
Abstract:
Adalimumab was the first fully humanized monoclonal antibody approved by the US Food and Drug Administration (FDA). Launched in 2003, the clinical efficacy and safety of adalimumab were assessed in various trials in rheumatoid arthritis, ankylosing spondylitis, psoriasis, psoriatic arthritis, Crohn’s Disease, and ulcerative colitis, among others. At the global level, adalimumab is a major sales success among biologicals. It is still the greatest blockbuster among monoclonal antibodies. The date of its patent expiration paves the road for several potential biosimilars in various markets worldwide. This article discusses the current situation of molecules that are the main candidates to become adalimumab biosimilars. In addition, it also addresses the production processes, clinical studies, and relevant regulatory issues relative to the approval of those molecules, which must meet the challenge of demonstrating similar efficacy and safety to Humira®, but at a lower cost.
Keywords::
Introduction
Tumor necrosis factor α (TNF-α) is an inflammatory cytokine produced by activated monocytes or macrophages. It is synthesized as a transmembrane TNF (tTNF) protein that is then cleaved into a soluble TNF (sTNF) molecule, and each form has biological functions of its own.Citation1
Biological agents that are able to bind to TNF-α molecules are known as TNF-α antagonists (anti-TNF). These agents were the first biologicals approved for the treatment of rheumatic diseases and are widely used in clinical practice. To induce their therapeutic effect, the currently commercialized anti-TNF drugs bind to both sTNF and tTNF, reducing the selectivity of the resulting effects.Citation2
The most widely used anti-TNF drugs are etanercept (Enbrel®; Immunex Corporation, Thousand Oaks, CA, USA), adalimumab (Humira®; AbbVie Inc., North Chicago, IL, USA), infliximab (Remicade®; Janssen Pharmaceutica, Beerse, Belgium), golimumab (Simponi®; Centocor Ortho Biotech Inc., Horsham, PA, USA), and certolizumab pegol (Cimzia®; Union Chimique Belge, Brussels, Belgium).Citation3
With the patent expiration dates of the main biologicals approaching, pharmaceutical companies worldwide are developing biosimilars, which are ultimately similar in quality, efficacy, and safety to the corresponding reference products according to the regulatory guidelines formulated by the European Medicines Agency (EMA), the US Food and Drug Administration (FDA), and the Central Drugs Standard Control Organization (CDSCO).Citation4 Despite inspection, some professionals are concerned with the clinical equivalence of biosimilars. The reason is that the reference products have undergone several changes since their original formulation, and thus the drugs currently administered to patients are not exactly identical but only similar to those approved by regulatory agencies.Citation5
Adalimumab (Humira®) is a fully humanized monoclonal antibody that specifically binds to TNF-α molecules. It was originally approved in the US by the FDA in 2002 for the treatment of rheumatoid arthritis (RA); later, it was also approved for other diseases, including juvenile idiopathic arthritis (JIA), ankylosing spondylitis (AS), psoriatic arthritis (PsA), Crohn’s disease (CD), and ulcerative colitis (UC).Citation6
This article discusses the current situation of molecules that are the main candidates to become adalimumab biosimilars. In addition, it also addresses the production processes, clinical studies, and relevant regulatory issues relative to the approval of those molecules, which must meet the challenge of demonstrating similar efficacy and safety to Humira®, but at a lower cost.
Adalimumab
Adalimumab was the first fully humanized monoclonal antibody approved by the FDA. Launched in 2003, its sales increased 20% per year, totaling USD 8.5 billion.Citation7 It was estimated that the net profit would reach USD 16 billion in 2016.Citation8
Adalimumab is a tetramer composed of two light kappa chains and two heavy immunoglobulin G1 (IgG1) chains, each of the latter containing one N-glycosylation site.Citation9 This biological is the result of a partnership that started in 1993 between BASF Bioresearch Corporation and Cambridge Antibody Technology. The drug was first discovered by means of the “phage display” technique and was named D2E7. Then, it underwent the manufacturing process at BASF Bioresearch Corporation and was developed by BASF Knoll. After Abbott Laboratories (Abbott Park, IL, USA) bought BASF, the former began manufacturing and commercializing the drug.
In January 2013, Abbott Laboratories was split into two independent companies, Abbott Laboratories and AbbVie Inc., and the latter was charged with the development and sale of Humira®.Citation10
The efficacy and safety of adalimumab were assessed in various experimental systems.Citation11 Its capacity to prevent polyarthritis was demonstrated in a rat model that mimicked the clinical and histopathological progression of human RA.Citation12 The rats treated with adalimumab did not develop signs of arthritis during the 11-week study period, while the control group developed erosive arthritis. The preliminary results of clinical trials of adalimumab in RA were disclosed in 1999.Citation13
According to the US Patent and Trademark Office, AbbVie Inc. requested a 326-day extension of the patent expiration date, which is February 9, 2016.Citation14 With the expiration date approaching, several pharmaceutical companies are investigating and developing biosimilar adalimumab candidates.Citation15
Clinical trials and main indications
A robust process of clinical development, including Phase II and III trials, was established for each of the diseases for which adalimumab was approved.
Rheumatoid arthritis
Humira® was approved by the FDA in December 2002 for the treatment of adults with moderate to severe RA and inadequate response to at least one disease-modifying antirheumatic drug (DMARD).Citation16
The efficacy and safety of adalimumab were assessed in ∼3,000 individuals with RA older than 18 years in five major multicenter randomized, double-blind, placebo-controlled trials. Currently, adalimumab is indicated in combination with methotrexate (MTX) to treat patients with moderate to severe RA and unsatisfactory previous responses to DMARDs or with active, severe, and progressive RA independently from previous use of other medications. The use is recommended in a single dose of 40 mg per subcutaneous route every other week.Citation17
The ARMADA clinical trial included 271 patients with moderate to severe active RA with no response to MTX.Citation18 In addition, the participants had not responded to treatment with at least one DMARD. The participants were given adalimumab 20 mg, 40 mg, and 80 mg per subcutaneous route every other week or placebo over 24 weeks.
Another study investigated the efficacy of adalimumab monotherapy in patients refractory to MTX.Citation19 Over 26 weeks, 54 volunteers were given adalimumab 20 mg every other week, 20 mg every week, 40 mg every other week, 40 mg every week, or placebo.
A third study investigated the efficacy of adalimumab to inhibit structural damage, to reduce the signs and symptoms of disease, and to improve the functional performance of patients with RA receiving concomitant MTX.Citation20 The study included 619 individuals with active RA who had not responded to MTX. The participants were randomized to receive adalimumab 40 mg every other week, 20 mg every week, or placebo combined with MTX over 52 weeks. After the first 52 weeks of treatment, 457 participants were included in an open-label extension study in which they were given 40 mg in alternate doses >10 years. The study assessed radiographic progression at week 52 using the modified total Sharp score (TSS), clinical progression at week 24, and physical function at week 52 based on the disability index of the Health Assessment Questionnaire. At week 52, there was significantly less radiographic progression, as measured by the change in TSS, in the patients receiving adalimumab 40 mg compared to the group given 20 mg weekly or placebo. The functional response was better in the patients given adalimumab in either regime compared to the placebo.Citation20
The clinical trial, Safety Trial of Adalimumab in Rheumatoid Arthritis, assessed the safety of adalimumab in combination with standard antirheumatic treatment in 636 patients with RA who were not adequately responding to these therapies.Citation21 Standard treatment included DMARDs, non-steroidal anti-inflammatory drugs (NSAIDs), low-dose corticosteroids, and/or analgesics. The participants were randomized to receive adalimumab 40 mg every other week or placebo over 24 weeks. At 24 weeks, there were no significant differences between the adalimumab and placebo groups in their respective rates of adverse events (86.5% vs 82.7%), serious adverse events (5.3% vs 6.9%), life-threatening events (11.9% vs 15.4%), or those leading to withdrawal (2.8% vs 2.2%). There were also no significant differences in the rates of infections (52.2% vs 49.4%) between the groups. The study showed that the addition of adalimumab 40 mg every other week to patients using concomitant standard antirheumatic therapy is well tolerated and provides significant improvements in signs and symptoms of RA.Citation21
The PREMIER study compared the efficacy and safety of adalimumab combined with MTX vs MTX monotherapy or adalimumab monotherapy in patients with early, active, and moderate-to-severe RA who had not previously received MTX treatment.Citation22 The sample consisted of 799 patients with <3 years of disease. Treatments included adalimumab 40 mg every other week plus MTX 20 mg every week, adalimumab 40 mg every other week combined with placebo every week, and weekly MTX in alternation with placebo for 2 years. The primary outcomes were American College of Rheumatology 50% improvement (ACR50) and change from baseline in modified TSS. A total of 539 volunteers completed the study.Citation22
Combination therapy was superior to both MTX and adalimumab monotherapy in all assessed parameters. There was significantly less radiographic progression at 6 months, 1 year, and 2 years among the patients receiving combination therapy compared to those given MTX or adalimumab monotherapy. In addition, at 2 years, the rate of disease progression in the group that was given combination therapy was half compared to that in the first year of treatment. Clinical remission, defined as Disease Activity Score below 2.6, was observed in 43% of the patients receiving combination therapy vs 23% in the group receiving adalimumab monotherapy and 21% in the group receiving MTX monotherapy. The response was maintained >2 years.
PsA and psoriasis
The use of adalimumab to reduce the signs and symptoms of arthritis in patients with PsA was approved in June 2005 by the EMA and in October of the same year by the FDA. Use in patients with severe psoriasis and chronic plaque psoriasis but without signs of arthritis was approved by the FDA in November 2007 and January 2008, respectively.Citation23
The effectiveness of adalimumab in the treatment of PsA was demonstrated in Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT),Citation24 which was published in 2005. A total of 315 patients with severe PsA and unsatisfactory response to NSAIDs were randomized to receive adalimumab 40 mg or placebo every other week for 24 weeks. One of the primary efficacy end points was ACR20 response at week 12; the results showed that 58% of the participants given adalimumab achieved ACR20 vs 14% in the placebo group. Secondary end points were measures of joint disease disability and quality of life in all the patients, along with the severity of skin disease evaluated at the end of the study. The results showed that adalimumab improved the skin lesions, as indicated by Psoriasis Area and Severity Index (PASI), which exhibited 75% improvement in 59% of the patients vs 1% in the placebo group. Based on the ADEPT data, Gladman et alCitation25 showed that treatment with adalimumab significantly reduced the overall risk of radiographic progression of PsA and provided the greatest radiographic benefit for patients with the greatest concentrations of C-reactive protein (CRP) at baseline.
A similar studyCitation26 conducted in 2007 corroborated the results of ADEPT. In this case, the patients were treated with adalimumab 40 mg per subcutaneous route every other week over 12 weeks of blinded therapy and 12 weeks of open-label therapy. The ACR20 response was attained by 39% of the participants given adalimumab vs 16% in the placebo group. PsA response criteria were attained by 51% of the participants given adalimumab vs 24% in the placebo group. In addition, the skin lesions exhibited significant improvement in the adalimumab group.
Adalimumab can also be used for psoriasis alone, ie, without signs of arthritis.Citation27–Citation30 The validity of this use was demonstrated by Gordon et alCitation27 in a multicenter, randomized, double-blind, placebo-controlled study that assessed 147 patients allocated into three groups. Use of adalimumab was associated with significant improvement of psoriasis, in addition to being well tolerated over 60 weeks. Corroborating those results, a Phase III study published in 2008Citation28 assessed the safety and efficacy of adalimumab in patients with moderate to severe psoriasis and found that 71% of the patients treated with adalimumab attained at least 75% improvement in PASI at week 16 vs 7% in the placebo group.
The CHAMPIONCitation29 study compared the efficacy of adalimumab vs MTX in the treatment of plaque psoriasis. A total of 271 patients were allocated to three groups: 108 patients received adalimumab 80 mg per subcutaneous route at week 0 and 40 mg every other week over the following 16 weeks, 110 patients received MTX 7.5 mg per oral route every week (eventually increased to up to 25 mg as needed), and 53 patients were given placebo. At week 16, 79.6% of the adalimumab-treated patients had achieved 75% improvement in PASI vs 35.5% in the MTX group and 18.9% in the placebo group. In addition to demonstrating the superiority of adalimumab in psoriasis, improvement occurred significantly faster. The efficacy of adalimumab was also demonstrated in patients with psoriasis with unsatisfactory responses to etanercept.Citation31,Citation32
Ankylosing spondylitis
Several articles published between 2002 and 2005 pointed to the efficacy of TNF antagonists in the treatment of AS.Citation33–Citation37 In June 2006, the EMA approved the use of adalimumab to reduce the signs and symptoms of AS in cases that do not respond to conventional therapy; the FDA approved the same indication the following month.
The efficacy of adalimumab in the treatment of AS was demonstrated in three randomized, double-blind, placebo-controlled trials.Citation38–Citation40 In 2006, Van Der Heijde et al published the results of the Adalimumab Trial Evaluating Long-Term Efficacy and Safety in AS (ATLAS) study with 315 patients. Two-thirds of the sample (206 patients) was randomized to receive adalimumab 40 mg per subcutaneous route every other week for 24 weeks. The primary efficacy end point was the percentage of patients with 20% response according to the Assessment in Ankylosing Spondylitis International Working Group criteria for improvement (ASAS20) at week 12. The results showed that 58.2% of the adalimumab-treated patients had attained ASAS20 vs 20.6% in the placebo group. The patients who did not achieve ASAS20 at week 12 were selected to continue treatment with the same dose of adalimumab every week for an additional 12 weeks. By week 24, 66.7% of the adalimumab-treated patients had achieved ASAS20 vs 0% in the placebo group. The ATLAS group continued the study, with the participants switched to an open-label trial for 2 years. At 24 months, 64.5% of the patients were ASAS20 responders, 50.6% were ASAS40 responders, and 33.5% had maintained ASAS-defined partial remission.
Another studyCitation39 demonstrated the efficacy of 12 weeks of treatment with adalimumab to reduce both spinal and sacroiliac joint inflammation, which are typical occurrences in AS, assessed using ASAS40; the response was maintained at week 52. The efficacy of adalimumab was also demonstrated in patients with spondyloarthritis without radiographically defined sacroiliitis.Citation38
Crohn’s disease
In February 2007, the FDA approved the use of adalimumab to reduce the signs and symptoms and induce and maintain clinical remission in adults with moderately to severely active CD and inadequate response to conventional therapy.Citation41 The indication also includes patients who do not benefit from treatment with infliximab. The EMA approved that indication the same year.Citation42
Three studies (CLASSIC-I,Citation43 CHARM,Citation44 and GAINCitation45) played crucial roles in the assessment of the therapeutic response to adalimumab in CD among adults.
In the CLASSIC-I trial, published in 2006 by Hanauer et al, 299 patients with moderate to severe CD were randomized to receive subcutaneous injections of adalimumab in three different doses or placebo at weeks 0 and 2. The rates of remission at week 4 (primary end point) in the adalimumab 40 mg/20 mg, 80 mg/40 mg, and 160 mg/80 mg groups were 18% (P=0.36), 24% (P=0.06), and 36% (P=0.001), respectively, and 12% in the placebo group. The CLASSIC-I trial confirmed the superiority of adalimumab vs placebo for treatment of CD, and the drug was well tolerated under such conditions. The CHARM trial was performed to accomplish the same aims but was designed to assess the long-term efficacy of adalimumab for treatment of CD. A total of 854 patients received adalimumab 80 mg and 40 mg at weeks 0 and 2, respectively. In a second stage, the participants were randomized to receive adalimumab 40 mg every other week, 40 mg every week, or placebo for 56 weeks. The rates of response (remission) at week 56 were similar between the groups treated with adalimumab weekly or every other week, and both were superior to placebo (36%, 41%, and 12%, respectively; P<0.001). Following on, the GAIN trial assessed 325 patients with CD who did not respond to infliximab. The participants received adalimumab 160 mg at week 0 and 80 mg at week 2. The rates of remission at week 4 (primary end point) were 21% in the adalimumab-treated patients vs 7% in the placebo group (P<0.001), demonstrating that this drug represents an option for patients who do not respond to other anti-TNF agents.
As in the case of adults, adalimumab is efficacious in the treatment of CD among children, including those without response to infliximab.Citation46,Citation47
Juvenile idiopathic arthritis
Adalimumab combined with MTX is indicated for the treatment of moderate to severe polyarticular JIA in patients aged 2 years or older and with unsatisfactory response to one or more DMARDs. In cases of adverse events with MTX, the biological can be used as monotherapy. Adalimumab has not yet been investigated in children <2 years old.Citation17
In the European Union (EU), Humira® was approved in 2008 for the treatment of children and adolescents aged 4–17 years with polyarticular JIA; in 2013, the age range was extended to 2–17 years old.Citation48
In the US, Humira® was approved in 2008 for patients older than 4 years with polyarticular JIA; the FDA approved extension to younger patients in October 2014.Citation48
The safety and efficacy of adalimumab in the treatment of JIA were mainly assessed in a multicenter, prospective, randomized, double-blind, placebo-controlled trial.Citation49 The sample included 171 children aged 4–17 years with active polyarticular JIA and with no response to treatment with NSAIDs. At the onset of the open-label phase, the participants were divided into two groups according to MTX use. All the patients were given 24 mg of adalimumab per square meter of body surface area, up to a maximum of 40 mg, every 2 weeks for 16 weeks. The participants were allowed to use NSAIDs and low-dose corticosteroids. Next, the patients who attained ACR pediatric 30% (ACR Pedi 30) were randomized to receive subcutaneous injections of adalimumab or placebo every other week for 32 weeks or up to aggravation of disease. During the double-blind phase, patients were monitored for disease flares. Patients who enrolled in the double-blind phase were eligible to receive open-label treatment with adalimumab in an extension phase of the study. At week 16, 64 out of 86 (74%) patients who were not using MTX and 80 out of 85 (94%) who were using MTX had achieved ACR Pedi 30 and were eligible for the double-blind phase. At week 48, the adalimumab-treated group exhibited better rates of ACR30, ACR50, ACR70, and ACR90 with or without concomitant use of MTX. Flares were less frequent in the adalimumab group compared to the placebo group. The response rates were sustained after 2 years of treatment. After 104 weeks of treatment, 40% of the patients achieved ACR Pedi 100. In addition, combination with MTX was associated with lower rates of anti-adalimumab antibody (AAA; 6% vs 26%). The most frequent adverse events were infections and injection-site reactions.
Adalimumab was also assessed in an open-label multicenter study conducted in 32 children aged 2–4 years (or older than 4 years with body weight <15 kg) with moderate to severe polyarticular JIA.Citation50 The study lasted 24 weeks, and most participants made concomitant use of MTX. The rates of ACR Pedi 30 at weeks 12 and 24 were 93.5% and 95%, respectively, and were sustained over 60 weeks. The study demonstrated that adalimumab is safe in children aged 2 years or older.
Children with JIA are predisposed to uveitis, while 20%–25% of the cases of uveitis in childhood are associated with JIA.Citation51,Citation52 Despite screening for uveitis in patients with JIA, and the current treatments, 10%–15% of the children with JIA and uveitis have bilateral visual loss followed by amaurosis.Citation53 Thus, more effective therapeutic options are necessary. Some studies with experimental models of autoimmune uveitis suggest that TNF-α plays an important role in the pathophysiology of eye inflammation.Citation54
Behçet’s disease
Adalimumab was the first biological approved for the treatment of Behçet’s disease (BD) in Japan in 2003; however, this indication has not yet been approved by the FDA or the EMA.Citation55
A multicenter study conducted in Japan in 2010 assessed the efficacy, safety, and pharmacokinetics (PKs) of adalimumab in patients with BD refractory to conventional therapies. A total of 20 patients were selected to receive adalimumab 160 mg at the onset of the study, 80 mg at week 2 and 40 mg from weeks 4 to 52.Citation56 The rate of complete symptom remission was 20% at week 24 and was sustained until week 52. No adverse events were observed, and no deaths occurred. The study showed that adalimumab is efficacious and safe for the treatment of BD.
Ulcerative colitis
The use of adalimumab in patients with moderately to severely active UC and resistant to conventional treatment was approved by the FDA in September 2012Citation57 and by the EMA in April 2012.Citation58
The efficacy and safety of adalimumab were demonstrated in two randomized, double-blind, placebo-controlled clinical trials, ie, ULTRA-1Citation59 and ULTRA-2.Citation60 In ULTRA-1, 390 patients were randomized to receive adalimumab (160 mg/80 mg) or placebo at weeks 0 and 2; a third arm consisted of patients who received 80 mg/40 mg at weeks 0 and 2. At week 8, partial response was achieved by 54.6% of the participants and complete remission of symptoms in 18.5%, which were significantly different compared to placebo in both arms of the study.Citation59 In ULTRA-2, 494 patients with UC were randomized to receive adalimumab 160 mg at week 0, 80 mg at week 2, and 40 mg every other week until the end of the study (52 weeks). At 8 weeks, ∼50.4% of the adalimumab-treated patients responded to treatment, while 16.5% achieved complete clinical remission. At week 52, ∼34.6% of the cases maintained the response to treatment and 17.3% maintained clinical remission.Citation60
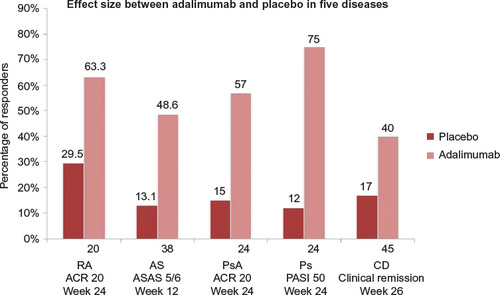
Following the initial approval of adalimumab for use in RA, the FDA and EMA approvals for other diseases described in the current article took place over more than a decade. Among the diseases mentioned in this article, it seems that the largest effect size corresponds to psoriasis, which may be the best sensitivity model for comparability studies of biosimilar candidates. shows different effect sizes of adalimumab.
Figure 1 Comparison of effect sizes between the efficacy of treatment with adalimumab and placebo in different diseases
Pharmacodynamics (PDs) and PKs
Adalimumab is a monoclonal antibody composed of variable IgG1 light and heavy chains (constant kappa regions).Citation61 It is fully humanized, which means that it does not have any nonhuman component or artificially fused peptide sequences.Citation62,Citation63 It binds to TNF-α, blocking its interaction with the membrane receptors p55 and p75.Citation64 Adalimumab has affinity and specificity for sTNF-α only, and not for lymphotoxins.Citation43
Studies conducted in patients with RA showed that adalimumab is able to induce significant reductions in the acute-phase reactant levels (such as CRP and fibrinogen), erythrocyte sedimentation rate, concentration of some interleukins (IL-1, IL-6, IL-8), concentration of cartilage and synovial remodeling markers, the amount of macrophage colony-stimulating factors, and the concentration of adhesion molecules that account for leukocyte migration, such as intercellular adhesion molecule 1 and vascular cell adhesion molecule 1.Citation65,Citation66
Similarly to infliximab, adalimumab is also able to induce lysis of TNF-α-expressing cells in the presence of complement. It is believed that this function is the main mechanism of action of TNF-α blockers in the pathophysiology of CD.Citation66–Citation69 Studies performed in animals showed that in the presence of adalimumab, activated mononuclear cells (monocytes with tTNF receptors and T lymphocytes) lead to apoptosis via the caspase pathway.Citation68,Citation69 In addition, in vitro studies showed that adalimumab influences the production of ILs by monocytes, resulting in reduced concentrations of IL-10 and IL-12, which inhibit the production of interferon-g.
The absolute number and density of epidermal Langerhans cells are reduced in patients with untreated psoriasis compared to patients without the disease. It is believed that Langerhans cells might have anti-inflammatory properties and that they are involved in the physiological differentiation of keratinocytes. Treatment with adalimumab restores the density of the epidermal Langerhans cells, and it is believed that this function is the main mechanism of action of this drug in plaque psoriasis.Citation66
Data on PKs of adalimumab in healthy volunteers and patients with RA have been published.Citation70,Citation71 The recommended dose of adalimumab in RA is 40 mg per subcutaneous route every other week.Citation72 In psoriasis, the initial dose is 80 mg per subcutaneous route, followed by 40 mg every other week starting 1 week after the initial dose.Citation73
The maximum serum concentration (Cmax) of the drug is 4.7±1.6 mg/mL; the concentration in the synovial fluid is 31%–96% of the serum concentration.Citation74 The time to attain Cmax is 131±56 hours. The bioavailability of adalimumab is 64%.Citation75 The drug elimination increases in the presence of AAA and is reduced in individuals older than 40 years. The distribution volume (Vd) is 4.7-6 L. The drug half-life (t1/2) is 2 weeks (10–20 days).Citation76
Immunogenicity, pharmacovigilance, and safety: Not all patients exhibit favorable responses to anti-TNF-α antibodies, exhibited as either primary (no response since the beginning of treatment) or secondary (loss of efficacy during treatment) failure. Immunogenicity has been described as an important mechanism in secondary failure, whereby AAA is produced, which might neutralize the therapeutic action of the drug.
Studies such as M06-827 and M02-433 found the development of AAA in 3.9% of the patients with UC and 2.6% of the patients with CD, respectively.Citation42 Production of AAA might be related to specific characteristics of patients, such as their genetic basis, comorbidities, and immunomodulatory therapy used concomitantly to adalimumab.Citation77
Formation of AAA reduces the drug bioavailability.Citation78 AAA is associated with reduced clinical response in patients with RA and PsA.Citation79 In patients with RA, combination with MTX seems to increase the available fraction of adalimumab, which suggests that MTX reduces AAA production in a dose-dependent manner.Citation80,Citation81 This association has not yet been confirmed in patients with PsA.Citation82 Other DMARDs do not seem to influence the potentiating effects of adalimumab and MTX in combination and might even be used in combination with adalimumab in the absence of MTX. The effect of the latter on the adalimumab concentration is lower, but is nonetheless superior to adalimumab monotherapy.Citation78,Citation79
In a prospective multicenter study, Jani et alCitation83 showed that the presence of AAA is associated with poorer response in RA. Thus, measurement of AAA might be useful to predict the response to long-term treatment with adalimumab and to establish the need to increase the dose of the drug or to change the therapeutic regimen.Citation83 Measurement of adalimumab and AAA in patients with SA seems to produce similar results compared to those of the study on RA.Citation77
One study assessed the cost–benefit relationship of individualized treatment of RA according to the measured serum drug concentration and showed that this strategy might be used in clinical practice.Citation84
According to the World Health Organization (WHO), pharmacovigilance is the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems.Citation85 The legislation in force in the EU was established in July 2012, and with it, EMA formulated a new set of pharmacovigilance guidelines in collaboration with the FDA that allow for more systematic and unified exchange of information on drug safety.Citation86,Citation87
Several programs were established based on the aforementioned guidelines, such as CARRA Consolidated Safety Registry, MyHumira, Alberta Biologics Pharmacosurveillance Program, and Adverse Event Reporting System.Citation88–Citation91
During more than 10 years of commercialization, clinical trials with long-term follow-up and recording of adverse events showed that adalimumab is associated with adverse effects, including severe infections, reactivation of the hepatitis B virus, allergic reactions, myocardial infarction, and autoimmune phenomena such as lupus-like syndrome, psoriasis, and neoplasms.Citation92,Citation93
Recent safety and efficacy studies showed that the presence of AAA might influence the efficacy of adalimumab, in addition to being correlated with autoimmune phenomena such as lupus-like syndrome.Citation94,Citation95 Pharmacovigilance is crucial in the case of biosimilars, because the process for their manufacture is different to that of the reference products. In addition, many adverse events are detected after long-term use of a given drug in large populations of patients. Thus, both prescribers and the pharmaceutical industry must be aware of the importance of post-market surveillance of biologicals and biosimilars.Citation96 A frequent doubt, particularly in emerging countries, is whether the plans for risk management of biosimilar manufacturers will be as rigorous as those required from manufacturers of innovative biologicals.
Manufacture of the reference product
In the first step of human monoclonal antibody production, several DNA sequences corresponding to the antibody variable fraction (Fv) heavy and light chains are inserted into phages to produce the desired molecule.Citation97 Next, phages with different genetic sequences undergo biopanning to select those with the highest affinity and specificity for TNF-α.Citation98,Citation99 The last step consists of guided selection, in which chains with high affinity and specificity for TNF-α are randomly combined with heavy and light chains in the Fv of anti-TNF-α monoclonal antibodies obtained from immunized mice, known as MAK195.Citation100 The result is a fully human antibody, but with the same affinity and binding properties as MAK195.Citation100
The molecule that results from guided selection, with more refined affinity and specificity, is known as D2E7.Citation97 The obtained Fv is added to the antibody constant fraction, and this gene sequence is introduced into Chinese hamster ovary (CHO) cells. The antibodies produced by CHO cells contain mainly oligosaccharides that terminate in Gal residues in position 0, 1, or 2. This glycosylation profile is very similar to the human profile, and for this reason, these cells are widely used for monoclonal antibody production,Citation101,Citation102 as one of the main causes of the immunogenicity of biologicals is thus avoided.Citation103
Following the insertion of the gene sequence in CHO cells, the more productive clone is selected for the master cell bank. The latter undergoes the upstream process, stages of fermentation, and cell propagation.Citation104 At this stage, the cells are transferred to bioreactors, which increase the cell density; this process is optimized through addition of production medium.Citation105 In the preclinical stage, adalimumab was produced in a single bioreactor with 1,000 L of cell culture; in just 9 years, it increased to two 6,000 L bioreactors that could yield 100 kg of active substance per year.Citation101
Next, the downstream process is performed, which consists of protein retrieval and purification of the product through several chromatography steps, treatment at low pH, and nanofiltration to inactivate and remove the virus so that the active substance is obtained.Citation104 Adalimumab is present in three active isoforms, which correspond to molecules carrying two, one, or no C-terminal lysines. The three components account for 85% of the material. Less than 15% of the active substance corresponds to acid groups, resulting mainly from fermentation.Citation9
The final steps include preparation of the pharmaceutical forms, packaging, and dosage to obtain the drug that will be commercialized.Citation104 Eighteen changes have already been made in the process of manufacture of adalimumab since it was first approved, which are somewhat welcome because they are often associated with improvement in the drug’s safety and efficacy.Citation5
From the analytic point of view, biologicals will always differ as to their impurity levels and glycosylation profiles, among other characteristics. The techniques available up to the current time are clearly limited as to their capacity to fully analyze such differences. In any case, the more sensitive the techniques are, the higher the odds of demonstrating differences between biosimilars and their reference products. A set of critical quality attributes should be established for each reference product, as should the acceptable margin of differences in such attributes between reference products and biosimilars.Citation106 In addition, it should be established which critical quality attributes most affect the efficacy and safety of each molecule. The current uncertainties as to the similarity relative to the aforementioned set of attributes might make regulatory agencies require more information from the manufacturers of a given biosimilar in the future, to check the extrapolation of indications.
Regulatory aspects
The EU formulated guidelines for the development of biosimilars of monoclonal antibodies in 2012, with special emphasis on clinical and nonclinical development.Citation107 CTP-13, the first biosimilar monoclonal antibody, produced by Celltrion company (Celltrion, Incheon, South Korea), was approved in 2014 by the EMA and is currently sold in >60 countries.Citation108
The FDA published a preliminary guide for a revision of the development and regulation of biosimilars. Although no definitive regulation has yet been established, Sandoz’s filgrastim (Zarzio®) was the first biosimilar approved in the US based on this preliminary legislation.Citation109
In India, biosimilars are known as “similar biologicals”, according to the guides formulated by the CDSCO, a branch of the Ministry of Health and Family Welfare, Government of India. Together with the Ministry of Health and Family Welfare, CDSCO established specific guidelines in 2012 listing the regulatory requirements for biosimilars to be approved for commercialization.Citation110
Considering the complexity of adalimumab and the limitations exhibited by the analytical techniques to establish whether a biosimilar is in fact an identical copy of its reference product, comparable clinical efficacy and safety must be demonstrated. To test the similarity between products, manufacturers must first perform the full physicochemical and biological characterization of biosimilars in comparison to the corresponding reference products. In addition, binding to cell receptors is assessed based on trials and studies conducted in animals, which further include the evaluation of PDs and toxicity. The methods used to establish the comparability between biosimilars and the corresponding reference products must be sufficiently selective and specific to be able to detect differences. The importance of such differences can only be established in preclinical and clinical studies. To ensure that any biosimilar entering the market is similar in efficacy and clinical safety to its reference product, the regulatory agencies established appropriate procedures for approval. Clinical PK and PD studies are mandatory and should be performed as either an independent phase or the initial stage of Phase III studies. In those studies, the route of administration and dose of the biosimilar must be the same as those of its reference product. Trials must necessarily contribute to detect differences between biosimilars and their reference products whenever they are present. Equivalence studies (those that require upper and lower comparative limits) are the preferred option to compare data. Non-inferiority tests might be used under certain circumstances, again, when they are justified. The margins of non-inferiority/equivalence must be previously specified and justified and then communicated to regulatory agencies based on their clinical relevance. Differences in the effects of treatment should be acceptable to the medical community and should not have any negative impacts on patient treatment.Citation111
The indications of the use of biosimilars should certainly reflect the results of clinical trials demonstrating their efficacy and safety. Although additional indications might be extrapolated according to the current guidelines (WHO), determining whether biosimilars are adequate to groups of related diseases (eg, inflammatory diseases) is a complex matter. The fact that a biosimilar meets the indications for a certain disease is no reason for it to be rated safe and efficacious for other indications just because they fall within the scope of its reference product. Therefore, the scientific evidence supporting extrapolations should be systematically and thoroughly evaluated.Citation112
Adalimumab biosimilars
Several companies are developing candidates for biosimilars of monoclonal antibodies and fusion proteins, the main focus falling on six reference products with substantial global sales, ie, adalimumab, bevacizumab, etanercept, infliximab, rituximab, and trastuzumab.Citation113
There are a few reports in clinical trial databases on the development of candidates to adalimumab biosimilars. A large part of the information on clinical studies is recorded and indexed on the website Clinicaltrials.gov, US National Institutes of Health. Trials relative to the molecules listed in were registered on that website up to the time this article was written.
Table 1 Status of biosimilars in development
The American multinational biopharmaceutical company Amgen is currently developing several biosimilar candidates and expects to launch five of them between 2017 and 2019.Citation114 According to the EU Clinical Trials Register, Amgen initiated a Phase III clinical trial of an adalimumab biosimilar candidate in patients with severe RA (ABP 501). Positive results were recently communicated that demonstrate equivalence to Humira® relative to efficacy, comparative safety, immunogenicity, and effectiveness. A multicenter, randomized, double-blind, active-controlled trial with parallel group design was conducted in 526 patients aged 18 years and older.Citation115,Citation116
The safety and PKs of Boehringer Ingelheim’s biosimilar candidate, BI695501, have already been investigated. The Phase I study, including 193 male participants aged 18–55 years, was completed in July 2012.Citation117,Citation118 The randomized, open-label study with parallel-arm design included an active comparator arm that tested the biosimilar candidate in a single dose. The study population was allocated into three groups: two were given Humira® and the third the biosimilar candidate.Citation119 The drug is currently undergoing Phase III studies.
Coherus and Pfizer, Inc. announced that their adalimumab biosimilar candidates, CHS-1420Citation120,Citation121 and PF-06410293,Citation122 respectively, achieved the primary end point of PK similarity relative to the reference product in clinical trials conducted in healthy individuals.
Sandoz, Novartis International AG’ division of generic drugs, started a Phase III study of its biosimilar candidate, GP2017.Citation123,Citation124 The multicenter, randomized, double-blind study is designed to demonstrate the candidate’s similarity, efficacy equivalence, and immunogenicity relative to its reference product (adalimumab) in patients with moderate to severe plaque psoriasis.Citation125
SB5, Samsung Bioepis’ Humira® biosimilar candidate, is currently being evaluated in a Phase III study. The randomized, double-blind study with parallel group design has aims to evaluate the safety, efficacy, PKs, immunogenicity, and tolerability of SB5 compared to Humira® in patients from Lithuania and Poland with moderate to severe RA.Citation126 Another adalimumab biosimilar candidate currently undergoing a Phase III clinical study is FKB327, by Fujifilm Kyowa Kirin Biologics Co. The randomized, blind, active-controlled study is designed to compare the biosimilar candidate efficacy and safety against Humira® in patients with moderate to severe inadequately controlled RA despite MTX use.Citation127
lists the laboratories that are currently developing adalimumab biosimilars worldwide.
Figure 2 Adalimumab biosimilars candidates.
Several adalimumab biosimilars are undergoing preclinical studies aiming at their molecular characterization and evaluation in in vitro and in vivo models that are summarized in .
Table 2 Biopharmaceuticals with similarity to adalimumab in preclinical phase
On December 9, 2014, in India, Cadila Healthcare Ltd. launched what it called the first adalimumab biosimilar, ZRC-3197.Citation128 This molecule was produced in genetically manipulated CHO cells containing adalimumab heavy and light chain genes (DE27). ZRC-3197 had primary and secondary structures similar to those of adalimumab and similar levels of purity and heterogeneity. The product was approved by the Indian drug control agency and is sold under the name Exemptia® for the treatment of RA, JIA, PsA, and AS (CTRI/2013/10/004040).Citation129 The corresponding clinical trial was conducted in India following the CDSCO (Ministry of Health and Family Welfare, Government of India) guidelines. According to the Indian regulatory agency, it followed the guidelines of the International Conference on Harmonization-Good Clinical Practice. The study consisted of a head-to-head comparison of ZRC-3197 and Humira®. According to the data available on the website of the Indian regulatory agency, the biosimilar exhibited high levels of similarity relative to efficacy, safety, and tolerability in patients with RA compared to the reference product. According to the Clinical Trial Registry of the National Institute of Medical Statistics of India, 120 patients were recruited for this Phase III study. The primary end points were PDs (efficacy) of adalimumab (ZRC-3197) and adalimumab in patients with RA compared to baseline, and the aim was to establish the proportion of patients who attained ACR20 in both groups on day 84. However, the data do not allow establishing whether the study was an equivalence or a non-inferiority study nor do they determine what margins the manufacturers and investigators defined for the assessment of comparative effects between the molecules. This information is essential to establish whether the sample size was adequate. These shortcomings notwithstanding, the product was extrapolated to all the indications of adalimumab. In addition, there is disagreement among authors as to whether patients with RA represent the most sensitive population or the best model for extrapolation of anti-TNF biosimilars to other diseases.Citation130–Citation133
Some authors believe that the product’s approval was not based on a regulatory process as rigorous as that required by the EU for clinical comparisons between biosimilars and their reference products.
Discussion
Adalimumab is a major sales success among biologicals at the global level. It is still the greatest blockbuster among monoclonal antibodies. The date of its patent expiration paves the road for several potential biosimilars in various markets worldwide.
Overall, it is expected that biosimilars will play a crucial role in the reduction of health costs and will improve the access to drugs able to save the lives of many patients, being an alternative to high-cost brand-name biologicals. However, the biosimilars must exhibit the same levels of safety and quality of the reference products.Citation133–Citation137 It is evident that with the development of more rigorous international regulations for the demonstration of biosimilarity, a lower number of candidates will be approved for commercialization, particularly in the European and North American markets. In addition, the vast amount of data available for adalimumab, accumulated over more than 10 years of commercialization, makes it very difficult for biosimilar manufacturers to demonstrate to the medical community that the safety profiles of their products are similar to that of the reference product relative to all of its approved indications. Although it might be demonstrated that a product has analytical similarity to its reference product, ie, similar biological, physical–chemical, and in vitro characteristics, which represent the basis for the extrapolation of indications applied by most regulatory agencies, a concern remains as to whether its comparative safety might be guaranteed based on a short-term clinical study with patients with a single disease model. It is frequently asserted that low-sensitivity clinical studies are less relevant than analytical tests for demonstrations of biosimilarity. When a biosimilar exhibits certain analytical characteristics, the clinical data are unlikely to detect deficiencies in efficacy and safety. However, clinically significant differences, especially in immunogenicity, detected in clinical trials seem to be more important than possible differences based on analytical and preclinical studies alone.
In contrast, any differences found either in vitro or in human beings during the development of biosimilars are sources of additional residual uncertainty. Thus, they might call for additional tests or eventually lead to decisions against complete or partial extrapolation.
The distribution across and penetration of monoclonal antibodies in the various tissues depends on certain PK properties, especially glycosylation. Variations in glycosylation patterns may modify the PK profile, influencing the biological activity of these molecules and resulting in different concentrations of their active principle in the main sites of action of the various indications.Citation138 Some evidence indicates that although the therapeutic target is the same as what is typical in some rheumatic conditions and CD, the pathophysiologic mechanisms and sites of action are quite different.Citation139,Citation140
From the clinical point of view, manufacturers of biologicals or biosimilars must make a continuous effort to develop more sensitive clinical tests to ensure that the effect sizes are very similar to those of the reference products relative to their various indications. In addition, to the maximum of the available scientific possibilities, they should include biomarkers based on the mechanisms of action of specific monoclonal antibodies in given stages of progression of the targeted diseases.
Comparison of the rates of placebo-adjusted responses in clinical trials of six indications of adalimumab in adults showed that RA was associated with one of the lowest effect sizes of treatment. Therefore, from the clinical point of view, RA seems to be a less sensitive model for detecting differences in efficacy between biosimilars and their reference products compared to other indications associated with greater treatment effect size, such as psoriasis and PsA.
Unfortunately, there is a lack of validated biomarkers to assess the response to and efficacy of anti-TNF agents. Neither the acute-phase proteins, such as CRP, nor anti-cyclic citrullinated peptide antibody or rheumatoid factor levels are clearly correlated with the effect of biologicals in the treatment of RA.
The FDA and EMA abbreviated paths represent a step forward in the demonstration of the biosimilarity of the copies relative to their reference products. The latest recommendations formulated by the EMA indicate that biosimilar candidates must include data supporting their extrapolation when there is evidence demonstrating different mechanisms of action in a given alleged indication.Citation107
The expectation of the international community is that the comparative efficacy and safety of many of the biosimilar candidates described in this article and many future candidates will be demonstrated and that those drugs will enter many markets around the world, thus promoting that which seems to be the most significant attribute of biosimilars, ie, increasing the access of patients to treatment with adalimumab thanks to the reduction of its cost.
Acknowledgments
The authors thank the support of Edumed Biotech staff for helping to review this article. This work was not produced with any financial support.
Disclosure
Valderílio Feijó Azevedo is a speaker for Pfizer, Inc., AbbVie Inc., Hoffman-La Roche Ltd., BMS, Celltrion, Janssen Pharmaceutica, Sanofi, Novartis International AG, AstraZeneca plc, and Union Chimique Belge. Doctor Valderílio Feijó Azevedo has produced graphic materials to AbbVie Inc., Janssen Pharmaceutica, BMS, Pfizer, Inc., and Novartis International AG. He also is part of the advisory board of AbbVie Inc., Janssen Pharmaceutica, BMS, Pfizer, Inc., Merck Serono, and AstraZeneca plc. Ludmila Della Coletta Troiano, Natalia Bassalobre Galli, Alais Kleinfelder, Nathan M Catolino, and Paulo Cesar Urbano Martins report no conflicts of interest in this work.
References
- Lis K, Kuzawińska O, Bałkowiec-Iskra E. State of the art paper tumor necrosis factor inhibitors – state of knowledge. Arch Med Sci. 2014;6:1175–1185.
- Agarwal SK. Biologic agents in rheumatoid arthritis: an update for managed care professionals. J Manag Care Pharm. 2011;17(9 Suppl B):14–18.
- Ma X, Xu S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep. 2013;1(2):177–184.
- Dörner T, Strand V, Castañeda-Hernández G, Ferraccioli G, Isaacs JD, Kvien TK, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–328.
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72(3):315–318.
- Balzola F, Cullen G, Hoentjen F, Ho GT, Russell R. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Inflamm Bowel Dis Monit. 2013;13:162.
- Abbot Imunology [webpage on the Internet]. The Humira Story or “How D2E7 Became a Star” 2013. Available from: http://www.phagedisplay.org/pdfs/Kamen_TheHumirastory.pdf. Accessed April 2015.
- FiercePharma [webpage on the Internet]. Humira biosimilars will gut AbbVie’s sales in 3 years 2015. Available from: http://www.fiercepharma.com/story/humira-biosimilars-will-gut-abbvies-sales-3-years-analyst-says/2015-02-10. Accessed March 2015.
- European Medicines Agency [webpage on the Internet]. Scientific discussion for the approval of Humira. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000481/WC500050867.pdf. Accessed April 2, 2015.
- AbbVie [webpage on the Internet]. A new beginning. Available from: http://www.abbvie.com/about-us/who-we-are.html. Accessed February 2015.
- Möller A, Emling F, Blohm D, Schlick E, Schollmeier K. Monoclonal antibodies to human tumor necrosis factor alpha: in vitro and in vivo application. Cytokine. 1990;2(3):162–169.
- Keffer J, Probert L, Caziaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025–4031.
- Kempeni J. Preliminary results of early clinical trials with the fully human anti-TNFalpha monoclonal antibody D2E7. Ann Rheum Dis. 1999;58(Suppl 1):170–172.
- Kazuo I [webpage on the Internet]. United States Patent and Trademark Office. Available from: http://www.uspto.gov/sites/default/files/web/offices/pac/dapp/opla/term/certs/6090382.pdf. Accessed February 2015.
- Siddiqui Z [webpage on the Internet]. India’s Cadila launches first cheaper copy of world’s top-selling drug 2014. Available from: http://www.reuters.com/article/2014/12/09/us-cadila-health-humira-india-idUSKBN0JN0X820141209. Accessed February 2015.
- U.S. Food and Drug Administration [webpage on the Internet]. Adalimumab Product Approval Information – Licensing Action. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm080610.htm. Accessed February 2015.
- European Medicines Agency [webpage on the Internet]. Anexo I – Resumo das Características do Medicamento: Humira 2010:1–29. Available from: http://www.ema.europa.eu/docs/pt_PT/document_library/EPAR_-_Product_Information/human/000481/WC500050870.pdf. Accessed April 2015.
- Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45.
- Van de Putte LB, Atkins C, Malaise M, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508–516.
- Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled. Arthritis Rheum. 2004;50(5):1400–1411.
- Furst DE, Schiff MH, Fleischmann RM, et al. Adalimumab, a fully human anti tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J Rheumatol. 2003;30(12):2563–2571.
- Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previo. Arthritis Rheum. 2006;54(1):26–37.
- U S Food and Drug Administration [webpage on the Internet]. FDA approves Humira to treat ulcerative colitis. FDA News Release 2012. Available from: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm321650.htm. Accessed January 2015.
- Mease PJ, Gladman DD, Ritchlin CT, et al; Adalimumab Effectiveness in Psoriatic Arthritis Trial Study Group. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52(10):3279–3289.
- Gladman DD, Mease PJ, Choy EH, Ritchlin CT, Perdok RJ, Sasso EH. Risk factors for radiographic progression in psoriatic arthritis: subanalysis of the randomized controlled trial ADEPT. Arthritis Res Ther. 2010;12(3):R113.
- Genovese MC, Mease PJ, Thomson GT, et al; M02-570 Study Group. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol. 2007;34(5):1040–1050.
- Gordon KB, Langley RG, Leonardi C, et al. Clinical response to adalimumab treatment in patients with moderate to severe psoriasis: double-blind, randomized controlled trial and open-label extension study. J Am Acad Dermatol. 2006;55(4):598–606.
- Menter A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol. 2008;58(1):106–115.
- Saurat JH, Stingl G, Dubertret L, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION). Br J Dermatol. 2008;158(3):558–566.
- Leonardi C, Langley RG, Papp K, et al. Adalimumab for treatment of moderate to severe chronic plaque psoriasis of the hands and feet: efficacy and safety results from REACH, a randomized, placebo-controlled, double-blind trial. Arch Dermatol. 2011;147(4):429–436.
- Bissonnette R, Bolduc C, Poulin Y, Guenther L, Lynde CW, Maari C. Efficacy and safety of adalimumab in patients with plaque psoriasis who have shown an unsatisfactory response to etanercept. J Am Acad Dermatol. 2010;63(2):228–234.
- Van Lümig PP, Lecluse LL, Driessen RJ, et al. Switching from etanercept to adalimumab is effective and safe: Results in 30 patients with psoriasis with primary failure, secondary failure or intolerance to etanercept. Br J Dermatol. 2010;163(4):838–846.
- Gorman JD, Sack KE, Davis JC. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. 2002;346(18):1349–1356.
- Brandt J, Khariouzov A, Listing J, et al. Six-month results of a double-blind, placebo-controlled trial of etanercept treatment in patients with active ankylosing spondylitis. Arthritis Rheum. 2003;48(6):1667–1675.
- Braun J, Brandt J, Listing J, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. 2002;359(9313):1187–1193.
- Braun J, Brandt J, Listing J, et al. Long-term efficacy and safety of infliximab in the treatment of ankylosing spondylitis: an open, observational, extension study of a three-month, randomized, placebo-controlled trial. Arthritis Rheum. 2003;48(8):2224–2233.
- Van Der Heijde D, Dijkmans B, Geusens P, et al; Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy Study Group. Efficacy and safety of infliximab in patients with ankylosing spondylitis: Results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum. 2005;52(2):582–591.
- Haibel H, Rudwaleit M, Listing J, et al. Efficacy of adalimumab in the treatment of axial spondyloarthritis without radiographically defined sacroiliitis: results of a twelve-week randomized, double-blind, placebo-controlled trial followed by an open-label extension up to week fifty-two. Arthritis Rheum. 2008;58(7):1981–1991.
- Lambert RGW, Salonen D, Rahman P, et al. Adalimumab significantly reduces both spinal and sacroiliac joint inflammation in patients with ankylosing spondylitis: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2007;56(12):4005–4014.
- Van Der Heijde D, Kivitz A, Schiff MH, et al; ATLAS Study Group. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: Results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006;54(7):2136–2146.
- U S Food and Drug Administration [webpage on the Internet]. FDA Approves New Treatment For Crohn’s Disease. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108852.htm. Accessed April 2015.
- European Medicines Agency [webpage on the Internet]. Assessment report: Humira 2012;44:3–65. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000481/WC500133080.pdf. Accessed April 2015.
- Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: The CLASSIC-I trial. Gastroenterology. 2006;130(2):323–332.
- Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology. 2007;132(1):52.
- Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146(12):829–838.
- Hyams JS, Markowitz J, Baldassano RN, et al. Safety and efficacy of adalimumab for moderate to severe Crohn’s disease in children. Gastroenterology. 2012;143(2):365–374.e2.
- Rosenbach Y, Hartman C, Shapiro R, Hirsch A, Avitzur Y, Shamir R. Adalimumab treatment in children with refractory Crohn’s disease. Dig Dis Sci. 2010;55(3):747–753.
- AbbVie’s. HUMIRA® (adalimumab) receives U.S. FDA approval for extension of polyarticular juvenile idiopathic arthritis indication to patients ages 2 and older 2014. Available from: http://www.prnewswire.com/news-releases/abbvies-humira-adalimumab-receives-us-fda-approval-for-extension-of-polyarticular-juvenile-idiopathic-arthritis-indication-to-patients-ages-2-and-older-278214991.html. Accessed February 2015.
- Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med. 2008;359(8):810–820.
- Kingsbury DJ, Bader-Meunier B, Patel G, Arora V, Kalabic J, Kupper H. Safety, effectiveness, and pharmacokinetics of adalimumab in children with polyarticular juvenile idiopathic arthritis aged 2 to 4 years. Clin Rheumatol. 2014;33(10):1433–1441.
- Edelsten C, Reddy MA, Stanford MR, Graham EM. Visual loss associated with pediatric uveitis in English primary and referral centers. Am J Ophthalmol. 2003;135(5):676–680.
- Smith JA, Mackensen F, Sen HN, et al. Epidemiology and course of disease in childhood uveitis. Ophthalmology. 2009;116(8):1544–1551.
- De Boer J, Wulffraat N, Rothova A. Visual loss in uveitis of childhood. Br J Ophthalmol. 2003;87(7):879–884.
- Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU). Prog Retin Eye Res. 2004;23(6):617–637.
- AbbVie GK [webpage on the Internet]. AbbVie and Eisai Announce HUMIRA® Pre-filled Syringe Has Received Approval for the Treatment of Intestinal Behçet’s Disease in Japan 2013. Available from: http://www.eisai.com/news/enews201330pdf.pdf. Accessed April 2015.
- Tanida S, Inoue N, Kobayashi K, et al. Adalimumab for the treatment of Japanese patients with intestinal Behçet’s diseases. Clin Gastroenterol Hepatol. 2014;13(5):940–948.
- U S Food and Drug Administration [webpage on the Internet]. FDA approves Humira to treat ulcerative colitis. FDA News Release 2012. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm321650.htm. Accessed April 2015.
- European Medicines Agency EMA [webpage on the Internet]. Humira: EPAR summary for the public 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000481/WC500050865.pdf. Accessed April 2015.
- Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780–787.
- Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257–265.e1–.e3.
- Bang LM, Keating GM. Adalimumab: a review of its use in rheumatoid arthritis. BioDrugs. 2004;18(2):121–139.
- Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45.
- Weisman MH, Moreland LW, Furst DE, et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther. 2003;25(6):1700–1721.
- Warner B, Harris AW. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;143(1):e42; author reply e42. Epub 2012 May 24.
- Mease PJ. Adalimumab in the treatment of arthritis. Ther Clin Risk Manag. 2007;3:133–148.
- Vena GA, Cassano N. Drug focus: adalimumab in the treatment of moderate to severe psoriasis. Biologics. 2007;1(2):93–103.
- Cassinotti A, Ardizzone A, Porro GB. Adalimumab for the treatment of Crohn ’ s disease. Biologics. 2008;2(4):763–777.
- Shen C, Assche GV, Colpaert S, et al. Adalimumab induces apoptosis of human monocytes: a comparative study with infliximab and etanercept. Aliment Pharmacol Ther. 2005;21(3):251–258.
- Shen C, Van Assche G, Rutgeerts P, Ceuppens JL. Caspase activation and apoptosis induction by adalimumab: demonstration in vitro and in vivo in a chimeric mouse model. Inflamm Bowel Dis. 2006;12(1):22–28.
- Michel MH, Moreland LW, Furst DE, et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther. 2003;25(6):1700–1721.
- Nestorov I. Clinical pharmacokinetics of TNF antagonists: how do they differ? Semin Arthritis Rheum. 2005;34(5 Suppl 1):12–18.
- Machado MA, Maciel AA, Lemos LL, et al. Adalimumabe no tratamento da artrite reumatoide: uma revisão sistemática e metanálise de ensaios clínicos randomizados [Adalimumab in rheumatoid arthritis treatment: a systematic review and meta-analysis of randomized clinical trials]. Rev Bras Reumatol. 2013;53:419–430. Portuguese.
- Brunton L, Chabner B, Bruce AB, Knollman B. Goodman Gilman’s Pharmacol. Basis of Therapeutics. 12th ed. New York, NY: McGraw Hill Education; 2010.
- Granneman RG, Zhang Y, Noertersheuser PA, Velagapudi RB, Awni WM. Pharmacokinetic/pharmacodynamic (PK/PD) relationships of adalimumab (HUMIRATM) in rheumatoid arthritis (RA) patients during phase II/III clinical trials. In: ACR Annual Scientific Meeting; November 2003; Orlando. Poster 256.
- Velagapudi RB. The effect of methotrexate (MTX) coadministration on the pharmacokinetics (PK) of adalimumab (HUMIRATM) following a single intravenous injection to rheumatoid arthritis patients. In: ACR Annual Scientific Meeting; November 2003; Orlando. Poster 258.
- Chen DM, Gordon KB, Leonardi CM. Adalimumab efficacy and safety in patients with moderate to severe chronic plaque psoriasis: preliminary findings from a 12-week dose ranging trial. J Am Acad Dermatol. 2004;50:491.
- Kneepkens EL, Wei JC, Nurmohamed MT, et al. Immunogenicity, adalimumab levels and clinical response in ankylosing spondylitis patients during 24 weeks of follow-up. Ann Rheum Dis. 2013;74(2):396–401.
- Bartelds GM, Krieckaert CL, Nurmohamed MT, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468.
- Vogelzang EH, Kneepkens EL, Nurmohamed MT, et al. Anti-adalimumab antibodies and adalimumab concentrations in psoriatic arthritis; an association with disease activity at 28 and 52 weeks of follow-up. Ann Rheum Dis. 2014;73(12):2178–2182.
- Pouw MF, Krieckaert CL, Nurmohamed MT, et al. Key findings towards optimising adalimumab treatment: the concentration-effect curve. Ann Rheum Dis. 2015;74(3):513–518.
- Burmester G-R, Kivitz AJ, Kupper H, et al. Efficacy and safety of ascending methotrexate dose in combination with adalimumab: the randomised CONCERTO trial. Ann Rheum Dis. 2015;74(6):1037–1044.
- Mease PJ. Spondyloarthritis: is methotrexate effective in psoriatic arthritis? Nat Rev Rheumatol. 2012;8(5):251–252.
- Jani M, Chinoy H, Warrens R, Griffiths C, Barton A. Effect of immunogenicity on efficacy of long-term treatment of rheumatoid arthritis with adalimumab. Lancet. 2014;68:1–16.
- Krieckaert CL, Nair SC, Nurmohamed MT, et al. Personalised treatment using serum drug levels of adalimumab in patients with rheumatoid arthritis: an evaluation of costs and effects. Ann Rheum Dis. 2015;74(2):361–368.
- World Health Organization [webpage on the Internet]. Pharmacovigilance guidelines 2010. Available from: http://www.who.int/medicines/areas/quality_safety/safety_efficacy/S.AfricaDraftGuidelines.pdf. Accessed December 2014.
- European Medicines Agency [webpage on the Internet]. Guidelines on good pharmacovigilance practices 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500143294.pdf. Accessed April 2015.
- U S Food and Drug Administration [webpage on the Internet]. FDA and European Medicines Agency strengthen collaboration in pharmacovigilance area; 2014. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm386372.htm. Accessed December 2014.
- Lionetti G, Kimura Y, Schanberg LE, et al. Using registries to identify adverse events in rheumatic diseases. Pediatrics. 2013;132(5):e1384–e1394.
- Martin L, Barnabe C [webpage on the Internet]. 15 Years of Managing Rheumatoid Arthritis in Alberta with Biologic Therapies: Program Infrastructure, Efficacy, Safety and Health Economics. Available from: http://www.departmentofmedicine.com/rounds/presentations/2014/managing_arthritis_alberta.pdf. Accessed December 2014.
- Humira [webpage on the Internet]. My Humira. Available from: https://www.humira.com/global/starting-humira. Accessed December 2014.
- U S Food and Drug Administration [webpage on the Internet]. Adverse events redefining drug safety 2014. Available from: http://adverseevents.com/research.php. Accessed December 2014.
- Pediatric Advisory Committee [webpage on the Internet]. Pediatric Postmarketing Adverse Event review 2009:2–16. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/PediatricAdvisoryCommittee/UCM191899.pdf. Accessed December 2014.
- Mendes D, Alves C, Batel-Marques F. Safety profiles of adalimumab, etanercept and infliximab: a pharmacovigilance study using a measure of disproportionality in a database of spontaneously reported adverse events. J Clin Pharm Ther. 2014;39(3):307–313.
- U S Food and Drug Administration [webpage on the Internet]. FDA Approves Humira to treat ulcerative colitis. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm321650.htm. Accessed April 2015.
- Bartelds GM, Krieckaret CL, Nurmohamed MT, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468.
- Kumar R, Singh J. Biosimilar drugs: current status. Int J Appl Basic Med Res. 2014;4(2):63–66.
- Martínez de Lagrán Z, Pérez-Barrio S, Díaz-Pérez JL. Adalimumab : la molécula y el proceso de obtención. Actas Dermosifiliogr. 2008;99:3–9.
- Azzazy HM, Highsmith WE. Phage display technology: clinical applications and recent innovations. Clin Bio Chem. 2002;35(6):425–445.
- Hoogenboom HR, de Bruine AP, Hufton SE, Hoet RM, Arends JW, Roovers RC. Antibody phage display techno- logy and its applications. Immunotechnology. 1998;4(1):1–20.
- Vispo NS, Yaquelin P. Bacteriófagos: de la terapia con fagos a la biología combinatoria. Biotecnología aplicada. 2001;18:135–147.
- George A. HUMIRA manufacturing: challenges and the path taken. GAb Abstr. 2004;3:15–17.
- Raju ST. Glycosylation variations with expression systems and their impact on biological activity of therapeutic immunoglobulins. Bioprocess Int. 2003;1:44–51.
- Umana P, Jean-Mairet J, Bailey JE. Tetracycline-regulated overexpression of glycosyltransferases in Chinese hamster ovary cells. Biotechnol Bioeng. 1999;65(5):542–549.
- Pimentel V, Gomes R [webpage on the Internet]. O desafio de adensar a cadeia de P & D de medicamentos biotecnológicos no Brasil 2010. Available from: http://www.bndes.gov.br/SiteBNDES/export/sites/default/bndes_pt/Galerias/Arquivos/conhecimento/bnset/set3805.pdf. Accessed April 2015.
- Rodrigues ME, Costa AR, Henriques M, Azeredo J, Oliveira R. Technological progresses in monoclonal antibody production systems. Biotechnol Prog. 2009;26(2):332–351.
- Rathore AS, Reason AJ, Weiskopf A. Defining critical quality attributes for monoclonal antibody therapeutic products. Bio Pharm Int. 2014;27(7):34–43.
- European Medicines Agency [webpage on the Internet]. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed April 2015.
- Beck A, Riechert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–623.
- Burki TK. First biosimilar drug approved in the USA. Lancet Oncol. 2015;16:161.
- Central Drugs Standard Control Organization (CDSCO). Guidelines on Similar Biologics: Regulatory Requirements for Marketing Authorization in India; 2012. Available from: http://dbtbiosafety.nic.in/Files%5CCDSCO-DBTSimilarBiologicsfinal.pdf. Accessed May 2015.
- Azevedo VF. Biosimilars require scientifically reliable comparative clinical data. Rev Bras Reum. 2013;53(1):129–131.
- Lee H. Is extrapolation of the safety and efficacy data in one indication to another appropriate for biosimilars? AAPS J. 2014;16(1):22–26.
- Reichert JM. Antibodies to watch in 2015. MAbs. 2015;7(1):1–8.
- Amgen [webpage on the Internet]. Positive Top-Line Results From Phase 3 Study Evaluating The Efficacy And Safety Of Biosimilar Candidate ABP 501 Compared With Adalimumab In Patients With Moderate-To-Severe Plaque Psoriasis. Available from: http://www.amgen.com/media/media_pr_detail.jsp?releaseID=1975377. Accessed May 2015.
- GaBi Online [webpage on the Internet]. Amgen to start phase III trial for biosimilar adalimumab; 2013. Available from: http://gabionline.net/Biosimilars/News/Amgen-to-start-phase-III-trial-for-biosimilar-adalimumab. Accessed February 2015.
- Clinical Trials [webpage on the Internet]. Study to Compare Efficacy and Safety of ABP 501 and Adalimumab (Humira®) in Adults With Moderate to Severe Plaque Psoriasis 2014. Available from: http://clinicaltrials.gov/ct2/show/NCT01970488. Accessed March 2015.
- Clinical Trials [webpage on the Internet]. Pharmacokinetics and safety study of BI 695501 in healthy subjects. Available from: http://clinicaltrials.gov/ct2/show/NCT01505491?term=BI+695501&rank=1. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Boehringer Ingelheim completes biosimilar adalimumab trial 2012. Available from: http://www.gabionline.net/Biosimilars/Research/Boehringer-Ingelheim-completes-biosimilar-adalimumab-trial. Accessed February 2015.
- Boehringer Ingelheim [homepage on the Internet]. Available from: https://www.boehringer-ingelheim.com/research_development/drug_discovery/pipeline.html. Accessed February 2015.
- Clinical Trials [webpage on the Internet]. Pfizer to start trial for biosimilar adalimumab; 2013. Available from: www.gabionline.net/Biosimilars/News/Pfizer-to-start-trial-for-biosimilar-adalimumab. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Adalimumab biosimilar has comparable pharmacokinetics to Humira. Available from: http://gabionline.net/layout/set/print/Biosimilars/Research/Adalimumab-biosimilar-has-comparable-pharmacokinetics-to-Humira. Accessed February 2015.
- Clinical Trials [webpage on the Internet]. Study of PF-06410293 and adalimumab in healthy subjects (REFLECTIONS B538-01). Available from: http://clinicaltrials.gov/ct2/show/NCT01870986?term=to+PF-06410293&rank=1. Accessed February 2015.
- Sandoz [webpage on the Internet]. Sandoz begins Phase II clinical trials for biosimilar adalimumab. Available from: www.biosimilarnews.com/sandoz-begins-phase-iii-clinical-trial-for-biosimilar-adalimumab. Accessed February 2015.
- Clinical Trials [webpage on the Internet]. Study to Demonstrate Equivalent Efficacy and to Compare Safety of Biosimilar Adalimumab (GP2017) and Humira (ADACCESS). Available from: https://clinicaltrials.gov/ct2/show/NCT02016105?term=gp2017&rank=1. Accessed February 2015.
- Kronthaler U, Hexal AG, Silva A, Poetzl J, Seliskar M. Similar preclinical pharmacokinetics of the proposed biosimilar adalimumab GP2017 and originator adalimumab upon single and multiple administrations. J Am Acad Dermatol. 2014;70(5):190.
- Clinical Trials [webpage on the Internet]. A Study Comparing SB5 to Humira® in Subjects With Moderate to Severe Rheumatoid Arthritis Despite Methotrexate Therapy. Available from: http://www.clinicaltrials.gov/ct2/show/NCT02167139?term=biosimilar&recr=Open&no_unk=Y&rank=8. Accessed February 2015.
- Clinicaltrials.gov [webpage on the Internet]. A Study to Compare FKB327 Efficacy and Safety With Humira® in Rheumatoid Arthritis Patients (ARABESC). Available from: https://www.clinicaltrials.gov/ct2/show/NCT02260791. Accessed February 2015.
- BioSpectrum Asia [webpage on the Internet]. India firm launches biosimilar for autoimmune disease 2014. Available from: http://www.biospectrumasia.com/biospectrum/news/219784/india-firm-launches-biosimilar-autoimmune-disease. Accessed March 2015.
- GaBi Online [webpage on the Internet]. Adalimumab similar biologic launched in India. Available from: http://www.gabionline.net/layout/set/print/Biosimilars/News/Adalimumab-similar-biologic-launched-in-India. Accessed February 2015.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation. J Clin Pharmacol. 2015;55(Suppl 3):123–132.
- Feagan BG, Choquette D, Ghosh S, et al. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42(4):177–183.
- Ebbers HC. Biosimilars: in support of extrapolation of indications. J Crohns Colitis. 2014;8(5):431–435.
- Simoens S. Biosimilar medicines and cost-effectiveness. Clinicoecon Outcomes Res. 2011;3:29–36.
- Mendes de Abreu M, Strand V, Levy RA, Araujo DV. Putting the value into biosimilar decision making: the judgment value criteria. Autoimmun Rev. 2014;13(6):678–684.
- Blackstone EA, Fuhr JP. Innovation and competition: will biosimilars succeed? Biotechnol Healthc. 2012;9(1):24–27.
- Ventola CL. Biosimilars part 1: proposed regulatory criteria for FDA approval. P T. 2013;38(5):270–287.
- Blackstone EA, Fuhr JP. The economics of biosimilars. Am Heal Drug Benefits. 2013;6(8):469–478.
- Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs. 2010;24(1):9–21.
- Fiorino G, Girolomoni G, Lapadula G, et al. The use of biosimilars in immune-mediated disease: a joint Italian Society of Rheumatology (SIR), Italian Society of Dermatology (SIDeMaST), and Italian Group of Inflammatory Bowel Disease (IG-IBD) position paper. Autoimmun Rev. 2014;13(7):751–755.
- Azevedo VF, de Souza Meirelles E, Kochen Jde A, et al. Recommendations on the use of biosimilars by the Brazilian Society of Rheumatology, Brazilian Society of Dermatology, Brazilian Federation of Gastroenterology and Brazilian Study Group on Inflammatory Bowel Disease-Focus on clinical evaluation of monoclonal antibodies and fusion proteins used in the treatment of autoimmune diseases. Autoimmun Rev. 2015;14(9):769–773.
- GaBi Online [webpage on the Internet]. The latest biosimilars agreements; 2014. Available from: www.gabionline.net/Biosimilars/News/The-latest-biosimilars-agreements. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Fujifilm and Kyowa launch biosimilars joint venture 2014. Available from: www.gabionline.net/Biosimilars/News/Fujifilm-and-Kyowa-launch-biosimilars-joint-venture. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Baxter and Momenta make biosimilars deal 2014. Available from: www.gabionline.net/Biosimilars/News/Baxter-and-Momenta-make-biosimilars-deal. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Oncobiologics and Viropro make biosimilar deal 2014. Available from: www.gabionline.net/Biosimilars/News/Oncobiologics-and-Viropro-make-biosimilar-deal. Accessed February 2015.
- NOMURA [webpage on the Internet]. Korean Biopharmaceuticals. Coming of age: generics-centred players flourishing, preparing to go global. Available from: http://www.nomura.com/europe/resources/upload/Korean_biopharmaceuticals.pdf. Accessed February 2015.
- GaBi Online [webpage on the Internet]. Adalimumab biosimilar has comparable pharmacokinetics to Humira 2014. Available from: http://www.gabionline.net/layout/set/print/Biosimilars/Research/Adalimumab-biosimilar-has-comparable-pharmacokinetics-to-Humira. Accessed April 2015.
- Clinical Trials [webpage on the Internet]. A Study Of PF-06410293 (Adalimumab-Pfizer) And Adalimumab (Humira) In Healthy Subjects 2014. Available from: https://clinicaltrials.gov/ct2/show/NCT02237729. Accessed April 2015.
- Clinical Trials [webpage on the Internet]. A Study to Compare FKB327 Efficacy and Safety With Humira® in Rheumatoid Arthritis Patients 2014. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02260791. Accessed April 2015.