registry, incorporating data from 21 countries, reported that 5.3% of its severely deficient patients had rare A1AT alleles Citation[14]. Another more recent study evaluating A1AT serum protein concentrations in different A1ATD phenotypes found 6.7% (111 out of 1664) of subjects with two deficiency alleles had a rare variant Citation[15]. They are known to be more common in certain regions of Italy, such as Sardinia, where there is a high prevalence of ‘Mmalton’. This mutation is characterized by the deletion of the entire codon for residue Phe52, leading to intracellular polymerization and lower serum levels Citation[16]. Results from the Italian registry show a medium to low prevalence of the common PiZZ genotype and Ferrarotti et al. raised the interesting question of whether rare variants are more prevalent in populations with a lower PiZ gene frequency Citation[17].
Screening
In 2003, a joint statement from the American Thoracic and European Respiratory Societies recommended genetic screening for all symptomatic adults with chronic obstructive pulmonary disease (COPD) or adult onset asthma with incompletely reversible airflow obstruction Citation[18]. Indeed, the number of patients being tested is increasing annually in the USA Citation[19]. However, the majority of severely A1AT-deficient patients predicted epidemiologically remain undiagnosed. The concept of population-based screening has been entertained, but at present, only targeted testing of high-risk groups is recommended Citation[18,20]. The benefits of early diagnosis can include smoking cessation advice, avoidance of occupational exposures and opportunities for preconception genetic counseling. However, before undertaking widespread population screening, these benefits must be weighed up with the cost and impact on services of diagnosing a genetic disease in potentially asymptomatic individuals.
The molecular pathophysiology of A1ATD
Serpins such as A1AT are structurally composed of three β-sheets (A–C) and eight or nine α-helices (A–I) with an exposed mobile reactive loop containing residues that act as pseudosubstrates for the targeted proteinase Citation[21,22]. In the case of A1AT, a methionine residue at position 358 in the polypeptide chain is critical for the interaction with NE. The process of proteinase binding brings about a conformational change within the A1AT protein, whereby the enzyme cleaves the reactive center loop, which moves to the opposite pole of the protein taking the tethered proteinase with it, before being inserted into β-sheet A. The structural deformation of the proteinase that occurs as a result is key to the inhibitory function of the serpin Citation[23]. Serpins have a metastable native state that becomes more stable during proteinase inhibition. This makes them prone to aberrant structural formation and protein misfolding as a result of genetic mutations Citation[24].
Polymerization
The Z mutation occurs owing to the substitution of lysine for glutamic acid at position 342 in the polypeptide chain. The resulting structural change promotes the interaction of the reactive center loop of one molecule and the gap in the β-sheet A of another, causing molecular linkage or so called ‘loop sheet polymerization’ Citation[25].
Yamasaki et al. challenged this polymerization theory when they used protein crystallography to assess the molecular structure of a stable dimer from plasma-derived antithrombin. They reported extensive ‘domain swapping’ of more than 50 residues and reported the intermolecular linkage included insertion of two β-strands (4A and 5A) of one molecule into the β-sheet A of another. They further demonstrated that A1AT polymers also formed via this domain-swapping mechanism. They postulated this as the reason for the hyperstability of serpin polymers Citation[26]. However, in this study, A1AT polymerization was induced by guanidine hydrochloride, and Ekeowa et al. have subsequently demonstrated that 2C1 (a monoclonal antibody that recognizes pathological polymers of A1AT Citation[27]) only recognizes heat-produced polymers not those produced by denaturants, questioning the validity of the data used to support this model of polymerization Citation[28]. In addition, Ekeowa et al. used other methods including the use of synthetic peptides, limited proteolysis and mass spectrometry to help define the mechanism of A1AT polymerization and concluded that the single stranded loop-sheet model remained the best explanation. However, Yamasaki et al. have subsequently crystallized trimeric A1AT polymers (recognized by 2C1) and have now reported that linkage involves domain swapping of carboxy-terminal residues Citation[29]. As the debate regarding the molecular basis of polymerization continues, clarification of the possible variation of linkage among the different mutations and serpins is needed if therapeutic polymerization prevention strategies are to be successful.
Intracellular accumulation of Z variant A1AT polymers within the endoplasmic reticulum (ER) of hepatocytes can lead to neonatal hepatitis, hepatic cirrhosis and hepatocellular carcinoma Citation[30]. The resulting lack of circulating A1AT predisposes individuals to proteolytic attack of their lung tissue, resulting predominantly in emphysema. However, this explanation of the pathogenesis of lung disease in A1ATD is oversimplified as we now recognize that the manifestation relies on a complex combination of both pathogenic mechanisms and other environmental and genetic factors.
Mechanisms of lung disease in A1ATD
Proteinase: antiproteinase theory
Our knowledge of the pathophysiology of emphysema in A1ATD started with the ‘proteinase:antiproteinase’ theory. Following the observations made by Laurell and Eriksson, investigation into why the inherited deficiency of A1AT would cause premature emphysema ensued. It was subsequently demonstrated that NE could reproduce changes in animal models suggestive of emphysema Citation[31]. The development of this animal model and the recognition of A1AT as an NE inhibitor formed the basis of the proteinase:antiproteinase hypothesis, whereby imbalance in favor of NE leads to excessive tissue destruction and hence emphysema Citation[32].
There is extensive literature on the role of NE in the pathogenesis of emphysema in A1ATD, but other proteinases may also be relevant. PR3 is also a serine proteinase found in the azurophil granules of neutrophils which also causes emphysema in animal models Citation[33]. PR3 activity has been shown to be greater than that of NE in sputum of A1AT-deficient individuals especially during exacerbations, suggesting a role in the pathophysiology of COPD in these individuals Citation[34]. Matrix metalloproteinases (MMPs; particularly MMP-12) and cysteine proteinases such as cathepsin B have also been implicated as having a direct role in proteolytic alveolar destruction Citation[35,36]. Interestingly, NE has been shown to upregulate MMP-2 and cathepsin B expression in vitro Citation[37] as well as inactivating their relevant inhibitors resulting in a proteinase cascade Citation[38]. A better understanding of the interaction of different proteinases and their inhibitors in the pathogenesis of emphysema is needed as this could have important ramifications with respect to developing novel therapeutic agents.
Chemotactic mediators
High neutrophil counts have been observed in bronchoalveolar lavage (BAL) specimens from A1AT-deficient patients Citation[39]. Neutrophils are key effector cells in airway inflammation and have the potential to accelerate parenchymal damage through release of their proteinases. Multiple factors affecting neutrophil recruitment and activation have been suggested. Leukotriene B4 (LTB4) is a potent neutrophil chemotactic mediator found in increased concentration in the sputum of A1AT-deficient patients Citation[40]. Furthermore, studies of sputum chemotactic activity identified both LTB4 and IL-8 as significant contributors to chemotaxis Citation[41], although only the former correlated with the total migratory potential. in vitro experiments demonstrated that LTB4 is released following the binding of alveolar macrophages and NE, supporting the concept that a proteinase imbalance could promote neutrophilic inflammation through the excess production of this chemokine Citation[42]. A reduction in sputum LTB4 concentrations in A1AT-deficient individuals occurs in response to augmentation therapy, confirming this as a likely mechanism Citation[43].
In a murine emphysema model, elastin degradation products found in BAL fluid have been shown to be chemotactic for monocytes. Using a monoclonal antibody to inhibit these elastin fragments eliminated the chemotaxis in vitro. Administration of the same monoclonal antibody at the time of cigarette exposure was also shown to reduce the accumulation of macrophages in the lung and abrogated the development of emphysema in vivo Citation[44]. Since elastin degradation is likely to be a key process in the development of emphysema in A1ATD, it is possible that elastin fragments also play a role in amplifying neutrophilic inflammation but direct evidence is lacking.
A1AT polymerization & the lung
Z A1AT polymers have been identified in the BAL fluid from A1AT-deficient patients Citation[45]. This extrahepatic polymerization may serve to further exacerbate the deficiency in these individuals, given the lack of functional antiproteinase activity of polymerized A1AT. The Z polymers have also been shown to activate neutrophils, manipulate neutrophil shape, promote adhesion and stimulate myeloperoxidase release in vitro Citation[46]. Mahadeva et al. subsequently demonstrated that polymers of A1AT colocalize with neutrophils in the interstitium of PiZ individuals, which may also add to the connective tissue degradation . Furthermore, Mahadeva et al. also showed that instilling polymers into the lungs of mice resulted in a significant neutrophil influx, which may reflect a direct or indirect chemoattractant process Citation[47].
Although the majority of A1AT is produced in the liver, it is also synthesized by other cells including bronchial and alveolar epithelial cells Citation[48], supported by the presence of Z A1AT polymers in the BAL fluid of a patient 9 years post-liver transplant Citation[49]. The potential proinflammatory properties of polymerized, locally produced Z A1AT in the lung may partly explain the progression of emphysema despite augmentation therapy in some individuals or the delay in efficacy seen in clinical trials Citation[50,51] by perpetuating the inflammation Citation[49]. Z polymerization is concentration dependent Citation[25] and since the A1AT concentration in the blood is higher than in the lung, it is likely that the blood is the site of highest polymer concentration. The addition of MM A1AT would therefore dilute the Z protein and reduce polymer formation, although this would also require clearance of lung tissue polymers before their proinflammatory effect would be lost. Whether this would result in total abrogation of the inflammatory process with time remains unknown.
The importance of recognizing these so-called gain of function effects within the lung, including the concept of ER stress (brought about by the accumulation of misfolded Z A1AT protein in the ER) and the activation of associated inflammatory signaling pathways, has been emphasized in recent years Citation[52]. ER stress can have multiple effects including NF-κB activation, promotion of ER-associated degradation and apoptosis Citation[53]. The contribution of local ER stress to the pathogenesis of emphysema in A1ATD is an important concept as even augmentation therapy would not prevent this local effect and combined therapy with small molecules to prevent intracellular accumulation in lung cells may be required.
Clinical manifestations of A1ATD in the lung
Emphysema
The premature onset of emphysema was the first identified clinical manifestation of A1ATD described in 1963. The classical distribution has a lower zone predominance Citation[54]; however, all zones can be affected. In one study of 102 A1AT-deficient patients, 65 (64%) had predominantly basal changes and 37 (36%) had a greater degree of apical emphysema Citation[55]. It was also noted in this study that basal emphysema had a greater association with forced expiratory volume in 1 s (FEV1) impairment than apical emphysema, confirming previous observations in usual COPD Citation[56].
Bronchiectasis
Earlier reports of the incidence of bronchiectasis in A1AT-deficient individuals were as high as 43%, although based on small study populations Citation[6,57]. In a larger study of 74 (PiZ) subjects, Parr et al. reported the incidence of ‘clinically relevant’ bronchiectasis to be 27%, which is similar to the reported incidence in usual COPD Citation[58]. Using a different approach, Cuvelier et al. looked at the frequency of deficiency alleles in 202 patients with known bronchiectasis compared with a control group and concluded that there was no association. They did, however, note a higher Z allele frequency in bronchiectasis associated with emphysema Citation[59]. Whether or not A1ATD is an independent risk factor for bronchiectasis or that bronchiectasis simply occurs in association with emphysema remains unresolved and requires a sufficiently powered case–control study with usual COPD Citation[60].
Small airways disease & bronchodilator reversibility
A1ATD can be associated with varying degrees of airflow obstruction, even varying within individual patients. A study of 1052 subjects with A1ATD from the National Heart, Lung and Blood Institute Registry found that 82% reported symptoms of wheeze without a cold and 70% described ‘attacks of wheezing’ associated with breathlessness. The onset of these attacks was around 31 years of age, therefore the initial diagnosis of asthma given in young A1AT-deficient individuals is unsurprising. The study found that 49% of patients demonstrated significant reversibility at some stage of their follow-up, and the average increase in the FEV1 was 382 ml (±180). Reversibility (defined as an FEV1 increase of ≥12% and at least 200 ml postbronchodilator) was even seen in 12.5 % of patients with a normal FEV1, suggesting bronchial hyper-responsiveness may be an early feature of the disease process Citation[61]. Analysis of data from the UK and USA A1AT registries has shown that bronchodilator reversibility is associated with a more rapid decline in FEV1 Citation[62,63]. The difficulty in distinguishing asthma and COPD in A1AT-deficient patients is particularly challenging, and recognizing overlap between the two conditions is important to ensure appropriate therapeutic strategies are used to prevent an accelerated decline in lung function Citation[64].
Clinical phenotypes
Emphysema and airflow obstruction often coexist; however, as seen in usual COPD, subsets of patients with either marked emphysema and preserved spirometry or severe airflow obstruction with relatively little parenchymal destruction are identifiable. The lung disease associated with A1ATD exhibits considerable heterogeneity, and the recognition of distinct clinical phenotypes is important as this may lead to more individualized therapy. This pathological disparity was first postulated by Parr et al., partly reflecting the distribution of the emphysema with apical disease having little effect on FEV1 Citation[55]. Holme and Stockley confirmed this in a prospective study of individuals identified with physiological discordance (normal FEV1 and low diffusing capacity and vice versa) Citation[65].
An observational study of 745 patients with severe A1ATD compared subjects with emphysema, chronic bronchitis and asthma overlap Citation[66]. This study found chronic bronchitis patients were younger with a lower number of pack years and had better preserved lung function. A greater proportion of asthma overlap patients were women (55.2%) compared with the emphysema group (34.8%). This highlights the importance of patient characterization in the study of A1ATD. The implication for the pathophysiology of the disease and role of A1AT remains currently unknown.
Emphysema risk in A1ATD
A multivariate analysis of 378 severely deficient PiZ patients identified male sex, asthma, chronic bronchitis, pneumonia and cigarette smoking as risk factors for severe airflow obstruction (FEV1 < 50%) Citation[67]. Another study looking at lung function in a cohort of 101 PiZ subjects found FEV1 decline to be more rapid in those with moderately severe disease, whereas carbon monoxide transfer coefficient declined quicker in severe disease. Within the moderately severe category, the rate of FEV1 decline was associated with bronchodilator reversibility, exacerbation frequency, low BMI and male sex Citation[63].
Considerable variability in the age of onset and severity of COPD in patients with A1ATD is well recognized. It is likely that this not only reflects environmental factors but also additional modifying genetic factors Citation[68]. In support of this concept, a study assessing familial aggregation of COPD characteristics in 52 A1AT-deficient individuals and 117 relatives found a trend towards lower FEV1 in PiMZ relatives of PiZ individuals with significant airflow obstruction compared with those without, suggesting the importance of other familial factors Citation[69]. Indeed in the study by Wood et al., physiological discordance was also noted in PiZZ siblings Citation[70]. Although there was concordance with gas transfer and emphysema on CT scan, the FEV1 valued showed no concordance, indicating the A1ATD and lung disease link is far from simple. One possible explanation for this familial effect is gene-by-environment interactions, whereby family members inherit differing susceptibility to certain environmental factors such as smoking. A study of airflow obstruction in 378 A1AT-deficient patients found the heritability of FEV1 to be greater only in analysis of smokers, suggesting the importance of at least gene-by-smoking interaction Citation[71]. The nonlinear relationship between smoking and FEV1 has recently been demonstrated in four large COPD study cohorts containing individuals with a wide range of airflow obstruction Citation[72]. The authors advocated the use of a piecewise linear approach to model smoking in preference to the traditional pack years method when looking for gene-by-smoking interactions.
Whether or not PiMZ individuals are at increased risk of COPD has remained controversial. Heterozygotes for the Z allele have an A1AT level of approximately 60% of normal, which is believed to be above the protective threshold. A meta-analysis found case–control studies often demonstrated an increased COPD risk to PiMZ individuals, whereas cross-sectional studies generally found no difference Citation[73]. A comparison between PiMM and PiMZ individuals from two large study populations containing a total of 4376 subjects demonstrated a 3.5% lower FEV1/forced vital capacity ratio in the MZ subjects, suggesting a possible minor susceptibility to airflow obstruction Citation[74]. It is possible that the presence of a small amount of proinflammatory Z polymers and the reduced function of Z A1AT could contribute to COPD risk in such individuals with a PiMZ phenotype Citation[75]. However, many MZ individuals are identified through family studies of A1AT-deficient subjects or those with a family history of COPD, which may bias data towards other gene/environment influences.
In recent years, the search for additional ‘susceptibility genes’ has advanced owing to genome-wide association studies using an international collaborative approach to increase statistical power. Suggested modifier genes in A1AT-deficient individuals so far include NOS3 Citation[76], GSTP1 Citation[77], IL-10 Citation[78], TNF-α Citation[79], IREB2 and CHRNA3 Citation[80]. Knowledge of how genetic and environmental interactions relate to phenotypes and severity of disease will improve our ability to assess risk of COPD in A1AT-deficient patients.
FEV1, out with the old & in with the new?: biomarkers & alternative methods of monitoring disease progression
Traditionally, FEV1 has been the gold standard for monitoring COPD progression; however, it has potential drawbacks. First, FEV1 is effort dependent and can be significantly affected by day-to-day variability. Second, the spirometric normal range is wide, and it is recognized that the emphysema component of COPD can exist with ‘normal’ FEV1 Citation[81].
The EXACTLE trial demonstrated that computed tomography (CT) densitometry is a more sensitive indicator of emphysema progression than physiological and health status parameters Citation[51]. A further post-hoc analysis of the data from this trial found that the lung density decline in A1ATD was reduced by intravenous augmentation with the greatest effect seen at the lung bases, questioning whether targeted sampling by this method is preferable to assessing the whole lung Citation[82]. The procurement of satisfactory CT data for such complex analytical methods is likely to hinder the involvement of smaller centers in future large-scale augmentation studies.
In recent years, there has been much interest in the development of simpler biomarkers as alternative outcome measures for use in studies of potential disease-modifying therapeutic agents, although validation remains a major challenge. As a significant amount of damage must be done to airways before FEV1 is altered, biomarkers that correlate with FEV1 may be deemed reflective rather than predictive. The ideal biomarker should be central to the pathogenic process, predict progression and be responsive to treatments or episodes known to influence this Citation[83]. The aim of the ECLIPSE study was to look for surrogate markers superior to FEV1 in the longitudinal evaluation of COPD patients; however, severe A1ATD was one of the exclusion criteria for enrolment Citation[84].
The measurement of elastin degradation products, such as desmosine/isodesmosine, has been investigated in both A1AT-deficient and usual COPD patients and does correlate with lung function Citation[85]. Fregonese et al. measured the urinary and plasma desmosine levels in 11 A1AT-deficient individuals over a period of 14 months. Both were significantly higher compared with healthy controls, but they found wide variation in urinary values over time Citation[86]. This was echoed in a study of 390 patients with usual COPD where urinary desmosine levels increased during exacerbations Citation[87]. Plasma levels are more stable for longitudinal studies and identify patients with increased elastin degradation and may facilitate recruitment for targeted therapies Citation[86,87].
Aα-Val360 (a NE-specific cleavage product of fibrinogen, which is generated when elastase is released) has been shown to correlate with severity of COPD as measured by FEV1 in A1AT-deficient patients and is a sensitive marker of exacerbations and response to A1AT replacement therapy Citation[88]. Whether it predicts progression in A1ATD remains unknown at present. Other suggested biomarkers in A1ATD include MMP-9 Citation[89] and leukotriene B4 Citation[43]. However, further longitudinal studies are needed to establish if any of these proposed biomarkers can satisfy all the criteria needed for the development of novel therapeutic strategies in A1ATD Citation[83]. It is possible that no single biomarker will be able to achieve this but alternative uses may include panels of biomarkers to help define phenotypic subgroups of patients, which is a concept that has been investigated in usual COPD Citation[90].
Treatment of A1AT-related COPD
The current mainstay of treatment of A1AT-related COPD is similar to that of usual COPD Citation[18]. This involves the use of inhaled bronchodilators and steroids, pulmonary rehabilitation, long-term oxygen therapy, antibiotics and/or steroids during exacerbations in addition to smoking cessation advice and preventative influenza and pneumonia vaccinations.
In the search for potential disease-modifying drugs for emphysema, interest in retinoids arose as all-trans-retinoic acid was shown to reverse emphysematous lung damage in rats Citation[91]. A randomized, double-blinded, placebo-controlled trial investigating the efficacy of palovarotene (an oral γ-selective retinoid agonist) in 262 patients with severe A1ATD did not, however, show any significant benefit with respect to changes in lung density or functional parameters after 1 year of therapy Citation[92].
The potential to reduce proteolytic lung damage through the use of synthetic NE inhibitors has also been explored. However, the lack of clinical benefit in usual COPD Citation[93] and other conditions such as cystic fibrosis Citation[94] and acute lung injury Citation[95] have meant that trials in A1ATD have not been undertaken.
The evidence for unilateral and bilateral lung volume reduction surgery (LVRS) is limited to a number of case series. Compared with the outcomes in non-A1ATD-related emphysema, the magnitude of improvement seen post-LVRS appears to be less and is not sustained for the same length of time Citation[96–98]. A recent case series assessing endobronchial valve placement as an alternative to surgical LVRS demonstrated a median FEV1 improvement of 0.575–0.905 l (p = 0.028) with an associated improvement in BODE index Citation[99]. Studies with larger numbers and longitudinal data will be needed before any firm conclusions can be drawn of long-term benefit but this approach may play a role in bridging younger patients to transplant. However, caution should be exercised as the results of a randomized trial of patients with usual COPD showed modest improvements in lung function following endobronchial valve placement at the expense of an increased risk of complications including hemoptysis, pneumonia and increased exacerbation frequency Citation[100].
Intravenous augmentation therapy: the current understanding
Intravenous administration of A1AT derived from pooled human plasma is only available in some countries as doubts remain over its efficacy and cost–effectiveness. The US FDA have approved the following products in the USA: Prolastin®, Prolastin-C® (Grifols, Barcelona, Spain), Aralast™, Aralast NP™ (Baxter, IL, USA), Zemaira® (CSL Behring, PA, USA) and GLASSIA™ (Baxter). Other products available elsewhere include Trypsone®/Trypsan® (Grifols) and Alfalastin® (LFB, Les Ulis, France).
Augmentation therapy can certainly increase and sustain serum levels above the accepted protective threshold (11 µM, 80 mg/dl or 57 mg/dl by nephelometry) at a dose of 60 mg/kg plasma-derived A1AT once weekly and increase the anti-NE capacity in the epithelial lining fluid of the lungs Citation[101].
Evidence regarding the clinical efficacy of augmentation therapy is less clearly defined. The majority of evidence is based upon observational cohort studies as only two small randomized controlled trials investigating this have been published to date. A reduction in mortality risk was reported in patients receiving augmentation therapy in the large observational study of 1129 participants followed up over a 3.5- to 7-year period. However, overall FEV1 decline was not altered, although in the subgroup with moderate FEV1 impairment (FEV1: 35–49% predicted), a significantly slower decline was seen in those receiving augmentation Citation[62]. However, it should also be recognized that FEV1 is generally a poor surrogate for emphysema. In addition, FEV1 decline is not linear and seems to be fastest in the moderately impaired range Citation[63]. This would be the range where evidence of efficacy (using FEV1 as the outcome) would be easiest to detect. Nevertheless, this range of FEV1 has been widely adopted as the indicator for augmentation therapy as a result of this observation. Other supportive evidence of efficacy comes from comparing the FEV1 decline in cohorts from countries with and without therapy Citation[102], sequential studies on FEV1 decline before and after therapy Citation[103], and meta-analyses of spirometric decline in A1ATD from other published cohorts Citation[104]. However, interpreting observational data can be influenced by healthcare delivery systems and other socioeconomic factors both between and within countries and nonlinearity of FEV1 decline and FEV1 remains relatively insensitive to the emphysema disease process. Not only can tissue destruction occur even when the FEV1 is stable Citation[105] but progressive loss of gas transfer can also be detected and seems to be greatest when the FEV1 is at its lowest Citation[63]. These data suggest that concentration of effort on measuring FEV1 (though simple) only provides a superficial view of the disease process and hence treatment decisions.
The first randomized control trial by Dirksen et al. involved four weekly infusions of either A1AT or albumin. The outcome measures included lung function and quantification of emphysema using CT densitometry. No significant effect on pulmonary function was seen, but CT data showed a trend towards protection against loss of lung tissue in the augmentation group Citation[50]. A similar trend was seen in the second randomized trial, which used a more conventional augmentation regimen involving weekly infusions of 60 mg/kg of Prolastin. The primary analysis of this study also failed to achieve conventional levels of statistical significance, although one of the analysis methods did. In addition, although there was no effect on exacerbation frequency, augmentation did reduce hospital admissions Citation[51]. Subsequent analysis of the CT data did indicate a significant benefit on lung density decline if only the lower zone (where the emphysema is classically localized) was assessed Citation[82]. In addition, despite the differences in methodology, an integrated analysis of the results of these two trials was performed and demonstrated a significant reduction in the decline of lung density in subjects treated with A1AT replacement (p = 0.006) Citation[106], suggesting that both individual studies were underpowered.
The acceptance of such data as evidence of efficacy, although not ideal, may be necessary given the challenges to performing a suitably powered double-blind randomized trial. These include the relative rarity of the disease, ethical justification for intravenous placebo arms, high cost and need for a long duration of follow-up Citation[106,107] and, importantly, experienced centers to ensure consistency of acquisition of densitometry data. A double-blinded, randomized trial of Zemaira (CSL Behring) versus placebo with a 2-year follow-up period has recently been closed to recruitment but the outcome has yet to be analyzed Citation[202].
Some studies have focused on the short-term effects of augmentation therapy by measuring markers of neutrophil recruitment, inflammation and proteinase activity. One such study demonstrated a significant reduction in the sputum concentrations of LTB4 (a neutrophil chemoattractant) in response to augmentation therapy Citation[43]. Whereas this is supportive of an anti-inflammatory effect, it cannot support clinical efficacy.
The variability of lung function and damage even in smokers with A1ATD indicates that management should be individualized. Smoking cessation slows or stops progression and physiology can decline even when the FEV1 remains stable. Thus, assuming augmentation therapy is effective, it will remain critical to assess progression accurately through a variety of methods and only instigate therapy when clear evidence of decline, above age-related changes, is shown to occur.
Exacerbations
Exacerbations in A1ATD are associated with greater inflammation and elastase activity than in usual COPD Citation[108]. These episodes relate to physiological decline even in patients on augmentation therapy Citation[109]. This suggests that such episodes should be treated perhaps with bolus intravenous augmentation or even inhaled A1AT to boost anti-elastase screen in the airways affected by the exacerbation. Although logical, the practicalities of designing and delivering such a study may prove insurmountable.
Novel therapeutic approaches
Inhaled replacement therapy
Intravenous augmentation via weekly infusions is both expensive and inconvenient for patients. This has promoted interest in finding alternative routes of administration such as inhalation. Superficially, apart from the convenience aspect, there may also be perceived benefits of delivery of A1AT directly to the target organ as only 2–3% of A1AT given intravenously reaches the lung Citation[110]. Preliminary studies demonstrated that the aerosol approach can successfully augment the anti-NE capacity of the respiratory epithelium and inhaled A1AT can pass into the lung interstitium as trace amounts can be detected in the systemic circulation Citation[110,111]. However, protecting against the interstitial damage leading to emphysema poses significant logistical problems. First, the A1AT has to reach the alveolar region in sufficient amounts and this is highly unlikely using conventional nebulizers particularly in areas already affected Citation[112,113]. It could be argued that it is more crucial to protect areas with the most preserved function, although particle size and dose remain challenges to overcome. Even if sufficient amounts of the drug can be deposited onto the alveolar surface, the epithelial integrity will largely prevent access to the interstitium Citation[114]. Nevertheless, it is possible that reducing the activity of elastase in the alveolar space could decrease LTB4 production by macrophages (see earlier). This in its own right would reduce polymorphonuclear neutrophil traffic and hence indirectly protect the interstitial connective tissue. Alternatively, nebulizer drug may be more beneficial for airways disease in those patients with little emphysema but acute or recurrent exacerbations. Indeed, a multicenter, double-blinded randomized controlled trial evaluating the safety and efficacy of inhaled human A1AT is currently underway Citation[203]. Interim reports indicate good safety and tolerability, although definitive results will not be available for approximately 12 months.
Recombinant/transgenic augmentation
The cost and finite supply of human plasma-derived A1AT products, and potential infection risks have prompted research into alternative recombinant and transgenic sources. The A1AT gene has been expressed in a large variety of hosts Citation[115]. Transgenic production of human A1AT has been achieved in mice, rats, rabbits, sheep and goats Citation[116–120]. Spencer et al. raised concern regarding antibody responses seen in some recipients due to impurities in the aerosolized human A1AT produced from the milk of transgenic sheep Citation[121]. The potential for immune-mediated responses to transgenic proteins has currently halted the clinical development of these products. Another issue that has been highlighted is the stability of these recombinant proteins Citation[115]. Problems with absent or aberrant glycosylation can lead to conformational abnormalities, protein misfolding and aggregation with subsequent functional impairment and exposure of immunogenic epitopes normally hidden. Lack of glycosylation also leads to the rapid clearance of recombinant glycoprotein from the circulation Citation[122]. The degree of sialylation of the terminal glycans of the A1AT glycoprotein is essential for its plasma survival Citation[123]. In order to achieve the necessary degree of sialylation in recombinant A1AT derived from an human embryonic cell line (PER.C6), Ross et al. have cotransfected the human A1AT cDNA with a single recombinant human sialyltransferase gene. They reported that the recombinant A1AT produced by selected cell subclones is pharmacokinetically similar to plasma-derived A1AT and has equivalent functional properties in vitro Citation[124]. This emerging technology may yet result in an effective transgenic form of A1AT, although caution and long-term safety studies will be essential.
Polymerization prevention strategies & chemical chaperones
The loop-sheet polymerization mechanism of the Z mutation promotes interaction between the reactive center loop of one A1AT molecule and the gap in the β-sheet A of another. The resulting polymers lead to aggregation in hepatocytes and subsequent plasma deficiency Citation[25]. A strategy to try and prevent polymerization has been the use of small peptides to compete with reactive loops and bind to β-sheet A Citation[125–129]. Although in vitro tests are promising, challenges to this approach being translated to humans have included how to deliver the peptides to the ER of hepatocytes and the fact that they inactivate Z A1AT Citation[127], therefore potentially worsening plasma deficiency, emphysema risk and progression. Nevertheless, such an approach would certainly be protective of the ER stress caused by polymerization and thought to predispose liver damage and failure.
The discovery of a distinct hydrophobic cavity in the crystal structure of A1AT that is only patent in its nonpolymerized form has provided a further target for drugs preventing polymerization Citation[130,131]. Mallya et al. have performed virtual ligand screening on 1.2 million small molecules to try and identify compounds that could target this cavity and prevent polymer formation in vitro. The leading identified compound named CG was shown to reduce the intracellular accumulation of Z A1AT by 70% in murine hepatoma cells Citation[132], although effects on A1AT function have not been reported.
An alternative approach is the use of synthetic chaperones with the aim of stabilizing the intermediates on the Z A1AT folding pathway and 4-phenylbutyric acid is the most studied chaperone to date. Animal studies were initially encouraging Citation[133] but unfortunately human trials have been disappointing so far. In a preliminary open-label study, ten patients with severe A1ATD were administered oral 4-phenylbutyric acid for 14 days, which caused considerable side effects and did not significantly alter their circulating A1AT level Citation[134].
Gene therapies
Interest in targeted gene therapies to treat A1ATD is gaining momentum. The classical approach of inserting normal genes into cells of patients with the genetic mutation is being investigated in the hope of achieving therapeutic A1AT levels and hence preserving lung function. The first clinical trial in humans involved delivery of the normal A1AT gene in a plasmid–cationic liposome complex into the nasal mucosa Citation[135]. Transient increases were seen in the A1AT levels in the nasal lavage specimens but they returned to baseline by day 14. The results using recombinant adeno-associated virus (rAAV) vectors have been more promising. A nonrandomized, open label, Phase II clinical trial evaluating the safety and efficacy of an rAAV vector is currently ongoing. The aim is to produce sustained serum A1AT levels above the protective threshold (>11 µM), which has not been achieved so far. No gene therapies are currently licensed for use in A1ATD. Details of all the clinical trials to date can be seen in Citation[135–139]. However, importantly, gene therapies utilizing this approach will not prevent the production of the defective A1AT Z protein and, therefore, will have no impact on A1AT-related liver disease caused by defective protein accumulating in hepatocytes.
Genetic approaches to preventing Z A1AT liver disease include strategies to inhibit the translation of the mutant gene. Initial attempts to do this have utilized ribozymes to cleave PiZ mRNA Citation[140,141]. In one animal study, a vector system based on simian virus 40 was used to deliver ribozyme directly to the liver of transgenic mice carrying the mutant human A1AT PiZ allele via portal vein catheters. This resulted in marked decreases of human A1AT mRNA and protein in the liver Citation[142]. More recently, studies have looked at the ability of siRNA to downregulate endogenous A1AT production. siRNAs are double-stranded RNA molecules that play a role in post-transcriptional gene silencing. Cruz et al. used adeno-associated virus vectors expressing siRNA packaged in AAV8 capsids to transduce the livers of transgenic mice. A significant reduction in circulating human Z A1AT levels and clearance of Z A1AT protein from hepatocytes was seen within 3 weeks of vector administration Citation[143]. in vivo toxicity has been seen in mice treated with high doses of short hairpin RNA vectors, highlighting the importance of preclinical studies in determining potential risks associated with gene therapies Citation[144]. Again, this approach, although protecting the liver, will result in a further reduction in plasma A1AT, reducing the potential protection of the lung.
For these reasons, the ideal gene therapy would not only knock down the PiZ protein but also increase M-A1AT protein production as a potential treatment for both liver and lung disease. Following reports that RNAi-associated toxicity can be avoided by using miRNAs as opposed to short hairpin RNAs, Mueller et al. created miRNAs that target the human A1AT gene. Using the same rAAV vector, they incorporated miRNA sequences that target the PiZ A1AT gene, while also driving the expression of a wild-type PiM gene (which is resistant to the miRNA-mediated knockdown). This had the desired effect in transgenic mice of reducing circulating Z A1AT and increasing secretion of M A1AT Citation[145]. Such an approach would therefore both protect the liver by switching off the Z protein translation and subsequent polymerization while secreting normal M protein to protect the lung. The complexities and safety of human therapy have yet to be addressed.
A final possibility involves the technique of gene correction. As the Z mutation is known, the addition of the correct M sequence as small DNA fragments lead to recombination and sequence correction. This approach has been used in several single-gene diseases and has been shown to be effective in cells from A1AT-deficient patients with the Z defect Citation[146]. Again, the practicalities and safety of human delivery and efficacy have yet to be addressed.
Stem cell
For a gene therapy to be successful, sustained gene expression is essential. Given their ability to self-renew and capacity for differentiation, the concept of using stem cells as a delivery platform is therefore an attractive prospect. In 2008, Wilson et al. described the approach of A1AT gene transfer by transplanting lentivirally transduced hemopoietic stem cells into mice, demonstrating that this can achieve sustained human A1AT expression in vivo Citation[147].
Other stem cell strategies have focused on utilizing mesenchymal stem cells. Ghaedi et al. differentiated mesenchymal stem cells to produce hepatocyte-like cells, which were subsequently genetically modified to provide a potential in vitro source of A1AT gene containing hepatocytes for autologous transplantation Citation[148]. Other studies have used lentiviral and rAAV vectors to transduce bone marrow and adipose-derived stem cells before transplanting them into mouse models with promising results Citation[149,150].
Induced pluripotent stem cells (IPSCs) are differentiated or ‘adult’ cells that are genetically reprogrammed to an embryonic-like state Citation[151]. The potential to generate unlimited cells for autologous cell-based treatments for a variety of diseases is an exciting prospect in this rapidly advancing field of research. Yusa et al. have successfully used a combination of zinc finger nucleases and piggyBac technology to correct the point mutation in human IPSCs derived from individuals with A1ATD Citation[152]. Furthermore, they demonstrated that these corrected IPSCs can differentiate into hepatocyte-like cells in vitro which can function in vivo when transplanted into the liver of a mouse model. This approach could potentially provide sustained A1AT gene expression. However, the risk of other mutations arising during prolonged culture of the IPSCs was highlighted and careful screening would be essential for their safe clinical use long term.
Expert commentary
As awareness of A1ATD increases, a growing number of people are being diagnosed with this hereditary condition and its associated complications. Since the original discovery of A1ATD and the classical association with basal panacinar emphysema, much has been learnt about the condition. We now have a better understanding of the complex interactions underlying the pathophysiology of lung disease in these individuals. We also recognize that presentation with lung or liver disease is variable and requires further epigenetic studies to identify relevant features. The variability of lung pathology (airways versus alveolar disease), distribution of emphysema and methods of determining progression will provide further insight into who, when and how to treat the lung disease. Currently, the mainstay of treatment for A1ATD-related lung disease is similar to that of usual COPD with the addition of intravenous augmentation (in some countries) but as we have described, new treatment paradigms are evolving.
Five-year view
In the next 5 years, new ways of generating and delivering A1AT need to be clarified for both efficacy and safety.
Alternative potential ways to treat the deficiency need exploration to determine methods of drug or gene-modifying treatment deliveries. Of importance will be genetic and biomarker studies that will predict individuals at risk for developing the clinical manifestations and progression in A1ATD. This will lead to more personalized treatment strategies.
Table 1. Current status of validation of selected biomarkers in α-1-antitrypsin deficiency.
Table 2. Clinical trials of gene therapy in α-1-antitrypsin deficiency utilizing different vectors of administration.
Key issues
• α-1-antitrypsin (A1AT) is a serine proteinase inhibitor that protects the lung from damage by proteolytic enzymes such as neutrophil elastase.
• A1AT deficiency (A1ATD) is an under-recognized hereditary disorder associated with the premature onset of chronic obstructive pulmonary disease. Genotyping is becoming the gold standard diagnostic test.
• The pathophysiology of lung damage in A1ATD is complex, involving the interaction of multiple inflammatory pathways.
• We are beginning to identify other genetic factors that may contribute to emphysema risk in A1ATD.
• Computed tomography densitometry is the most sensitive measure of emphysema and its progression.
• There is growing interest in the development of biomarkers in A1ATD. The most promising candidates include desmosine, Aα-Val360, matrix metalloproteinase 9 and leukotriene B4, but further investigation and validation is still needed.
• There remains a lack of suitably powered double-blind randomized trials proving efficacy of intravenous augmentation. Supportive evidence is largely based on observational cohort studies.
• New paradigms of treatment are continuing to evolve including inhaled A1AT replacement, the use of recombinant/transgenic sources of A1AT for augmentation, polymerization prevention strategies and gene and stem cell therapies.
References
- Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. Scand. J. Clin. Lab. Invest. 15, 132–140 (1963).
- Brantly M, Nukiwa T, Crystal RG. Molecular basis of α-1-antitrypsin deficiency. Am. J. Med. 84(6A), 13–31 (1988).
- Fagerhol MK, Laurell CB. The polymorphism of ‘prealbumins’ and α-1-antitrypsin in human sera. Clin. Chim. Acta 16(2), 199–203 (1967).
- Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in α 1-antitrypsin deficiency. N. Engl. J. Med. 314(12), 736–739 (1986).
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe α 1-antitrypsin deficiency. Treatment and review of the literature. Arch. Dermatol. 123(12), 1655–1661 (1987).
- Shin MS, Ho KJ. Bronchiectasis in patients with α 1-antitrypsin deficiency. A rare occurrence? Chest 104(5), 1384–1386 (1993).
- Mahr AD, Edberg JC, Stone JH et al. α1-antitrypsin deficiency-related alleles Z and S and the risk of Wegener’s granulomatosis. Arthritis Rheum. 62(12), 3760–3767 (2010).
- Guidelines for the approach to the patient with severe hereditary α-1-antitrypsin deficiency. American Thoracic Society. Am. Rev. Respir. Dis. 140, 1494–1497 (1989).
- Hutchison DC. α 1-antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respir. Med. 92(3), 367–377 (1998).
- Blanco I, Fernández E, Bustillo EF. α-1-antitrypsin PI phenotypes S and Z in Europe: an analysis of the published surveys. Clin. Genet. 60(1), 31–41 (2001).
- de Serres FJ, Blanco I. Prevalence of a1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther. Adv. Respir. Dis. 6(5), 277–295 (2012).
- Miravitlles M, Herr C, Ferrarotti I et al. Laboratory testing of individuals with severe α1-antitrypsin deficiency in three European centres. Eur. Respir. J. 35(5), 960–968 (2010).
- Rodriguez-Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare α-1-antitrypsin variants: are they really so rare? Ther. Adv. Respir. Dis. 6(2), 79–85 (2012).
- Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P; Alpha One International Registry (AIR) group. Ongoing research in Europe: Alpha One International Registry (AIR) objectives and development. Eur. Respir. J. 29(3), 582–586 (2007).
- Bornhorst JA, Greene CM, Ashwood ER, Grenache DG. α 1-antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest 143(4), 1000–1008 (2013).
- Curiel DT, Holmes MD, Okayama H et al. Molecular basis of the liver and lung disease associated with the α 1-antitrypsin deficiency allele Mmalton. J. Biol. Chem. 264(23), 13938–13945 (1989).
- Ferrarotti I, Baccheschi J, Zorzetto M et al. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for α1-antitrypsin deficiency. J. Med. Genet. 42(3), 282–287 (2005).
- American Thoracic Society/European Respiratory Society Statement: standards for the diagnosis and management of individuals with α-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 168, 818–900 (2003).
- Campos M, Shmuels D, Walsh J. Detection of α-1 antitrypsin deficiency in the US. Am. J. Med. 125(7), 623–624 (2012).
- de Serres FJ, Blanco I, Fernández-Bustillo E. Estimating the risk for α-1 antitrypsin deficiency among COPD patients: evidence supporting targeted screening. COPD 3(3), 133–139 (2006).
- Davies MJ, Lomas DA. The molecular aetiology of the serpinopathies. Int. J. Biochem. Cell Biol. 40(6-7), 1273–1286 (2008).
- Whisstock J, Skinner R, Lesk AM. An atlas of serpin conformations. Trends Biochem. Sci. 23(2), 63–67 (1998).
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature 407(6806), 923–926 (2000).
- Whisstock JC, Bottomley SP. Molecular gymnastics: serpin structure, folding and misfolding. Curr. Opin. Struct. Biol. 16(6), 761–768 (2006).
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z α 1-antitrypsin accumulation in the liver. Nature 357(6379), 605–607 (1992).
- Yamasaki M, Li W, Johnson DJ, Huntington JA. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 455(7217), 1255–1258 (2008).
- Miranda E, Pérez J, Ekeowa UI et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with α1-antitrypsin deficiency. Hepatology 52(3), 1078–1088 (2010).
- Ekeowa UI, Freeke J, Miranda E et al. Defining the mechanism of polymerization in the serpinopathies. Proc. Natl Acad. Sci. USA 107(40), 17146–17151 (2010).
- Yamasaki M, Sendall TJ, Pearce MC, Whisstock JC, Huntington JA. Molecular basis of a1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 12(10), 1011–1017 (2011).
- Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu. Rev. Biochem. 78, 147–176 (2009).
- Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. The induction of pulmonary emphysema with human leukocyte elastase. Am. Rev. Respir. Dis. 116(3), 469–475 (1977).
- Turino GM. The origins of a concept: the protease-antiprotease imbalance hypothesis. Chest 122(3), 1058–1060 (2002).
- Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Invest. 82(6), 1963–1973 (1988).
- Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with α-1-antitrypsin deficiency and COPD. Eur. Respir. J. 41(5), 1042–1050 (2013).
- Churg A, Zhou S, Wright JL. Series ‘matrix metalloproteinases in lung health and disease’: matrix metalloproteinases in COPD. Eur. Respir. J. 39(1), 197–209 (2012).
- Lesser M, Padilla ML, Cardozo C. Induction of emphysema in hamsters by intratracheal instillation of cathepsin B. Am. Rev. Respir. Dis. 145(3), 661–668 (1992).
- Geraghty P, Rogan MP, Greene CM et al. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. J. Immunol. 178(9), 5871–5878 (2007).
- Sullivan A, Stockley RA. Proteinases in COPD. In: Recent Advances in the Pathophysiology of COPD. Hansel TT, Barnes PJ (Eds). Birkhauser Verlag, Basel, Switzerland (2004).
- Morrison HM, Kramps JA, Burnett D, Stockley RA. Lung lavage fluid from patients with α 1-proteinase inhibitor deficiency or chronic obstructive bronchitis: anti-elastase function and cell profile. Clin. Sci. 72(3), 373–381 (1987).
- Hill AT, Bayley DL, Campbell EJ, Hill SL, Stockley RA. Airways inflammation in chronic bronchitis: the effects of smoking and α1-antitrypsin deficiency. Eur. Respir. J. 15(5), 886–890 (2000).
- Woolhouse IS, Bayley DL, Stockley RA. Sputum chemotactic activity in chronic obstructive pulmonary disease: effect of α(1)-antitrypsin deficiency and the role of leukotriene B(4) and interleukin 8. Thorax 57(8), 709–714 (2002).
- Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG. Neutrophil accumulation in the lung in α 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J. Clin. Invest. 88(3), 891–897 (1991).
- Stockley RA, Bayley DL, Unsal I, Dowson LJ. The effect of augmentation therapy on bronchial inflammation in α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 165(11), 1494–1498 (2002).
- Houghton AM, Quintero PA, Perkins DL et al. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Invest. 116(3), 753–759 (2006).
- Elliott PR, Bilton D, Lomas DA. Lung polymers in Z α1-antitrypsin deficiency-related emphysema. Am. J. Respir. Cell Mol. Biol. 18(5), 670–674 (1998).
- Parmar JS, Mahadeva R, Reed BJ et al. Polymers of α(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am. J. Respir. Cell Mol. Biol. 26(6), 723–730 (2002).
- Mahadeva R, Atkinson C, Li Z et al. Polymers of Z α1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am. J. Pathol. 166(2), 377–386 (2005).
- Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in α1-antitrypsin deficiency: molecular and cellular insights. Eur. Respir. J. 34(2), 475–488 (2009).
- Mulgrew AT, Taggart CC, Lawless MW et al. Z α1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest 125(5), 1952–1957 (2004).
- Dirksen A, Dijkman JH, Madsen F et al. A randomized clinical trial of α(1)-antitrypsin augmentation therapy. Am. J. Respir. Crit. Care Med. 160(5 Pt 1), 1468–1472 (1999).
- Dirksen A, Piitulainen E, Parr DG et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in α1-antitrypsin deficiency. Eur. Respir. J. 33(6), 1345–1353 (2009).
- Greene CM, Hassan T, Molloy K, McElvaney NG. The role of proteases, endoplasmic reticulum stress and SERPINA1 heterozygosity in lung disease and a-1 anti-trypsin deficiency. Expert Rev. Respir. Med. 5(3), 395–411 (2011).
- Greene CM, McElvaney NG. Z a-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 1(5), 94–101 (2010).
- Gishen P, Saunders AJ, Tobin MJ, Hutchison DC. α 1-antitrypsin deficiency: the radiological features of pulmonary emphysema in subjects of Pi type Z and Pi type SZ: a survey by the British Thoracic Association. Clin. Radiol. 33(4), 371–377 (1982).
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in α1-antitrypsin deficiency influences lung function impairment. Am. J. Respir. Crit. Care Med. 170(11), 1172–1178 (2004).
- Gurney JW, Jones KK, Robbins RA et al. Regional distribution of emphysema: correlation of high-resolution CT with pulmonary function tests in unselected smokers. Radiology 183(2), 457–463 (1992).
- King MA, Stone JA, Diaz PT, Mueller CF, Becker WJ, Gadek JE. α 1-antitrypsin deficiency: evaluation of bronchiectasis with CT. Radiology 199(1), 137–141 (1996).
- Parr DG, Guest PG, Reynolds JH, Dowson LJ, Stockley RA. Prevalence and impact of bronchiectasis in α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 176(12), 1215–1221 (2007).
- Cuvelier A, Muir JF, Hellot MF et al. Distribution of α(1)-antitrypsin alleles in patients with bronchiectasis. Chest 117(2), 415–419 (2000).
- Chan ED, Iseman MD. Significance of bronchiectasis in patients with α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 178(2), 208; author reply 208–208; author reply 209 (2008).
- Eden E, Hammel J, Rouhani FN et al. Asthma features in severe α1-antitrypsin deficiency: experience of the National Heart, Lung, and Blood Institute Registry. Chest 123(3), 765–771 (2003).
- Survival and FEV1 decline in individuals with severe deficiency of α1-antitrypsin; the Alpha-1-Antitrypsin Deficiency Registry Study Group. Am. J. Respir. Crit. Care Med. 158, 49–59 (1998).
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in α1-antitrypsin deficiency. Eur. Respir. J. 33(6), 1338–1344 (2009).
- Eden E. Asthma and COPD in α-1 antitrypsin deficiency. Evidence for the Dutch hypothesis. COPD 7(5), 366–374 (2010).
- Holme J, Stockley RA. Radiologic and clinical features of COPD patients with discordant pulmonary physiology: lessons from α1-antitrypsin deficiency. Chest 132(3), 909–915 (2007).
- Piras B, Ferrarotti I, Lara B et al. Clinical phenotypes of Italian and Spanish patients with α-1-antitrypsin deficiency. Eur. Respir. J. doi:10.1183/09031936.00104712 (2012) (Epub ahead of print).
- Demeo DL, Sandhaus RA, Barker AF et al. Determinants of airflow obstruction in severe α-1-antitrypsin deficiency. Thorax 62(9), 806–813 (2007).
- DeMeo DL, Silverman EK. α1-antitrypsin deficiency. 2: genetic aspects of α(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax 59(3), 259–264 (2004).
- Silverman EK, Province MA, Rao DC, Pierce JA, Campbell EJ. A family study of the variability of pulmonary function in α 1-antitrypsin deficiency. Quantitative phenotypes. Am. Rev. Respir. Dis. 142(5), 1015–1021 (1990).
- Wood AM, Needham M, Simmonds MJ, Newby PR, Gough SC, Stockley RA. Phenotypic differences in α 1 antitrypsin-deficient sibling pairs may relate to genetic variation. COPD 5(6), 353–359 (2008).
- DeMeo DL, Campbell EJ, Brantly ML et al. Heritability of lung function in severe α-1 antitrypsin deficiency. Hum. Hered. 67(1), 38–45 (2009).
- Castaldi PJ, Demeo DL, Hersh CP et al.; AATGM Investigators; ICGN Investigators. Impact of non-linear smoking effects on the identification of gene-by-smoking interactions in COPD genetics studies. Thorax 66(10), 903–909 (2011).
- Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. Chronic obstructive pulmonary disease in α1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax 59(10), 843–849 (2004).
- Sørheim IC, Bakke P, Gulsvik A et al. a1-Antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohorts. Chest 138(5), 1125–1132 (2010).
- Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z-type α 1-antitrypsin is less competent than M1-type α 1-antitrypsin as an inhibitor of neutrophil elastase. J. Clin. Invest. 80(5), 1366–1374 (1987).
- Novoradovsky A, Brantly ML, Waclawiw MA et al. Endothelial nitric oxide synthase as a potential susceptibility gene in the pathogenesis of emphysema in α1-antitrypsin deficiency. Am. J. Respir. Cell Mol. Biol. 20(3), 441–447 (1999).
- Rodriguez F, de la Roza C, Jardi R, Schaper M, Vidal R, Miravitlles M. Glutathione S-transferase P1 and lung function in patients with α1-antitrypsin deficiency and COPD. Chest 127(5), 1537–1543 (2005).
- Demeo DL, Campbell EJ, Barker AF et al. IL10 polymorphisms are associated with airflow obstruction in severe α1-antitrypsin deficiency. Am. J. Respir. Cell Mol. Biol. 38(1), 114–120 (2008).
- Wood AM, Simmonds MJ, Bayley DL, Newby PR, Gough SC, Stockley RA. The TNFα gene relates to clinical phenotype in α-1-antitrypsin deficiency. Respir. Res. 9, 52 (2008).
- Kim WJ, Wood AM, Barker AF et al. Association of IREB2 and CHRNA3 polymorphisms with airflow obstruction in severe α-1 antitrypsin deficiency. Respir. Res. 13, 16 (2012).
- O’Brien C, Guest PJ, Hill SL, Stockley RA. Physiological and radiological characterisation of patients diagnosed with chronic obstructive pulmonary disease in primary care. Thorax 55(8), 635–642 (2000).
- Parr DG, Dirksen A, Piitulainen E, Deng C, Wencker M, Stockley RA. Exploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study of augmentation therapy in α 1-antitrypsin deficiency. Respir. Res. 10, 75 (2009).
- Stockley RA. Biomarkers in COPD: time for a deep breath. Thorax 62(8), 657–660 (2007).
- Vestbo J, Anderson W, Coxson HO et al.; ECLIPSE investigators. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur. Respir. J. 31(4), 869–873 (2008).
- Lindberg CA, Engström G, de Verdier MG et al. Total desmosines in plasma and urine correlate with lung function. Eur. Respir. J. 39(4), 839–845 (2012).
- Fregonese L, Ferrari F, Fumagalli M, Luisetti M, Stolk J, Iadarola P. Long-term variability of desmosine/isodesmosine as biomarker in α-1-antitrypsin deficiency-related COPD. COPD 8(5), 329–333 (2011).
- Huang JT, Chaudhuri R, Albarbarawi O et al. Clinical validity of plasma and urinary desmosine as biomarkers for chronic obstructive pulmonary disease. Thorax 67(6), 502–508 (2012).
- Carter RI, Mumford RA, Treonze KM et al. The fibrinogen cleavage product Aa-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 66(8), 686–691 (2011).
- Omachi TA, Eisner MD, Rames A, Markovtsova L, Blanc PD. Matrix metalloproteinase-9 predicts pulmonary status declines in a1-antitrypsin deficiency. Respir. Res. 12, 35 (2011).
- Agustí A, Edwards LD, Rennard SI et al.; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS ONE 7(5), e37483 (2012).
- Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat. Med. 3(6), 675–677 (1997).
- Stolk J, Stockley RA, Stoel BC et al. Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor. Eur. Respir. J. 40(2), 306–312 (2012).
- Vogelmeier C, Aquino TO, O’Brien CD, Perrett J, Gunawardena KA. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 9(2), 111–120 (2012).
- Elborn JS, Perrett J, Forsman-Semb K, Marks-Konczalik J, Gunawardena K, Entwistle N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur. Respir. J. 40(4), 969–976 (2012).
- Zeiher BG, Artigas A, Vincent JL et al.; STRIVE Study Group. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit. Care Med. 32(8), 1695–1702 (2004).
- Dauriat G, Mal H, Jebrak G et al. Functional results of unilateral lung volume reduction surgery in α1-antitrypsin deficient patients. Int. J. Chron. Obstruct. Pulmon. Dis. 1(2), 201–206 (2006).
- Stoller JK, Gildea TR, Ries AL, Meli YM, Karafa MT; National Emphysema Treatment Trial Research Group. Lung volume reduction surgery in patients with emphysema and α-1 antitrypsin deficiency. Ann. Thorac. Surg. 83(1), 241–251 (2007).
- Tutic M, Bloch KE, Lardinois D, Brack T, Russi EW, Weder W. Long-term results after lung volume reduction surgery in patients with α1-antitrypsin deficiency. J. Thorac. Cardiovasc. Surg. 128(3), 408–413 (2004).
- Tuohy MM, Remund KF, Hilfiker R, Murphy DT, Murray JG, Egan JJ. Endobronchial valve deployment in severe a-1 antitrypsin deficiency emphysema: a case series. Clin. Respir. J. 7(1), 45–52 (2013).
- Sciurba FC, Ernst A, Herth FJ et al.; VENT Study Research Group. A randomized study of endobronchial valves for advanced emphysema. N. Engl. J. Med. 363(13), 1233–1244 (2010).
- Wewers MD, Casolaro MA, Sellers SE et al. Replacement therapy for α 1-antitrypsin deficiency associated with emphysema. N. Engl. J. Med. 316(17), 1055–1062 (1987).
- Seersholm N, Wencker M, Banik N et al. Does α1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary α1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) α1-AT study group. Eur. Respir. J. 10(10), 2260–2263 (1997).
- Wencker M, Fuhrmann B, Banik N, Konietzko N; Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen. Longitudinal follow-up of patients with α(1)-protease inhibitor deficiency before and during therapy with IV α(1)-protease inhibitor. Chest 119(3), 737–744 (2001).
- Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for α1 antitrypsin deficiency: a meta-analysis. COPD 6(3), 177–184 (2009).
- Clark KD, Wardrobe-Wong N, Elliott JJ, Gill PT, Tait NP, Snashall PD. Patterns of lung disease in a ‘normal’ smoking population: are emphysema and airflow obstruction found together? Chest 120(3), 743–747 (2001).
- Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of a-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir. Res. 11, 136 (2010).
- Tonelli AR, Brantly ML. Augmentation therapy in α-1 antitrypsin deficiency: advances and controversies. Ther. Adv. Respir. Dis. 4(5), 289–312 (2010).
- Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with α(1)-antitrypsin deficiency (PiZ). Am. J. Respir. Crit. Care Med. 160(6), 1968–1975 (1999).
- Wencker M, Denker J, Konietzko N. Serial measurements of FEV1 over 12 years in a patient with α-1-protease inhibitor deficiency: influence of augmentation therapy and infections. Respiration. 61(4), 195–198 (1994).
- Hubbard RC, Crystal RG. Strategies for aerosol therapy of α 1-antitrypsin deficiency by the aerosol route. Lung 168(Suppl.), 565–578 (1990).
- Hubbard RC, Brantly ML, Sellers SE, Mitchell ME, Crystal RG. Anti-neutrophil-elastase defenses of the lower respiratory tract in α 1-antitrypsin deficiency directly augmented with an aerosol of α 1-antitrypsin. Ann. Intern. Med. 111(3), 206–212 (1989).
- Brand P, Beckmann H, Maas Enriquez M et al. Peripheral deposition of α1-protease inhibitor using commercial inhalation devices. Eur. Respir. J. 22(2), 263–267 (2003).
- Kropp J, Wencker M, Hotze A et al. Inhalation of [123I] α1-protease inhibitor: toward a new therapeutic concept of α1-protease inhibitor deficiency? J. Nucl. Med. 42(5), 744–751 (2001).
- Gorin AB, Stewart PA. Differential permeability of endothelial and epithelial barriers to albumin flux. J. Appl. Physiol. 47(6), 1315–1324 (1979).
- Karnaukhova E, Ophir Y, Golding B. Recombinant human α-1 proteinase inhibitor: towards therapeutic use. Amino Acids 30(4), 317–332 (2006).
- Archibald AL, McClenaghan M, Hornsey V, Simons JP, Clark AJ. High-level expression of biologically active human α 1-antitrypsin in the milk of transgenic mice. Proc. Natl Acad. Sci. USA 87(13), 5178–5182 (1990).
- Massoud M, Bischoff R, Dalemans W et al. Expression of active recombinant human α 1-antitrypsin in transgenic rabbits. J. Biotechnol. 18(3), 193–203 (1991).
- Tsymbalenko NV, Golinskii GF, Gaitskhoki VS. [Expression of the human α-1-antitrypsin gene in transgenic rats]. Biull. Eksp. Biol. Med. 120(7), 81–83 (1995).
- Wright G, Carver A, Cottom D et al. High level expression of active human α-1-antitrypsin in the milk of transgenic sheep. Biotechnology (NY) 9(9), 830–834 (1991).
- Ziomek CA. Commercialization of proteins produced in the mammary gland. Theriogenology 49(1), 139–144 (1998).
- Spencer LT, Humphries JE, Brantly ML; Transgenic Human Alpha 1-Antitrypsin Study Group. Antibody response to aerosolized transgenic human α1-antitrypsin. N. Engl. J. Med. 352(19), 2030–2031 (2005).
- Casolaro MA, Fells G, Wewers M et al. Augmentation of lung antineutrophil elastase capacity with recombinant human α-1-antitrypsin. J. Appl. Physiol. 63(5), 2015–2023 (1987).
- Yu SD, Gan JC. Effects of progressive desialylation on the survival of human plasma α1-anti-trypsin in rat circulation. Int. J. Biochem. 9(2), 107–115 (1978).
- Ross D, Brown T, Harper R et al. Production and characterization of a novel human recombinant α-1-antitrypsin in PER.C6 cells. J. Biotechnol. doi:10.1016/j.jbiotec.2012.09.018 (2012) (Epub ahead of print).
- Chang YP, Mahadeva R, Chang WS, Shukla A, Dafforn TR, Chu YH. Identification of a 4-mer peptide inhibitor that effectively blocks the polymerization of pathogenic Z α1-antitrypsin. Am. J. Respir. Cell Mol. Biol. 35(5), 540–548 (2006).
- Chang YP, Mahadeva R, Chang WS, Lin SC, Chu YH. Small-molecule peptides inhibit Z α1-antitrypsin polymerization. J. Cell. Mol. Med. 13(8B), 2304–2316 (2009).
- Mahadeva R, Dafforn TR, Carrell RW, Lomas DA. 6-mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerization. Implications for the prevention of Z α(1)-antitrypsin-related cirrhosis. J. Biol. Chem. 277(9), 6771–6774 (2002).
- Parfrey H, Dafforn TR, Belorgey D, Lomas DA, Mahadeva R. Inhibiting polymerization: new therapeutic strategies for Z α1-antitrypsin-related emphysema. Am. J. Respir. Cell Mol. Biol. 31(2), 133–139 (2004).
- Zhou A, Stein PE, Huntington JA, Sivasothy P, Lomas DA, Carrell RW. How small peptides block and reverse serpin polymerisation. J. Mol. Biol. 342, 931–941 (2004).
- Elliott PR, Pei XY, Dafforn TR, Lomas DA. Topography of a 2.0 A structure of α1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 9(7), 1274–1281 (2000).
- Parfrey H, Mahadeva R, Ravenhill NA et al. Targeting a surface cavity of α 1-antitrypsin to prevent conformational disease. J. Biol. Chem. 278(35), 33060–33066 (2003).
- Mallya M, Phillips RL, Saldanha SA et al. Small molecules block the polymerization of Z α1-antitrypsin and increase the clearance of intracellular aggregates. J. Med. Chem. 50(22), 5357–5363 (2007).
- Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant α 1-antitrypsin (α 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in α 1-AT deficiency. Proc. Natl Acad. Sci. USA 97(4), 1796–1801 (2000).
- Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum α-1-antitrypsin in patients with α-1-antitrypsin deficiency: a preliminary study. J. Pediatr. Gastroenterol. Nutr. 39(1), 34–37 (2004).
- Brigham KL, Lane KB, Meyrick B et al. Transfection of nasal mucosa with a normal α1-antitrypsin gene in α1-antitrypsin-deficient subjects: comparison with protein therapy. Hum. Gene Ther. 11(7), 1023–1032 (2000).
- Brantly ML, Spencer LT, Humphries M et al. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 αl-antitrypsin (AAT) vector in AAT-deficient adults. Hum. Gene Ther. 17(12), 1177–1186 (2006).
- Brantly ML, Chulay JD, Wang L et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl Acad. Sci. USA 106(38), 16363–16368 (2009).
- Chulay JD, Ye GJ, Thomas DL et al. Preclinical evaluation of a recombinant adeno-associated virus vector expressing human α-1 antitrypsin made using a recombinant herpes simplex virus production method. Hum. Gene Ther. 22(2), 155–165 (2011).
- Flotte TR, Trapnell BC, Humphries M et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing a1-antitrypsin: interim results. Hum. Gene Ther. 22(10), 1239–1247 (2011).
- Ozaki I, Zern MA, Liu S, Wei DL, Pomerantz RJ, Duan L. Ribozyme-mediated specific gene replacement of the α1-antitrypsin gene in human hepatoma cells. J. Hepatol. 31(1), 53–60 (1999).
- Zern MA, Ozaki I, Duan L, Pomerantz R, Liu SL, Strayer DS. A novel SV40-based vector successfully transduces and expresses an α 1-antitrypsin ribozyme in a human hepatoma-derived cell line. Gene Ther. 6(1), 114–120 (1999).
- Duan YY, Wu J, Zhu JL et al. Gene therapy for human α1-antitrypsin deficiency in an animal model using SV40-derived vectors. Gastroenterology 127(4), 1222–1232 (2004).
- Cruz PE, Mueller C, Cossette TL et al. in vivo post-transcriptional gene silencing of α-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab. Invest. 87(9), 893–902 (2007).
- Grimm D, Streetz KL, Jopling CL et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441(7092), 537–541 (2006).
- Mueller C, Tang Q, Gruntman A et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol. Ther. 20(3), 590–600 (2012).
- McNab GL, Ahmad A, Mistry D, Stockley RA. Modification of gene expression and increase in α1-antitrypsin (α1-AT) secretion after homologous recombination in α1-AT-deficient monocytes. Hum. Gene Ther. 18(11), 1171–1177 (2007).
- Wilson AA, Kwok LW, Hovav AH et al. Sustained expression of α1-antitrypsin after transplantation of manipulated hematopoietic stem cells. Am. J. Respir. Cell Mol. Biol. 39(2), 133–141 (2008).
- Ghaedi M, Lotfi AS, Soleimani M. Establishment of lentiviral-vector-mediated model of human α-1 antitrypsin delivery into hepatocyte-like cells differentiated from mesenchymal stem cells. Tissue Cell 42(3), 181–189 (2010).
- Li H, Lu Y, Witek RP et al. Ex vivo transduction and transplantation of bone marrow cells for liver gene delivery of α1-antitrypsin. Mol. Ther. 18(8), 1553–1558 (2010).
- Li H, Zhang B, Lu Y, Jorgensen M, Petersen B, Song S. Adipose tissue-derived mesenchymal stem cell-based liver gene delivery. J. Hepatol. 54(5), 930–938 (2011).
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4), 663–676 (2006).
- Yusa K, Rashid ST, Strick-Marchand H et al. Targeted gene correction of a1-antitrypsin deficiency in induced pluripotent stem cells. Nature 478(7369), 391–394 (2011).
- Wood AM, Stockley RA. Alpha one antitrypsin deficiency: from gene to treatment. Respiration 74(5), 481–492 (2007).
Websites
- Entrez SNP. www.ncbi.nlm.nih.gov/projects/SNP (Accessed 7 November 2012)
- Zemaira® in subjects with emphysema due to α1-proteinase inhibitor (API) deficiency. http://clinicaltrials.gov/ct2/show/NCT00261833 (Accessed 12 November 2012)
- International study evaluating the safety and efficacy of inhaled, human, α-1 antitrypsin (AAT) in α-1 antitrypsin deficient patients with emphysema. http://clinicaltrials.gov/show/NCT01217671 (Accessed 12 November 2012)
Recent advances in α-1-antitrypsin deficiency-related lung disease
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/expertrespiratory. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, [email protected]. For technical assistance, contact [email protected]. American Medical Association's Physician's Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the AMA PRA CME credit certificate and present it to your national medical association for review.
Activity Evaluation: Where 1 is strongly disagree and 5 is strongly agree
1. You are seeing a 30-year-old woman who was recently diagnosed with alpha-1-antitrypsin deficiency (A1ATD). What should you consider regarding the pathophysiology of this disease?
□ A Most patients with severe deficiency of A1AT have the PiZZ genotype
□ B Bronchoalveolar lavage usually yields normal counts of inflammatory cells among patients with A1ATD
□ C Leukotriene B4 (LTB4) levels are reduced in the sputum of patients with A1ATD
□ D Elastin degradation products function as the principal trigger for the inflammatory response in A1ATD
2. What should you expect regarding clinical manifestations of A1ATD in this patient?
□ A A1ATD primarily affects the apices of the lungs
□ B Apical emphysema is predictive of a lower forced expiratory volume in 1 second (FEV1) vs basilar emphysema
□ C Only 5% of patients demonstrate bronchodilator reversibility at any point in their disease
□ D Bronchodilator reversibility is associated with a more rapid decline in FEV1
3. Which of the following options is the best means to predict the progression of emphysema in this patient?
□ A Computed tomography (CT) densitometry
□ B Her age
□ C Her current treatment regimen
□ D Pulmonary function testing
4. Which of the following statements regarding the treatment of this patient is most accurate?
□ A Intravenous augmentation therapy with A1AT has stronger evidence of physiological vs clinical benefits
□ B Retinoids are a promising line of therapy
□ C Lung volume reduction surgery appears particularly effective for patients with A1ATD
□ D Currently available commercial gene therapy can improve both pulmonary and liver outcomes among patients with A1ATD