
Abstract
Oxidative stress, which can cause DNA damage, can both activate TNF-R1 directly in the absence of TNF stimulation and phosphorylate c-Abl, thus promoting its cytoplasmic translocation. Persistent cytoplasmic localization of c-Abl has been associated with cellular transformation. c-Abl phosphorylates OTULIN at tyrosine 56, thereby disrupting its relationship with LUBAC. OTULIN-released LUBAC interacts with SPATA2 and is recruited to the TNF-R1sc, facilitating SPATA2-CYLD interaction. All these interactions are required for the activation of IKKβ to stimulate NF-κB transcriptional activity following genotoxic stress. IKKβ also induces the critical phosphorylation of CYLD at serine 568 to increase its deubiquitinating (DUB) activity required for the termination of signaling cascades. Contrary to the widespread belief that CYLD is an absolute tumor suppressor, CYLD initiates and terminates NF-κB activity by alternately using its oncoprotein and tumor suppressor activities, respectively. If IKKβ fails to achieve the DUB activity-inducing phosphorylation at serine 568, CYLD would operate in a sustained mode of oncogenic activity. The resulting dysregulated NF-κB activation and other accompanying pathologies will disrupt cellular homeostasis in favor of transformation.
Plain Summary
•Oxidative stress can cause both DNA damage and ligand-free TNFR stimulation.
•DNA damage disrupts the OTULIN-LUBAC interaction and initiates NF-kB transcription via the LUBAC-SPATA2-CYLD-mediated IKKβ activation pathway.
•IKKβ also terminates the NF-κB signaling by activating the deubiquitinase function of CYLD through Serine 568 phosphorylation.
•Following oxidative DNA damage, failure to phosphorylate CYLD at serine 568 could initiate a myriad of signaling that can trigger a series of feedback cycles that can force uncontrolled cell proliferation.
Introduction
There is extensive evidence indicating that reactive oxygen species (ROS)-induced DNA damage accumulation leads to genomic instability, which is important for malignant transformation.Citation1 DNA damage can also be caused by a variety of factors other than oxidative stress, including exogenous agents such as ultraviolet light and ionizing radiation, as well as endogenous triggers such as errors in DNA replication (replication stress). Unrepaired damage to DNA can eventually cause cell death if the damage is too extensive. In case of the damage is tolerable, it can be retained and potentially transmitted to future progeny if the cell maintains its ability to divide.Citation1,Citation2 As evidence, the vast majority of human cancers contain pathological changes that exhibit persistent DNA damage and genomic instability.Citation1 Several characteristic alterations occur during the transformation of a cell, including the capacity to proliferate autonomously, escape apoptosis, invade surrounding tissues, and metastasize to distant sites. Since many of these properties accompany the dysregulated activation of nuclear factor-kappa B (NF-kB), a transcription factor is accepted as one of the leading mediators capable of inducing cell transformation. Consistently, NF-κB is aberrantly activated in tumor cells to promote the cellular advantage in survival and proliferation.Citation3
Nuclear Processes Due to Oxidative DNA Damage
The genome of eukaryotic cells is under continuous attack from a variety of endogenous and exogenous DNA-damaging agents, which lead to many types of DNA lesions. ROS are not known to directly cause DNA double-strand breaks (DSBs). Instead, DSBs could be generated if two single-strand breaks (SSBs) oppose each other on complementary strands during an intermediate step in a repair process.Citation4 The major mechanism that cells use to repair oxidative damage lesions is base excision repair (BER). In other words, many, but not all, of the DNA lesions repaired by BER, are products of ROS attack. DNA polymerase-β (Pol-β) is a key enzyme implicated in the BER pathway, where it mainly repairs SSBs.Citation5,Citation6
Despite its error-prone nature, non-homologous end-joining (NHEJ) is the predominant form of DSB repair in human somatic cells.Citation7 Ku70 and Ku80, important components of NHEJ, likely influence BER, independent of the Ku heterodimer. Ku80 actively repairs the base lesion, most probably through facilitating the accumulation of other BER components. Remarkably, if Ku80 has no impact, Ku70 interferes with the repair of these lesions.Citation4 On the other hand, Pol-β of the BER pathway physically interacts with Ku70 of the NHEJ pathway, and this association is enhanced by DNA damage. When SSB occurs, Ku70 binds to Pol-β and promotes its polymerase activity, thus accelerating BER. Reciprocally, when DSB occur, Pol-β binds to Ku70 and participates in NHEJ to promote repair of the DSB lesions. Thus, two-way crosstalk between Ku70 and Pol-β modulates DNA repair through BER and NHEJ.Citation7
The ataxia-telangiectasia-mutated (ATM) is activated by oxidative stress. Interestingly, oxidation of ATM directly induces ATM activation in the absence of DNA break and the Mre11-Rad50-Nbs1 (MRN) complex.Citation8 ATM phosphorylates histone H2AX at serine 139 (Ser139) into γ-H2AX around the DSB site. The activated ATM also phosphorylates the scaffolding subunit of PP2A, PR65, at Ser401, leading to the disassociation of the ATM-PR65 complex.Citation9 Furthermore, Ser401 phosphorylation of PR65 could result in the dissociation of the holoenzyme and causes its translocation to the cytoplasm, leaving the catalytic domain of PP2A, PP2A(C), in the nucleusCitation8,Citation9 (). Catalytically inactive PP2A may associate with CYLD in the cytoplasm,Citation10 which may indirectly regulate the phosphorylation of Akt. Oxidative stress-induced activation of ATMCitation11 and DNA-PKCitation12 results in direct Akt phosphorylation. Altogether, DNA damage-induced signaling may promote AKT activity and prosurvival signaling by increasing phosphorylation and reducing dephosphorylation. On the other hand, PP2A(C), retained in the nucleus, directly interacts with γ-H2AX and dephosphorylates it and potentially other factors at the DNA break, causing an inefficient repair.Citation13 Thus, premature dephosphorylation of γH2AX could facilitate DNA replication and increase mutations and genomic instability.Citation14
Figure 1 Oxidative stress-induced DNA damage may stimulate the assembly of DNAPKsome formation and activation of Mps1, which phosphorylates c-Abl at Threonine 735 (T735), promoting its cytoplasmic translocation. c-Abl phosphorylates the Tyrosine 56 (Y56) in the PIM domain of OTULIN, disrupting its association with LUBAC. The released LUBAC interacts with SPATA2 and is involved in the TNF-R1-mediated signaling pathway, which could also be activated by oxidative stress. Oxidative stress may also directly activate ATM, which phosphorylate the scaffolding subunit of PP2A, PR65, at Serine 401 (S401). This phosphorylation disassembles the holoenzyme, causing the translocation of phosphorylated PR65 to the cytoplasm and retention of the catalytic subunit, PP2A(C), in the nucleus. P2A(C) interacts with and dephosphorylates γ-H2AX. In case of severe DNA damage, nuclear ATM activation may lead to the phosphorylation of c-Abl at S465, which may interfere with DNAPKsome formation and facilitate apoptotic processes.
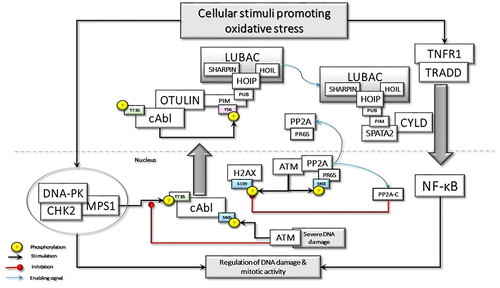
ATM also contributes to the oxidative BER pathway by activating DNA glycosylases that recognize base damage.Citation15 Importantly, ATM was shown to activate the Ku70/80 heterodimer in regions of oxidatively damaged DNA. Thus, ATM also plays a role in the regulation of NHEJ in response to oxidative stress.Citation16 Classical NHEJ is initiated by DSB recruitment of the Ku70/80 heterodimer, which constrains the DSB and engages with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the DNA-PK complex.Citation2 Ku70/Ku80 constitutively interacts with checkpoint kinase 2 (CHK2), suggesting that CHK2 may be recruited to DNA-PK through an interaction with the Ku heterodimer in response to DNA damage.Citation17 Another important component of the DNA-PK complex is the Monopolar spindle 1 (MPS1, also known as TTK), an important mitotic checkpoint kinase, which regulates the expression of DNA-PK.Citation18 Furthermore, CHK2 interacts with the MPS1 in response to DNA damage; subsequently, MPS1 can directly phosphorylate CHK2 at threonine 68 (Thr68).Citation19 Interestingly, CHK2 can also phosphorylate MPS1 at Thr288 and can regulate the stability of the kinase, thus forming a positive feedback loop.Citation20 The activated MPS1 as a part of BER machinery can colocalize with MDM2 and phosphorylate it, which in turn promotes histone H2B ubiquitination and chromatin relaxation in response to oxidative stress.Citation21 Altogether, several key checkpoint proteins, including DNA-PK, CHK2, and Mps1, which are important for mitotic activity, form a phosphorylation-specific large nuclear complex in response to oxidative DNA damage.Citation22 I would like to suggest calling this complex as DNAPKsome, which arises in response to oxidative DNA damage.
Cytoplasmic c-Abl-Induced Significant Alterations Prior to NF-κB Activation
Remarkably, upon exposure to oxidative DNA damage, activated MPS1, as a component of DNAPKsome, phosphorylates c-Abl, a non-receptor tyrosine kinase, at Thr735 and promotes its cytoplasmic translocationCitation23 (). Cytoplasmic c-Abl is considered an inducer of cellular transformation by promoting cell proliferation and inhibiting cell death.Citation24,Citation25 c-Abl interacts with Mucin 1 (MUC1) and phosphorylates it at Tyrosine 60 (Tyr60), which blocks c-Abl signaling to the nucleus and the apoptotic response. MUC1 is an oncoprotein, which is overexpressed by most human carcinomas and blocks the induction of apoptosis by genotoxic agents.Citation26 Transcription of the ARF (p19) locus is repressed by binding of the Cdc6 replication-licensing factor. c-Abl activates the ubiquitin ligase, CUL-4, which promotes the nuclear export of Cdc6 and relieves repression of ARF transcription. The ARF tumor suppressor stabilizes and activates p53 by directly inhibiting MDM2. However, cytoplasmic c-Abl-MUC1 interaction abrogates the c-Abl-driven CUL-4 activation, thus suppressing the ARF-mediated activation of the p53 signaling pathway.Citation27
c-Abl when expressed in the nucleus inhibits NF-κB transcriptional activity, whereas cytosolic c-Abl is unable to share the same function.Citation28 ATM kinase interacts and activates c-Abl in response to DNA damage in the nucleus.Citation29 c-Abl is phosphorylated by ATM at serine-465 in response to severe DNA damage, resulting in c-Abl activation.Citation30 ATM-activated c-Abl, in turn, phosphorylates DNA-PKcs, which inhibits the ability of DNA-PK to form a complex with DNA.Citation31 In addition, nuclear c-Abl stabilizes the histone deacetylase, HDAC1,Citation32 which is a negative regulator of inducible NF-κB activity.Citation33 Taken together, nuclear c-Abl harms DNAPKsome formation and NF-κB transcription.
Cytoplasmic translocation of c-Abl may interfere with the ATM-mediated DNA damage response (DDR). Following DNA damage, H3K9me3 histone tagged nucleosomes are recognized by the TIP60/KAT5 acetyltransferase.Citation34 TIP60 is activated by c-Abl-mediated phosphorylation. In turn, activated Tip60 acetylates and facilitates the binding of ATM to H3K9me3 on chromatin, which may promote ATM-mediated phosphorylation of downstream effectors.Citation35 However, c-Abl is not an obligatory upstream activator for ATM functions; instead, modifications between c-Abl, Tip60, and ATM may be important for the activation of their pro-apoptotic function.Citation36 Taken together, DNAPKsome-mediated cytoplasmic transfer of c-Abl would be a barrier against the apoptotic effect of ATM ().
Another consequence of the DNAPKsome-mediated cytoplasmic accumulation of c-Abl as a result of genotoxic stress is OTULIN phosphorylation at Tyr56, which may have critical outcomesCitation37 (). OTULIN is a deubiquitinating (DUB) enzyme with high activity and unique specificity for methionine 1 (Met1)-linked polyubiquitin chain, which is assembled by the linear ubiquitin chain assembly complex (LUBAC). LUBAC is a multi-subunit E3 ligase consisting of HOIP, HOIL-1, and SHARPIN. LUBAC function is required for the full activation of the inhibitor κB (IκB) kinase (IKK) complex and hence the productive inflammatory signaling pathway.Citation38 The N-terminus of OTULIN contains the PUB-interacting motif (PIM) that mediates the interaction with the PUB domain of the catalytic HOIP subunit and includes the Tyr56 phosphorylation site.Citation39
OTULIN is implicated in angiogenesis and Wnt signaling. LUBAC-induced Met1-ubiquitination suppresses canonical Wnt signaling.Citation40 c-Abl-driven Tyr56 phosphorylation within the PIM domain of OTULIN terminates its interaction with the catalytic HOIP component of LUBAC upon genotoxic stress. Subsequently, OTULIN interacts with β-catenin, inhibiting its linear ubiquitination, thereby robust activation of Wnt/β-catenin signaling.Citation37 Linear ubiquitination has also been demonstrated to play an important role in the maintenance of proteasome function. OTULIN deficiency leads to excessive linear ubiquitination on proteasome subunits, which disrupts proteasome assembly and function.Citation41 Therefore, Ty56 phosphorylation of OTULIN may increase its DUB activity on the proteasome, potentiating the ubiquitin-proteasome system (UPS) activity that may have profound effects on oncogenesis.Citation42
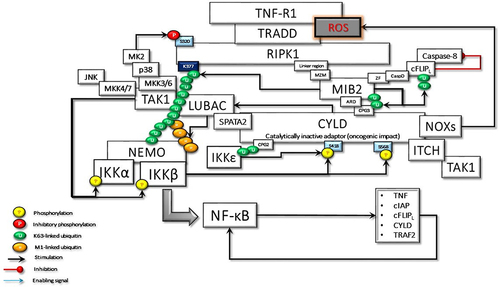
Cylindromatosis protein (CYLD), another DUB, is recruited to LUBAC via the adaptor SPATA2, which binds to both CYLD and HOIP via PUB-PIM interactions. Since SPATA2 and OTULIN bind to the same PUB domain of HOIP, their binding is mutually exclusive.Citation43–45 Therefore, the OTULIN-HOIP relationship, which is disrupted as a result of genotoxicity, may increase the CYLD-SPATA2-HOIP association. In other words, DNAPKsome-induced cytoplasmic c-Abl activity may promote the interaction between CYLD and LUBAC. CYLD-LUBAC association is crucial for NF-kB activation and cell-fate determination by interacting with regulatory proteinsCitation46 ().
The CYLD gene encodes a 956 amino acid protein with several functional domains: three N‐terminal cytoskeleton-associated protein glycine-rich (CAP-Gly) domains and a C‐terminal ubiquitin‐specific catalytic domain [or ubiquitin-specific protease (USP) domain].Citation47 The first and second CAP-Gly domains interact with microtubules.Citation48 Third CAP-Gly (CAP-Gly3) interacts with several important effectors, including NF-κB essential modulator (NEMO),Citation49 mind bomb homolog 2 (MIB2), an E3 ligase,Citation50 and Aurora-B, a mitotic kinase.Citation10 Remarkably, in a recent study, Elliott et al reported that CAP-Gly2 and CAP-Gly3, in addition to microtubule-binding activities, are novel ubiquitin-binding domains (UBDs) of CYLD. Thus, they may increase CYLD activity through the linkage preference to lysine 63 (K63)-linked ubiquitin chains.Citation45
CYLD is regulated by post-translational mechanisms. IKKβ and IKKε, one of the non-canonical IKKs, have been shown to phosphorylate CYLD at a cluster of serine residues located in the region between the second and third CAP-Gly domains, around Ser418.Citation45,Citation51,Citation52 Furthermore, a novel and atypical phosphorylation site, Ser568 in the linker between the CAP-Gly3 and the USP domain of CYLD, is demonstrated. Intriguingly, IKKϵ-mediated Ser418 phosphorylation alone can potentiate LUBAC activity by reducing its autoubiquitination through the stimulation of CYLD. In contrast, UBD activity of CAP-Gly3 and phosphorylation of both Ser418 and Ser568 are indispensable for full CYLD DUB activity on K63-linked ubiquitin chains.Citation45 Altogether, while UBD and Ser418 phosphorylation-driven CYLD activity facilitate the recruitment of LUBAC and the deposition of Met1-ubiquitin, Ser568 phosphorylation of CYLD stimulates its DUB activity and preferentially hydrolyzes K63-ubiquitin chains at signaling complexes.
Until recently, OTULIN has been considered a negative regulator of NF-κB signaling in response to tumor necrosis factor receptor 1 (TNF-PP6R1) activation.Citation38 However, the notion that OTULIN functions primarily by preventing LUBAC-mediated activation of proinflammatory NF-κB or MAPK signaling has recently been challenged.Citation53 OTULIN supports rather than counteracts LUBAC function by preventing its autoubiquitination. Thus, OTULIN stabilizes TNF-R1-associated complex I, which is required for receptor-interacting protein kinase 1 (RIPK1) scaffold function and NF-kB activation.Citation53,Citation54
Since two different DUBs, OTULIN and CYLD, can activate the LUBAC function after TNFR stimulation and only one of them can bind to LUBAC, the question is, what determines the choice of one of them. TNF is a physiologically important pro-inflammatory cytokine for the function of the innate immune system. Through the stimulation of pattern recognition receptors, it increases the expression of genes required to control tissue inflammation and injury. However, it is also involved in the pathophysiology of many inflammatory disorders. Therefore, changes in TNF concentration, tissue and cell type, TNF receptor distribution, and duration of TNF stimulation will determine whether the reaction will be physiological or pathological as a result of a complex interaction.Citation55
The increased TNF/TNF-R1 signaling was shown to be sufficient to induce genotoxicity. Elevation of intracellular reactive oxygen and nitrogen species and redox imbalance may be responsible for TNF-α/TNF-R1 signalling-induced DNA breaks.Citation56 Interestingly, TNFR signaling can be activated independently of TNF-α in the presence of oxidative stress by self-dimerization of the receptor. Moreover, TNF-α can induce stronger downstream signaling to NF-κB in the presence of ROS.Citation57 Taken together, it seems reasonable to assume that OTULIN functions in the TNFR signaling complex in response to physiological TNFα stimulation that does not accompany by oxidative base damage. CYLD has been shown to be activated following DNA damage to suppress tumorigenesis.Citation58 Considering that, following genotoxicity, CYLD is activated and cytoplasmic transfer of c-Abl enables CYLD-LUBAC interaction, CYLD will take over the OTULIN function in the case of pathological genotoxicity ().
Cytoplasmic NF-κB Activation Signaling Pathway Following DNA Damage
Multiple early reports have been published suggesting that DNA damage can activate NF-κB. Genotoxic stress-induced NF-κB activation shares some features with the mode of NF-κB stimulation by TNF-R1.Citation59 Although TNF-R1-mediated NF-κB stimulation is relatively well defined, the mechanisms to describe the NF-κB signaling pathway in response to DNA damage are complicated and remain to be elucidated. Previous studies suggested that NF-κB activation by genotoxic stress is also mediated by cell surface receptors, such as TNF-R1 and IL-1 receptors.Citation60 Nevertheless, the activation of NF-κB, after intertwined molecular interactions for the repair and survival of cells exposed to genotoxic stimuli, induces a large number of inflammatory genes, including those encoding TNF-α.Citation60,Citation61 Increased TNF expression by the ATM-centered NF-κB activation in response to DNA damage triggers a second wave of NF-κB activation mediated by autocrine TNF-TNFR1 signaling. This second wave may determine cell fate according to the extent of DNA damage.Citation61
Soluble TNF-activated TNF-R1 recruits the TNFR1-associated death domain (TRADD) protein, which in turn recruits TNF Receptor Associated Factor 2 (TRAF2) and RIPK1 to form the membrane-bound pro-survival complex I.Citation62 TRAF2 is an E3 Ub ligase required for itself and K63-linked ubiquitination of RIPK1. In unstimulated cells, cytosolic TRAF2-interacts with TRAF-interacting protein (TRIP/TRAIP), which is a negative regulator of TRAF2-mediated NF-κB activation.Citation63 Following TNF stimulation, TRADD-associated TRAF2 binds to sphingosine kinase 1 (SpK1) that generates the pro-survival lipid mediator sphingosine-1-phosphate (S1P). S1P specifically binds to the N-terminal RING domain and stimulates TRAF2 E3 ligase activity by removing TRIPCitation64 ().
Figure 2 TRADD in the TNFR1sc recruits the TRAF2, c-IAPs, and RIPK1 through dead domain (DD) interactions. Membrane-associated TRAF2 comes close to sphingosine kinase (SpK1) and binds with it. SpK1 may activate the E3 ligase function of TRAF2 through its product sphingosine-1-phosphate (SP1). Subsequently, K63-linked autoubiquitination of TRAF2 promotes the recruitment of downstream effector kinases, such as MEKK1 and TANK. TRAF2-TANK interaction stimulates the activation of IKKϵ and TBK1. IKKϵ phosphorylates TRAF2 at Ser11, which may promote the interaction of ubiquitinated TRAF2 with the UBA domain of c-IAPs. Then, the stabilized TRAF2-c-IAP interaction drives the K63-linked ubiquitination of IKKϵ, RIPK1, and TRADD for the recruitment of LUBAC.
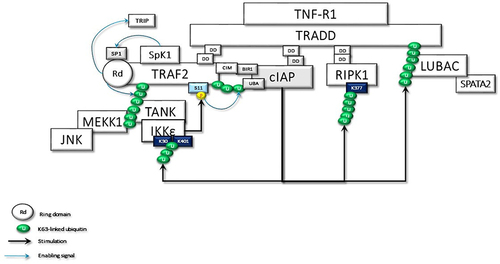
TNF-α-induced activation of c-Jun N-terminal kinase (JNK), but not that of IKK, depends on the integrity of the TRAF2 E3 ligase activity. K63-linked ubiquitin chains on autoubiquitinated TRAF2 have been reported to interact with components of the JNK signaling pathway, including the MAPK kinase kinases, such as MEKK1.Citation65 TRAF2-MEKK1-MEKK7 signaling and activation of JNKCitation66 is critical for TNF regulation of the AP-1 group of transcription factors.Citation67 K63-linked ubiquitin chains at TRAF2 also create a platform for TRAF family member-associated NF-κB activator (TANK).Citation68 Thus, TRAF2 forms a complex with TANK, and two noncanonical IKKs, TBK1 and IKKεCitation68 ().
The BIR1 domain of cellular inhibitor of apoptosis proteins (cIAPs) is required for TNF signaling and readily associated with the cIAP-interacting motif (CIM) of TRAF2 immediately after TNF-R1 signaling.Citation69 However, the UBA domain of c-IAPs binds only ubiquitinated TRAF2. cIAP1/TRAF2 E3 ligase complex is responsible for IKKε ubiquitination. K63-linkage-specific ubiquitination of IKKε at K30 and K401, which is required for its kinase activity, is essential for IKKε-mediated cell transformation.Citation70 Activated IKKε phosphorylates TRAF2 on Ser11, leading to the stabilization of TRAF2, which is required for the recruitment of the UBA domain of cIAP. UBA-mediated TRAF2 binding facilitates cIAP-mediated K63-linked ubiquitination of RIPK1 at K377.Citation71 Thus, TRAF2-cIAP interaction represses RIPK1 kinase activity, which is believed to promote malignant transformation by the RIPK1-mediated NF-κB activationCitation72 ().
Under resting conditions, CYLD interacts with TRAF2.Citation51 Following stimulation, LUBAC is recruited to the TNF-R-signaling complex (TNF-Rsc) through TRADD, TRAF2, and c-IAP interaction and c-IAP-generated ubiquitin chainsCitation73 (). cIAP-driven ubiquitination and activation of IKKϵ promote its homo-dimerization.Citation70,Citation74 The dimerized ubiquitinated IKKϵ most likely interacts with the second CAP-Gly domain (amino acid 232–303) of CYLD monomers, one of the UBDs, upstream of the TRAF2 binding site (amino acid 453–457).Citation75 Subsequently, IKKε can phosphorylate a cluster of serines between residues 418 and 444 of CYLDCitation52 ().
Figure 3 The activated and ubiquitinated IKKϵ may interact with the second CAP-Gly UBD domain of TRAF2-associated CYLD. This interaction enables IKKϵ to phosphorylate Ser418, which may induce the dimerization of CYLD. Then, the dimerized CYLD is associated with the SPATA2-LUBAC complex. Thus, IKKϵ phosphorylation promotes the LUBAC-associated transportation of CYLD to TNF-Rsc. Furthermore, the dimerization of CYLD also may induce the interaction with MIB2, NEMO, and ITCH. In addition to augmenting protein interactions through scaffolding activity, CYLD protects the MIB2 and LUBAC degradation caused by autoubiquitination.
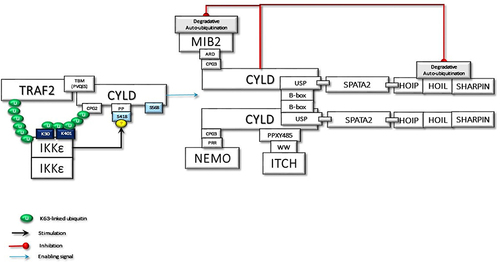
SPATA2 is a LUBAC-associated component of TNF-Rsc and has an important function in regulating the outcome of TNFR signaling through recruitment of CYLD to TNF-Rsc. The B-box-mediated dimerization of CYLD is essential for SPATA2 interaction. Thus, CYLD and SPATA2 form a highly stable heterotetramer.Citation43 Phosphorylation-mediated allosteric changes of a protein can reason to dimerization, which facilitates the binding of many response regulators to their partners.Citation76 Therefore, homo-dimerized IKKϵ-mediated Ser418 phosphorylation could promote dimerization of CYLD and thus facilitate the interaction of CYLD with SPATA2. This association could be considered an important step that can trigger the linear ubiquitination required for NF-κB activation. Since the autoubiquitination of LUBAC inhibits its activity in TNF-Rsc, a signal is required for LUBAC to be active. CYLD-SPATA2 association, possibly by inhibiting LUBAC autoubiquitination, operates as a triggering mechanism for LUBAC activity, thus promoting linear ubiquitination.Citation45 CYLD dimerization not only increases LUBAC activity in TNF-Rsc but also creates a bonding opportunity for the increasing amount of proteins ().
MIB2, RING-type Ub-E3 ligase, interacts with RIPK1 through its MZM region only when RIPK1 is recruited to TNF-R1. Therefore, MIB2 may sense the activity status of RIPK1. The MIB2-RIPK1 interaction will have crucial implications for NF-κB activation and cell fate determination. First, MIB2 ubiquitinates RIPK1 at K377 and K634, contributing to the suppression of the RIPK1 kinase activity-dependent cytotoxic function.Citation77 Second, MIB2 binds and polyubiquitinates the caspase-like domain of the long-form of cellular FLICE-inhibitory protein (cFLIPL), a catalytically inactive homolog of caspase 8. Although caspase 8 and ubiquitinated cFLIPL are recruited to the RIPK1/FADD complex, ubiquitinated cFLIPL suppresses TNF-induced apoptosis by inhibiting the formation of a proper oligomer with caspase-8.Citation78 Third, MIB2 is an E3 ligase, known to ubiquitylate itself. CYLD may interact with the ubiquitinated MIB2, which may facilitate the binding of the third CAP-Gly domain of CYLD with the ankyrin repeat domain (ARD) of MIB2. Deubiquitinases and E3 ligases frequently interact. MIB2 is not a deubiquitination target of CYLD but rather is stabilized by CYLD in a non-catalytic-dependent manner (). Furthermore, while the expression of CYLD alone inhibits LUBAC-stimulated NF-κB activation, CYLD association with MIB2 rescues the NF-κB activityCitation50,Citation79 (). Fourth, K63-linked ubiquitin chains on MIB2 recruit ubiquitin-binding TAB proteins along with transforming growth factor-β-activated kinase 1 (TAK1) and NEMO (IKKγ) to support NF-κB activating signal transduction. MIB2-TAK1 interaction results in increased kinase activity of TAK1 following its phosphorylation.Citation80 Several lysine residues of TAK1 are potential sites for polyubiquitination. MIB2 may directly stimulate K63-linked polyubiquitination of TAK1 at K562, which has been reported to be required for the autophosphorylation of TAK1 at Thr187Citation81 ().
Figure 4 The CYLD-SPATA2-LUBAC association enhances the linear ubiquitination function of LUBAC. Activated LUBAC, associated with K63 linked ubiquitin chains on RIPK1, adds Met1-linked ubiquitin chains on K63-linked ubiquitin chains. K63/Met1-hybrid ubiquitin chains on RIPK1 recruit NEMO for the activation of canonical IKKs. RIPK1 in the TNF-R1sc interacts with the MZM motif of MIB2 through its linker region. Thus, activated MIB2 autoubiquitinates itself with K63-linked chains, which facilitates its interaction with the third CAP-Gly domain of CYLD by its ankyrin repeat domain (ARD). MIB2 also interacts by its zinc finger (ZF) motif to the caspase domain (CasD) of c-FLIPL and then decorates it with K63-linked ubiquitin chains. Furthermore, CYLD-associated LUBAC conjugates M1-linked ubiquitination chains on c-FLIPL. Thus, the hybrid K63/Met1 ubiquitin chains stabilize c-FLIPL to inhibit the caspase-8 activity. In addition to the scaffolding activities, K63-linked and Met1-linked poly-ubiquitin chains on RIPK1 block its kinase activity. K63-linked ubiquitin chains on MIB2 interact with TAB proteins for the recruitment and the activating phosphorylation of TAK1, which is also associated with CYLD-bound ITCH. Following TAB-driven interaction with TAK1, MIB2 may ubiquitinate the K562 residue, which may promote autophosphorylation of TAK1 at T187 and subsequent phosphorylation at T189 (significance was denoted by an asterisk (*) and it has been moved to a free area and detailed in order not to confuse the main shape further in the figure). Besides K63-linked poly-ubiquitin chains on RIPK1, K63-linked chains on MIB2 may also provide support for the activation of CYLD-associated NEMO by providing an additional scaffold. PLK1 interaction with CYLD and RIPK1 may alter its mitotic activity as detailed in the text.
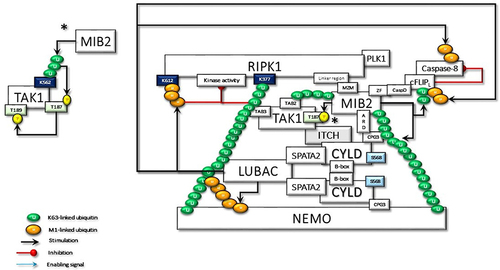
The dimerized CYLD directly interacts with NEMO with its third CAP-Gly domain,Citation49,Citation82 which may facilitate the association of MIB2 with NEMO. In addition, the PPXY motif of CYLD interacts with the WW domain of another important E3 ligase, ITCH,Citation83 which is also associated with TAK1Citation84 and c-FLIPLCitation85 ( and ).
MIB2-dependent recruitment of CYLD to RIPK1 may also facilitate SPATA2-mediated transport of the LUBAC as well.Citation43,Citation86 Subsequently, LUBAC conjugates M1-linked ubiquitin chains on cFLIPL, further stabilizing it to protect cells from TNFα-induced apoptosis.Citation87 Moreover, CYLD-MIB2-RIPK1 interaction may facilitate HOIL-mediated direct interaction of RIPK1 with LUBAC.Citation88 Consequently, LUBAC-induced linear ubiquitination of RIPK1 on K612 and caspase-8 may restrict necroptosis by limiting the formation of RIPK1/RIPK3/MLKL containing complexCitation89,Citation90 ().
The ubiquitination of RIPK1 at K377 is indispensable for the activation of NF-κB. K63-linked poly-ubiquitinated RIPK1 interacts with the polyubiquitin binding adaptors TAB2/TAB3, which promotes the recruitment and activation of TAK1 kinase.Citation91 TAK1-driven p38/MK2 kinase cascade directly phosphorylates RIPK1 at Ser321, thus blocking the RIPK1 kinase-dependent apoptosis and necroptosis.Citation92 In addition, the K63-linked ubiquitin chains facilitate the recruitment of LUBAC, enabling the deposition of Met1-Ub on the existing K63-ubiquitin chains.Citation93 In turn, the K63-Ub and Met1-Ub facilitate the recruitment of NEMO-canonical IKK kinase subunits of the IKKα and IKKβ complex. TAK1-catalyzes the phosphorylation of IKKβ at Ser177, which is a priming event that enables IKKβ to activate itself by phosphorylating Ser181.Citation94 Activated IKKβ phosphorylates the NF-κB inhibitor IkBα, which results in proteasomal degradation of IκBα and subsequent nuclear translocation of NF-κB and activation of NF-κB target genes.Citation77 NFκB regulates more than 500 genes involved in inflammation, cellular transformation, survival, proliferation, angiogenesis, invasion, and metastasis (). In cases where NF-κB activity cannot be controlled, increased genetic expressions of some proteins, including, TNFα, TRAF2, c-IAPs, Bcl2, CYLD, and c-FLIPL, could stimulate NF-κB activity, thus promoting a positive feedback loop.Citation95–97
Figure 5 The activated TAK1 phosphorylates IKKα and IKKβ complexed with NEMO, which is required for the NF-κB transcriptional activity. In the absence of regulation to terminate the signaling pathway, NFK stimulates the expression of many genes, some of which promote persistent activity. IKKβ also phosphorylates Ser568 of CYLD to stimulate its DUB activity. The activated CYLD deconjugates K63-linked ubiquitin chains that trigger a cascade of reactions to terminate signal transduction.
Termination of signaling is a crucially important factor to prevent the inappropriate activity of NF-κB. In addition to its essential role in stimulating NF-κB activity, even termination of this activity has been shown to require the function of IKKβ. IKKβ–mediated phosphorylation of CYLD at Ser568,Citation45 which resides in the third CAP-Gly domain of CYLD, increases its DUB activity, promoting deconjugation of K63-linked ubiquitin chains on NEMO, TRAF2, and MIB2 (). Moreover, in response to oxidative stress, IKKβ was found to translocate into the nucleus and directly phosphorylate ATM, promoting DNA repair. This mechanism would be beneficial for the maintenance of homeostasis by repairing DNA damage that occurs in normal cells. However, the same mechanism may also enable cancer cells to resist chemotherapy.Citation98
Figure 6 The activated CYLD behaves as a tumor suppressor and deubiquitinates the K63-linked ubiquitin chains on TRAF2, RIPK1, and MIB2. Loss of K63-linked chains reason for the stimulation of the K48-linked degradative ubiquitination of TAK1 and c-FLIPL by ITCH, and CYLD by MIB2, thus terminating CYLD-mediated signaling events.
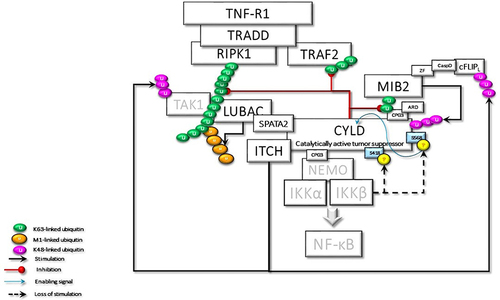
IKKβ-stimulated activation of CYLD may enable the CYLD-ITCH complex to promote proteasomal degradation. CYLD-ITCH complex may sequentially cleave K63-linked ubiquitin chains and catalyze the degradative K48-linked polyubiquitin chain on TAK1Citation83 and c-FLIPCitation85 to terminate TAK1 and c-FLIP-mediated pro-survival signals. Similarly, following CYLD-mediated cleavage of K63 ubiquitin chains, MIB2 catalyzes K48 ubiquitin chains on CYLD for degradation, thus completely terminating the CYLD activity.Citation50 Altogether, while CYLD initially functions as a bridging and adaptor protein for signal progression independent of its DUB activity, it eventually terminates signaling by supporting the degradation of key molecules, including itself through its DUB activity ().
Novel CYLD-Centered Model of Malignant Transformation
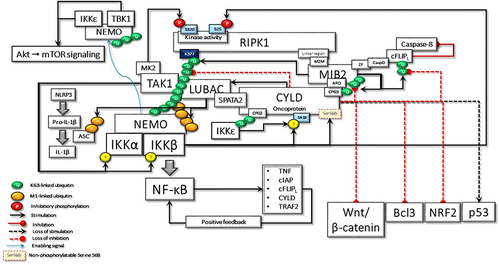
In addition to its role in innate immunity, IKKε, an upstream regulator of the transcription factor NF-κB, is also accepted as an oncogene that is amplified and overexpressed in up to 30% of breast cancers.Citation99 According to a highly regarded study from Cantley Lab,Citation52 TRAF2-interacted and c-IAP-ubiquitinated IKKϵCitation70,Citation71 phosphorylates CYLD at Ser418, contributing to cell transformation. In fact, besides IKKϵ, canonical IKKs can also cause CYLD Ser418 phosphorylation.Citation51 Consistent with these early reports, all subsequent studies, particularly, Elliot et al reaffirmed that Ser418 phosphorylation inhibits CYLD DUB activity.Citation44 They also demonstrated that phosphorylation of a hitherto unknown atypical serine residue, Ser568, increases CYLD’s DUB activity toward K63-linked ubiquitin chains. It is therefore questionable why the Ser568 residue might have been overlooked in previous studies. The explanation is most likely that while results from previous studies were obtained with transformed cell lines, the recent study used CYLD KO U2OS/NOD2 cells for reconstitution of wild-type CYLD expression. Taken together, there might be a possibility that malignant cells were unable to express phosphorylatable Ser568.
Serine is a high probability target of mutations; therefore, it mutates very often and evolves very quickly. Encoded by two separate codon sets, AGY and TCN, serine is unique among the 20 amino acids; therefore, it will be more easily reached from another amino acid after mutation.Citation100 Phosphoserines tend to occur in intrinsically disordered regions (IDRs), which play essential roles in a wide range of biological processes and can function as linear motifs, linkers, or entropic chains.Citation101 IDRs are integral parts of many cancer-associated proteins and can be direct targets of cancer-inducing mutations.Citation102 Since Ser568 is the unique phosphorylatable site within the linker region (IDR) between third CAP-Gly and the USP domain of CYLD,Citation45 it could be considered a residue with high mutability. Another mechanism that may affect the phosphorylation of CYLD Ser568 is the methylation of cytosines within serine codons. The differentially methylated cytosines may lead to alternative splicing mechanisms resulting in the expression of alternative or less sensitive proteins.Citation103 So, in the light of the data summarized above, let us try to evaluate the alterations that might occur when a cell that could not phosphorylate CYLD Ser568 is exposed to oxidative stress and genotoxicity ().
Cytosolic c-Abl phosphorylates Tyr56 of OTULIN, facilitating SPATA2-LUBAC interaction. LUBAC-free OTULIN may increase Wnt-β-catenin signaling and UPS activity.
Cytosolic c-Abl interacts with MUC1, which blocks nuclear targeting of c-Abl and therefore the apoptotic response to genotoxic stimuli. Furthermore, c-Abl may alter cell polarity, and facilitate epithelial-to-mesenchymal transition (EMT) program, invasion, or growth.Citation104
Cytosolic c-Abl blocks the ARF-mediated p53 activation, bypassing p53-mediated tumor suppression.
While CYLD could not exert DUB activity, its scaffolding function leads to uncontrolled NF-κB transcription. Thus, overexpression of CYLD and anti-apoptotic proteins may increase cell survival as a result of positive feedback signaling.
CYLD directly interacts with and deubiquitinates p53, promoting DNA damage-induced p53 stabilization and activation in response to oxidative stress-induced genotoxicity.Citation58 The stabilized p53 transcription factor is a key tumor suppressor and maintains genomic stability after cellular stresses.Citation105 The lack of CYLD catalytic DUB activity may impair the tumor suppressor function of p53.
Cancer cells, even in oxygen-rich conditions, prefer the less efficient use of glycolysis to metabolize glucose, a prominent metabolic change called the Warburg effect or aerobic glycolysis. Therefore, active metabolic reprogramming by altered oncoproteins and tumor suppressors is considered a key hallmark of cancer “even Warburg did not anticipate”.Citation106 p53 regulates cellular metabolism; it represses glycolysis and the synthesis of lipids and nucleotides that all contribute to its tumor-suppressive function.Citation107 p53 can limit the activity of phosphofructokinase-1 (PFK-1), the rate-limiting enzyme in glycolysis.Citation108 p53 suppresses transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) as well, which is also a rate-limiting enzyme that promotes glycolysis.Citation109 p53 stabilization by catalytically active CYLD was reported to suppress glycolysis through the inhibition of PFK1 and PFKFB3 transcriptions.Citation110 Furthermore, CYLD interacts with Fizzy-related protein 1 (FZR1, also named Cdh1), an important component of the anaphase-promoting complex/cyclosome (APC/C), a ubiquitin E3 ligase, and enhances FZR1 activity, which promotes the degradation of PFKFB3, cyclin B1, and cyclin D1.Citation110 Taken together, it would not be difficult to speculate that CYLD, unable to exploit its catalytic activity, may support the operation of metabolic reprogramming.
Loss of CYLD DUB activity increases the expression of proto-oncogene Bcl-3-mediated pro-survival, pro-inflammatory, and genes that control cell cycle activity.Citation111,Citation112
Loss of CYLD DUB activity increases the nuclear translocation of the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2).Citation113 NRF2 is the major mediator of oxidative stress responses and DDR pathways stimulate NRF2 signaling. The increased NRF2 signaling plays important role in cancer initiation, progression, metabolic reprogramming, metastasis, and resistance to therapy. Intriguingly, similar to CYLD, NRF2 has been increasingly believed to operate as both a tumor suppressor and an oncoprotein.Citation114
NF-κB signaling has been reported to increase the expression of nicotinamide adenine dinucleotide phosphate oxidases (NOXs).Citation115 Interestingly, the DUB activity of CYLD negatively regulates NOXO1 protein expression,Citation116 Therefore, the loss of CYLD DUB activity may potentiate NOX-mediated ROS generation, thus contributing to the oncogenesis.Citation117
CYLD interacts with PLK1 and regulates mitotic entry.Citation118 Furthermore, active PLK1 is recruited into the RIPK1-Caspase 8 complex, which cleaves PLK1 during mitosis. Since caspase 8 activity is blocked, active PLK1 accumulates, resulting in defects in chromosome congression, segregation, and higher aneuploidy.Citation119 Thus, unregulated PLK1 activity may favor tumor evolution, drug resistance, and risk for tumor relapse.
In addition to the resistance to cell death, CYLD-MIB2 interaction may also increase Notch signaling, which may impart a stem-like phenotype.Citation79
CYLD binds tubulin primarily through its first CAP-Gly domain and promotes tubulin polymerization into microtubules, which is important for cell migration.Citation48 CYLD is also associated with end binding protein (EB1). They act in concert to regulate microtubule nucleation at the centrosome, and microtubule growth at the cell periphery, thus further contributing to the directional cell migration.Citation120 Importantly, this function of CYLD is independent of its DUB activity.
The overexpression of Aurora B is a trigger for tumorigenesis and has been associated with a poor prognosis for cancer. The ubiquitin E3-ligase protein Skp2, which is also a cell-cycle regulatory protein, is required for the activation of Aurora B through K63-linked ubiquitination.Citation121 CYLD interacts with the catalytic domain of Aurora B, inhibiting its activity. The third CAP-Gly domain and the deubiquitinase domain are required for CYLD to inhibit Aurora B activity. CYLD also interacts with the scaffolding subunit of PP2A and promotes inactive PP2A/Aurora B interaction.Citation10 EB1 is an Aurora-B-interacting protein as well, and it stimulates Aurora-B activity through antagonizing its dephosphorylation/inactivation by PP2A.Citation122 Taken together, due to EB1 interaction and lack of DUB activity of CYLD, the decreased K63-linked deubiquitination, and dephosphorylation of Aurora B would be critical for cell proliferation toward malignancy.
CYLD suppresses Wnt signaling through deubiquitination of K63-linked ubiquitin chains on Dvl, an adapter protein that transduces proximal Wnt signals.Citation123 Consistently, loss of CYLD catalytic activity has been reported to fuel tumorigenesis and aggressiveness through hyperactivation of the Wnt pathway.Citation124
The localization of cell polarity protein disheveled (Dvl) at the cell cortex is important for spindle orientation. Cortical polarity proteins can recruit the nuclear mitotic apparatus (NuMA) protein, which can generate pulling forces on astral microtubules to rotate the spindle. CYLD, through removing K63-linked polyubiquitin chains, stabilizes astral microtubules and stimulates the formation of the Dvl-NuMA-dynein/dynactin complex at the cell cortex, thereby promoting proper spindle orientation.Citation125 Thus, the loss of CYLD DUB activity may lead to misoriented cell division in epithelial cells, which is also important for carcinogenesis.
CYLD also binds HDAC6 through its first CAP-Gly domain and inhibits HDAC6 activity through its DUB function, leading to acetylated α-tubulin around the nucleus.Citation126 HDAC6 is a microtubule-associated deacetylase and HDAC6-mediated deacetylation regulates microtubule-dependent cell motility.Citation127 In the case of CYLD DUB activity loss, elevated HDAC6 activity, and increased deacetylation of microtubules, may promote tumor formation and oncogenic transformation by facilitating anchorage-independent proliferation, which allows cells to survive by escaping anoikis.Citation128
LUBAC-mediated linear ubiquitination may also participate in the NLRP3 inflammasome activation pathway-mediated IL-1β secretion by promoting caspase-1 activation.Citation129 IL-1β is claimed as a cancer marker by some, due to its pleiotropic effects on immune cells, angiogenesis, cancer cell proliferation, migration, and metastasis.Citation130
TBK1 and IKKϵ synergize with TANK to promote interaction with the canonical IKKs. The TANK binding domain within NEMO is required for the proper functioning of these IKK subunits.Citation131 K63-linked polyubiquitin chains on NEMO and TANK link TAK1 and the canonical IKK complex to TBK1/IKKε, enabling Ser172 phosphorylation and activation.Citation132 Sustained activation of noncanonical IKKs, IKKϵ and TBK1, also promotes the oncogenic phenotype.Citation133 They can activate Akt, which is involved in several critical cellular pathways including survival, proliferation, invasion, apoptosis, and angiogenesis,Citation134 by direct phosphorylation on both Thr308 and Ser473.Citation135 In addition to the Akt-mediated mTORC1 signaling axis, IKKϵ and TBK1 can also positively regulate mTORC1 activity.Citation136,Citation137 Furthermore, they directly phosphorylate and activate mTORC2 which controls cellular metabolism, proliferation, and survival.Citation138 Importantly, uninterrupted mTOR activation has been recalling Blagosklonny’s “hyperfunction theory”.Citation139,Citation140 The tight relationships between oxidative stress, DNA damage, ROS, and mTOR are critical to the concept of hyperfunction. Thus, a defective CYLD-mediated “hyperfunctional” state following oxidative DNA damage could be considered to force senescence in postmitotic cells, while providing support for malignant transformation in cells with mitotic activity. Altogether, non-canonical IKK-driven deregulated mTOR signaling contributes significantly to carcinogenesis with its myriad functions.Citation141
In addition to NF-κB activating function, IKKβ promotes tumoral transformation by phosphorylating several other proteins that regulate many cellular processes from the cell cycle to metabolism and differentiation.Citation142
NF-κB is a key positive regulator of programmed cell death ligand (PD-L1) expression in cancer.Citation143 NF-κB directly induces PD-L1 gene transcription by binding to its promoter, and it can also regulate PD-L1 post-transcriptionally through indirect pathways. Thus, cancer cells exploit the PD-L1-driven inhibitory pathway to their benefit as a key mechanism of immune evasion.Citation144
Continuous linear ubiquitination of NEMO increases TRAF3 association and disrupts the MAVS-TRAF3 complex formation that inhibits type I Interferons (IFNs) activation.Citation145 IFNs are key regulators of natural host defense against viral infection and cancer.Citation146
Oxidative stress-induced DNA breaks and subsequent abnormal use of the non-conservative NHEJ DNA repair through the activation of the NF-κB pathway may increase genome instability and favor transformation.Citation147,Citation148
Oxidative stress-induced direct activation of ATM may reason in the dissociation of the PP2A holoenzyme, which may promote the translocation of the scaffolding unit to the cytoplasm and the retention of the catalytic unit in the nucleus. Direct interaction of PP2A(C) with γ-H2AX causes premature dephosphorylation and consequent defects in DNA repair, which may increase the mutation rate.
Suppression of CYLD DUB activity and TAK1-facilitated activation of JNK stimulates the promoter activation of AP-1, leading to the transcription of genes important for cell proliferation, such as cyclin D1 and c-Myc.Citation149 c-Myc-driven epigenetic reprogramming promotes the formation, and maintenance of tumor-initiating cells and their attainment of metastatic capacity.Citation150
Finally, evidence directly suggesting a relationship between loss of CYLD DUB activity and carcinogenesis came from a remarkable recent study. The researchers reported that transgenic mice lacking the deubiquitinase function of CYLD spontaneously develop tumors of various origins.Citation151
Figure 7 In the case of hypothetical loss of IKKβ-mediated Ser568 phosphorylation, CYLD behaves as an oncoprotein, keeping its scaffolding activity. Here, some of the important cyclical activities that can lead to cell transformation due to the loss of CYLD DUB activity are summarized: i) uncontrolled NF-κB activation due to positive feedback regulation; ii) RIPK1 kinase activity-dependent and -independent resistance to apoptosis; iii) IKKϵ- and TBK1-stimulated increased Akt and mTOR signaling; iv) LUBAC-induced increased secretion of IL-1β; v) Bcl3-driven increased transcriptional activity; vi) NRF2-driven increased transcriptional activity; vii) increased Wnt-β-catenin signaling; viii) loss of p53-mediated transcriptional activity; ix) MIB2-c-FLIPL-mediated caspase 8 inhibition; x) Increased Akt-mTOR signaling; xi) increased IL-1β secretion.
Treatment Option for This Novel Postulate
Tyrosine kinase inhibitors (TKIs), with multiple targets, including c-Abl, are the major medicines for targeted therapy of cancer. Imatinib is a first-generation BCR-ABL TKI, which selectively targets the ATP binding site of the BCR-ABL protein. It was approved in 2001 by the FDA for treating chronic myelogenous leukemia. Since abnormally activated Abl kinases are implicated in a variety of pathologies, including various solid tumors, inflammatory disorders, neurodegenerative diseases, and even COVID-19, targeting Abl kinases with small molecule inhibitors is considered an option to treat different pathologies with hyperactive c-Abl.Citation152,Citation153 However, while treatment with these kinase inhibitors has a predominant role on the cytoplasmic c-Abl, they cannot provide its nuclear localization.Citation154 If the hypothetical mechanisms operate as described, cytoplasmic translocation of c-Abl would be indispensable for triggering CYLD-mediated pathological outcomes. Therefore, the design of small molecules and future perspectives for the application of drugs targeting Thr735 of c-Abl could interfere with its cytosolic functions. This approach can also re-establish the apoptotic response in cancer cells by increasing ATM-c-Abl interaction.Citation155
Conclusion
Contrary to the general belief that CYLD is an absolute tumor suppressor, it could operate both as a tumor suppressor and an oncoprotein in a context-dependent manner. Physiological alternately regulation of these two activities is crucially important for the maintenance of cellular homeostasis. If one of these features dominates independently of the other, the pathological outcome would be inevitable. Most cylindroma lesions in which CYLD cannot be expressed are benign. Contrary to common belief, lack of CYLD, which would also cause concomitant loss of CYLD-mediated oncogenic activity, may limit tumor growth and prevent progression to malignancy. In the case of non-phosphorylatable CYLD Ser568, oxidative stress that can lead to DNA damage could enable the malignant transformation of the cell as a result of the self-sustaining signaling network of the resistance to apoptosis, accelerated mitotic activity, and other concomitant unregulated processes.
Disclosure
The author reports no conflicts of interest in this work.
References
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science (80-). 2008;319(5868):1352–1355. doi:10.1126/science.1140735
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi:10.1038/nature08467
- Lin Y, Bai L, Chen W, Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14(1):45–55. doi:10.1517/14728220903431069
- Li H, Marple T, Hasty P. Ku80-deleted cells are defective at base excision repair. Mutat Res Mol Mech Mutagen. 2013;745–746:16–25. doi:10.1016/j.mrfmmm.2013.03.010
- David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi:10.1038/nature05978
- Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2008;30(1):2–10. doi:10.1093/carcin/bgn250
- Xia W, Ci S, Li M, et al. Two‐way crosstalk between BER and c‐NHEJ repair pathway is mediated by Pol‐β and Ku70. FASEB J. 2019;33(11):11668–11681. doi:10.1096/fj.201900308R
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science (80-). 2010;330(6003):517–521. doi:10.1126/science.1192912
- Amrita Sule SE, Golding SF, Farhan JW, et al. ATM phosphorylates PP2A subunit A resulting in nuclear export and spatiotemporal regulation of the DNA damage response. bioRxiv Prepr. 2021. doi:10.1101/2021.08.29.458108
- Sun L, Gao J, Huo L, et al. Tumour suppressor CYLD is a negative regulator of the mitotic kinase Aurora-B. J Pathol. 2010;221(4):425–432. doi:10.1002/path.2723
- Golding SE, Rosenberg E, Valerie N, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8(10):2894–2902. doi:10.1158/1535-7163.MCT-09-0519
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBα/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30(2):203–213. doi:10.1016/j.molcel.2008.02.024
- Chowdhury D, Keogh M-C, Ishii H, Peterson CL, Buratowski S, Lieberman J. γ-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20(5):801–809. doi:10.1016/j.molcel.2005.10.003
- Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi:10.1038/35077232
- Chou W-C, Wang H-C, Wong F-H, et al. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2008;27(23):3140–3150. doi:10.1038/emboj.2008.229
- Kuhne C. Repair of a minimal DNA double-strand break by NHEJ requires DNA-PKcs and is controlled by the ATM/ATR checkpoint. Nucleic Acids Res. 2003;31(24):7227–7237. doi:10.1093/nar/gkg937
- Shang Z-F, Huang B, Xu Q-Z, et al. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 phosphorylation in response to DNA damage. Cancer Res. 2010;70(9):3657–3666. doi:10.1158/0008-5472.CAN-09-3362
- Maachani UB, Kramp T, Hanson R, et al. Targeting MPS1 enhances radiosensitization of human glioblastoma by modulating DNA repair proteins. Mol Cancer Res. 2015;13(5):852–862. doi:10.1158/1541-7786.MCR-14-0462-T
- Wei J-H, Chou Y-F, Ou Y-H, et al. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J Biol Chem. 2005;280(9):7748–7757. doi:10.1074/jbc.M410152200
- Yeh C-W, Yu Z-C, Chen P-H, Cheng Y-C, Shieh S-Y. Phosphorylation at threonine 288 by cell cycle Checkpoint Kinase 2 (CHK2) controls human monopolar Spindle 1 (Mps1) kinetochore localization. J Biol Chem. 2014;289(22):15319–15327. doi:10.1074/jbc.M114.552273
- Yu Z-C, Huang Y-F, Shieh S-Y. Requirement for human Mps1/TTK in oxidative DNA damage repair and cell survival through MDM2 phosphorylation. Nucleic Acids Res. 2016;44(3):1133–1150. doi:10.1093/nar/gkv1173
- Shang Z, Yu L, Lin Y-F, Matsunaga S, Shen C-Y, Chen BPC. DNA-PKcs activates the Chk2–Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis. 2014;3(2):e85–e85. doi:10.1038/oncsis.2013.49
- Nihira K, Taira N, Miki Y, Yoshida K. TTK/Mps1 controls nuclear targeting of c-Abl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene. 2008;27(58):7285–7295. doi:10.1038/onc.2008.334
- Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev. 1995;9(21):2569–2582. doi:10.1101/gad.9.21.2569
- Wang JYJ. The capable ABL: what is its biological function? Mol Cell Biol. 2014;34(7):1188–1197. doi:10.1128/MCB.01454-13
- Raina D, Ahmad R, Kumar S, et al. MUC1 oncoprotein blocks nuclear targeting of c-Abl in the apoptotic response to DNA damage. EMBO J. 2006;25(16):3774–3783. doi:10.1038/sj.emboj.7601263
- Raina D, Ahmad R, Chen D, Kumar S, Kharbanda S, Kufe DW. Muc1 oncoprotein suppresses activation of the ARF-MDM2-p53 pathway. Cancer Biol Ther. 2008;7(12):1959–1967. doi:10.4161/cbt.7.12.6956
- Kawai H, Nie L, Yuan Z-M. Inactivation of NF-κB-dependent cell survival, a novel mechanism for the proapoptotic function of c-Abl. Mol Cell Biol. 2002;22(17):6079–6088. doi:10.1128/MCB.22.17.6079-6088.2002
- Shafman T, Khanna KK, Kedar P, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387(6632):520–523. doi:10.1038/387520a0
- Shaul Y, Ben-Yehoyada M. Role of c-Abl in the DNA damage stress response. Cell Res. 2005;15(1):33–35. doi:10.1038/sj.cr.7290261
- Kharbanda S, Pandey P, Jin S, et al. Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature. 1997;386(6626):732–735. doi:10.1038/386732a0
- Aoyama K, Yamaguchi N, Yuki R, et al. c-Abl induces stabilization of histone deacetylase 1 (HDAC1) in a kinase activity-dependent manner. Cell Biol Int. 2015;39(4):446–456. doi:10.1002/cbin.10413
- Liberatore RA, Goff SP, Nunes I. NF-κB activity is constitutively elevated in c-Abl null fibroblasts. Proc Natl Acad Sci. 2009;106(42):17823–17828. doi:10.1073/pnas.0905935106
- Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11(11):1376–1382. doi:10.1038/ncb1982
- Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817. doi:10.1016/j.molcel.2017.05.015
- Jiang Z, Kamath R, Jin S, Balasubramani M, Pandita TK, Rajasekaran B. Tip60-mediated acetylation activates transcription independent apoptotic activity of Abl. Mol Cancer. 2011;10(1):88. doi:10.1186/1476-4598-10-88
- Wang W, Li M, Ponnusamy S, et al. ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nat Commun. 2020;11(1):3965. doi:10.1038/s41467-020-17770-9
- Keusekotten K, Elliott PR, Glockner L, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. 2013;153(6):1312–1326. doi:10.1016/j.cell.2013.05.014
- Elliott PR, Nielsen SV, Marco-Casanova P, et al. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol Cell. 2014;54(3):335–348. doi:10.1016/j.molcel.2014.03.018
- Takiuchi T, Nakagawa T, Tamiya H, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells. 2014;19(3):254–272. doi:10.1111/gtc.12128
- Tao P, Wang S, Ozen S, et al. Deubiquitination of proteasome subunits by OTULIN regulates type I IFN production. Sci Adv. 2021;7:47. doi:10.1126/sciadv.abi6794
- Di Costanzo A, Del Gaudio N, Conte L, Altucci L. The ubiquitin proteasome system in hematological malignancies: new insight into its functional role and therapeutic options. Cancers. 2020;12(7):1898. doi:10.3390/cancers12071898
- Elliott PR, Leske D, Hrdinka M, et al. SPATA2 Links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol Cell. 2016;63(6):990–1005. doi:10.1016/j.molcel.2016.08.001
- Douglas T, Saleh M. Post-translational modification of OTULIN regulates ubiquitin dynamics and cell death. Cell Rep. 2019;29(11):3652–3663.e5. doi:10.1016/j.celrep.2019.11.014
- Elliott PR, Leske D, Wagstaff J, et al. Regulation of CYLD activity and specificity by phosphorylation and ubiquitin-binding CAP-Gly domains. Cell Rep. 2021;37(1):109777. doi:10.1016/j.celrep.2021.109777
- Hrdinka M, Gyrd-Hansen M. The Met1-linked ubiquitin machinery: emerging themes of (De)regulation. Mol Cell. 2017;68(2):265–280. doi:10.1016/j.molcel.2017.09.001
- Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25(2):160–165. doi:10.1038/76006
- Gao J, Huo L, Sun X, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283(14):8802–8809. doi:10.1074/jbc.M708470200
- Saito K, Kigawa T, Koshiba S, et al. The CAP-Gly domain of CYLD associates with the proline-rich sequence in NEMO/IKKγ. Structure. 2004;12(9):1719–1728. doi:10.1016/j.str.2004.07.012
- Uematsu A, Kido K, Takahashi H, et al. The E3 ubiquitin ligase MIB2 enhances inflammation by degrading the deubiquitinating enzyme CYLD. J Biol Chem. 2019;294(38):14135–14148. doi:10.1074/jbc.RA119.010119
- Reiley W, Zhang M, Wu X, Granger E, Sun S-C. Regulation of the deubiquitinating enzyme CYLD by IκB kinase gamma-dependent phosphorylation. Mol Cell Biol. 2005;25(10):3886–3895. doi:10.1128/MCB.25.10.3886-3895.2005
- Hutti JE, Shen RR, Abbott DW, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKɛ promotes cell transformation. Mol Cell. 2009;34(4):461–472. doi:10.1016/j.molcel.2009.04.031
- Heger K, Wickliffe KE, Ndoja A, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature. 2018;559(7712):120–124. doi:10.1038/s41586-018-0256-2
- Weinelt N, van Wijk SJL. Ubiquitin-dependent and -independent functions of OTULIN in cell fate control and beyond. Cell Death Differ. 2021;28(2):493–504. doi:10.1038/s41418-020-00675-x
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science (80-). 2002;296(5573):1634–1635. doi:10.1126/science.1071924
- Westbrook AM, Wei B, Hacke K, Xia M, Braun J, Schiestl RH. The role of tumour necrosis factor- and tumour necrosis factor receptor signalling in inflammation-associated systemic genotoxicity. Mutagenesis. 2012;27(1):77–86. doi:10.1093/mutage/ger063
- Ozsoy HZ, Sivasubramanian N, Wieder ED, Pedersen S, Mann DL. Oxidative stress promotes ligand-independent and enhanced ligand-dependent tumor necrosis factor receptor signaling. J Biol Chem. 2008;283(34):23419–23428. doi:10.1074/jbc.M802967200
- Fernández-Majada V, Welz P-S, Ermolaeva MA, et al. The tumour suppressor CYLD regulates the p53 DNA damage response. Nat Commun. 2016;7(1):12508. doi:10.1038/ncomms12508
- McCool KW, Miyamoto S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246(1):311–326. doi:10.1111/j.1600-065X.2012.01101.x
- Hur GM, Lewis J, Yang Q, et al. The death domain kinase RIP has an essential role in DNA damage-induced NF-κB activation. Genes Dev. 2003;17(7):873–882. doi:10.1101/gad.1062403
- Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145(1):92–103. doi:10.1016/j.cell.2011.02.023
- Blackwell K, Zhang L, Thomas GS, Sun S, Nakano H, Habelhah H. TRAF2 phosphorylation modulates tumor necrosis factor alpha-induced gene expression and cell resistance to apoptosis. Mol Cell Biol. 2009;29(2):303–314. doi:10.1128/MCB.00699-08
- Park E-S, Choi S, Shin B, et al. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-Sphingosine 1-Phosphate (S1P) interaction. J Biol Chem. 2015;290(15):9660–9673. doi:10.1074/jbc.M114.609685
- Alvarez SE, Harikumar KB, Hait NC, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–1088. doi:10.1038/nature09128
- Shi C-S, Kehrl JH. Activation of stress-activated protein Kinase/c-Jun N-terminal kinase, but not NF-κB, by the Tumor Necrosis Factor (TNF) receptor 1 through a TNF receptor-associated factor 2- and germinal center kinase related-dependent pathway. J Biol Chem. 1997;272(51):32102–32107. doi:10.1074/jbc.272.51.32102
- Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15(11):1419–1426. doi:10.1101/gad.888501
- Ventura -J-J, Kennedy NJ, Lamb JA, Flavell RA, Davis RJ. c-Jun NH 2 -terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor. Mol Cell Biol. 2003;23(8):2871–2882. doi:10.1128/MCB.23.8.2871-2882.2003
- Pomerantz JL. NF-kappa B activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18(23):6694–6704. doi:10.1093/emboj/18.23.6694
- Vince JE, Pantaki D, Feltham R, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (TNF) to efficiently activate NF-κB and to prevent TNF-induced apoptosis. J Biol Chem. 2009;284(51):35906–35915. doi:10.1074/jbc.M109.072256
- Zhou AY, Shen RR, Kim E, et al. IKKε-mediated tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep. 2013;3(3):724–733. doi:10.1016/j.celrep.2013.01.031
- Shen RR, Zhou AY, Kim E, Lim E, Habelhah H, Hahn WC. IκB kinase ε phosphorylates TRAF2 to promote mammary epithelial cell transformation. Mol Cell Biol. 2012;32(23):4756–4768. doi:10.1128/MCB.00468-12
- Annibaldi A, Wicky John S, Vanden Berghe T, et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol Cell. 2018;69(4):566–580.e5. doi:10.1016/j.molcel.2018.01.027
- Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36(5):831–844. doi:10.1016/j.molcel.2009.10.013
- Nakatsu Y, Matsuoka M, Chang T-H, et al. Functionally distinct effects of the C-Terminal regions of IKKε and TBK1 on Type I IFN production. PLoS One. 2014;9(4):e94999. doi:10.1371/journal.pone.0094999
- Sun S-C. CYLD: a tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differ. 2010;17(1):25–34. doi:10.1038/cdd.2009.43
- Marié I, Smith E, Prakash A, Levy DE. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol. 2000;20(23):8803–8814. doi:10.1128/MCB.20.23.8803-8814.2000
- Feltham R, Jamal K, Tenev T, et al. Mind bomb regulates cell death during TNF signaling by suppressing RIPK1ʹs cytotoxic potential. Cell Rep. 2018;23(2):470–484. doi:10.1016/j.celrep.2018.03.054
- Nakabayashi O, Takahashi H, Moriwaki K, et al. MIND bomb 2 prevents RIPK1 kinase activity-dependent and -independent apoptosis through ubiquitylation of cFLIPL. Commun Biol. 2021;4(1):80. doi:10.1038/s42003-020-01603-y
- Rajan N, Elliott RJR, Smith A, et al. The cylindromatosis gene product, CYLD, interacts with MIB2 to regulate Notch signalling. Oncotarget. 2014;5(23):12126–12140. doi:10.18632/oncotarget.2573
- Stempin CC, Chi L, Giraldo-Vela JP, High AA, Häcker H, Redecke V. The E3 ubiquitin ligase mind bomb-2 (MIB2) protein controls B-cell CLL/lymphoma 10 (BCL10)-dependent NF-κB activation. J Biol Chem. 2011;286(43):37147–37157. doi:10.1074/jbc.M111.263384
- Chen I-T, Hsu P-H, Hsu W-C, Chen N-J, Tseng P-H. Polyubiquitination of transforming growth factor β-activated Kinase 1 (TAK1) at Lysine 562 residue regulates TLR4-mediated JNK and p38 MAPK Activation. Sci Rep. 2015;5(1):12300. doi:10.1038/srep12300
- Zhao Y, Ma CA, Wu L, et al. CYLD and the NEMO zinc finger regulate tumor necrosis factor signaling and early embryogenesis. J Biol Chem. 2015;290(36):22076–22084. doi:10.1074/jbc.M115.658096
- Ahmed N, Zeng M, Sinha I, et al. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat Immunol. 2011;12(12):1176–1183. doi:10.1038/ni.2157
- Moreno-García ME, Sommer K, Rincon-Arano H, et al. Kinase-Independent Feedback of the TAK1/TAB1 Complex on BCL10 Turnover and NF-κB Activation. Mol Cell Biol. 2013;33(6):1149–1163. doi:10.1128/MCB.06407-11
- Le Clorennec C, Lazrek Y, Dubreuil O, et al. ITCH-dependent proteasomal degradation of c-FLIP induced by the anti-HER3 antibody 9F7-F11 promotes DR5/caspase 8-mediated apoptosis of tumor cells. Cell Commun Signal. 2019;17(1):106. doi:10.1186/s12964-019-0413-8
- Wei R, Xu LW, Liu J, et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev. 2017;31(11):1162–1176. doi:10.1101/gad.299776.117
- Tang Y, Joo D, Liu G, et al. Linear ubiquitination of cFLIP induced by LUBAC contributes to TNFα-induced apoptosis. J Biol Chem. 2018;293(52):20062–20072. doi:10.1074/jbc.RA118.005449
- Peltzer N, Darding M, Montinaro A, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature. 2018;557(7703):112–117. doi:10.1038/s41586-018-0064-8
- Lafont E, Kantari‐Mimoun C, Draber P, et al. The linear ubiquitin chain assembly complex regulates TRAIL ‐induced gene activation and cell death. EMBO J. 2017;36(9):1147–1166. doi:10.15252/embj.201695699
- Tu H, Tang Y, Zhang J, et al. Linear ubiquitination of RIPK1 on Lys612 regulates systemic inflammation via preventing cell death. J Immunol. 2021;207(2):602–612. doi:10.4049/jimmunol.2100299
- Kanayama A, Seth RB, Sun L, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15(4):535–548. doi:10.1016/j.molcel.2004.08.008
- Jaco I, Annibaldi A, Lalaoui N, et al. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell. 2017;66(5):698–710.e5. doi:10.1016/j.molcel.2017.05.003
- Emmerich CH, Ordureau A, Strickson S, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci. 2013;110(38):15247–15252. doi:10.1073/pnas.1314715110
- Zhang J, Clark K, Lawrence T, Peggie MW, Cohen P. An unexpected twist to the activation of IKKβ: TAK1 primes IKKβ for activation by autophosphorylation. Biochem J. 2014;461(3):531–537. doi:10.1042/BJ20140444
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424(6950):793–796. doi:10.1038/nature01803
- Lim JW, Kim H, Kim KH. Expression of Ku70 and Ku80 mediated by NF-κB and Cyclooxygenase-2 is related to proliferation of human gastric cancer cells. J Biol Chem. 2002;277(48):46093–46100. doi:10.1074/jbc.M206603200
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-κB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–5305. doi:10.1128/MCB.21.16.5299-5305.2001
- Sakamoto K, Hikiba Y, Nakagawa H, et al. Promotion of DNA repair by nuclear IKKβ phosphorylation of ATM in response to genotoxic stimuli. Oncogene. 2013;32(14):1854–1862. doi:10.1038/onc.2012.192
- Boehm JS, Zhao JJ, Yao J, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. doi:10.1016/j.cell.2007.03.052
- Creixell P, Schoof EM, Tan CSH, Linding R. Mutational properties of amino acid residues: implications for evolvability of phosphorylatable residues. Philos Trans R Soc B Biol Sci. 2012;367(1602):2584–2593. doi:10.1098/rstb.2012.0076
- Mészáros B, Hajdu-Soltész B, Zeke A, Dosztányi Z. Mutations of intrinsically disordered protein regions can drive cancer but lack therapeutic strategies. Biomolecules. 2021;11(3):381. doi:10.3390/biom11030381
- Pajkos M, Zeke A, Dosztányi Z. Ancient evolutionary origin of intrinsically disordered cancer risk regions. Biomolecules. 2020;10(8):1115. doi:10.3390/biom10081115
- Asselman J, De Coninck DI, Beert E, et al. Bisulfite sequencing with daphnia highlights a role for epigenetics in regulating stress response to microcystis through preferential differential methylation of serine and threonine amino acids. Environ Sci Technol. 2017;51(2):924–931. doi:10.1021/acs.est.6b03870
- Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13(8):559–571. doi:10.1038/nrc3563
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi:10.1038/nrc3711
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi:10.1016/j.ccr.2012.02.014
- Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. 2021. doi:10.1016/j.semcancer.2021.03.010
- Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi:10.1016/j.cell.2006.05.036
- Franklin DA, He Y, Leslie PL, et al. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. 2016;6(1):38067. doi:10.1038/srep38067
- Wang L, Lin Y, Zhou X, et al. CYLD deficiency enhances metabolic reprogramming and tumor progression in nasopharyngeal carcinoma via PFKFB3. Cancer Lett. 2022;532:215586. doi:10.1016/j.canlet.2022.215586
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-κB signaling. Cell. 2006;125(4):665–677. doi:10.1016/j.cell.2006.03.041
- Chang T-P, Vancurova I. Bcl3 regulates pro-survival and pro-inflammatory gene expression in cutaneous T-cell lymphoma. Biochim Biophys Acta - Mol Cell Res. 2014;1843(11):2620–2630. doi:10.1016/j.bbamcr.2014.07.012
- Erol A. IKK-mediated CYLD phosphorylation and cellular redox activity. Mol Med. 2022;28(1):14. doi:10.1186/s10020-022-00439-y
- Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43. doi:10.1016/j.ccell.2018.03.022
- Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-κB in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2010;396(4):901–907. doi:10.1016/j.bbrc.2010.05.019
- Haq S, Sarodaya N, Karapurkar JK, et al. CYLD destabilizes NoxO1 protein by promoting ubiquitination and regulates prostate cancer progression. Cancer Lett. 2022;525:146–157. doi:10.1016/j.canlet.2021.10.032
- Skonieczna M, Hejmo T, Poterala-Hejmo A, Cieslar-Pobuda A, Buldak RJ. NADPH oxidases: insights into selected functions and mechanisms of action in cancer and stem cells. Oxid Med Cell Longev. 2017;2017:1–15. doi:10.1155/2017/9420539
- Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci. 2007;104(21):8869–8874. doi:10.1073/pnas.0703268104
- Liccardi G, Ramos Garcia L, Tenev T, et al. RIPK1 and Caspase-8 ensure chromosome stability independently of their role in cell death and inflammation. Mol Cell. 2019;73(3):413–428.e7. doi:10.1016/j.molcel.2018.11.010
- li D, Gao J, Yang Y, et al. CYLD coordinates with EB1 to regulate microtubule dynamics and cell migration. Cell Cycle. 2014;13(6):974–983. doi:10.4161/cc.27838
- Wu J, Huang Y-F, Zhou X-K, et al. Skp2 is required for Aurora B activation in cell mitosis and spindle checkpoint. Cell Cycle. 2015;14(24):3877–3884. doi:10.1080/15384101.2015.1120916
- Sun L, Gao J, Dong X, et al. EB1 promotes Aurora-B kinase activity through blocking its inactivation by protein phosphatase 2A. Proc Natl Acad Sci. 2008;105(20):7153–7158. doi:10.1073/pnas.0710018105
- Tauriello DVF, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/β-Catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell. 2010;37(5):607–619. doi:10.1016/j.molcel.2010.01.035
- van Andel H, Kocemba KA, de Haan-Kramer A, et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene. 2017;36(15):2105–2115. doi:10.1038/onc.2016.368
- Yang Y, Liu M, Li D, et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proc Natl Acad Sci. 2014;111(6):2158–2163. doi:10.1073/pnas.1319341111
- Wickström SA, Masoumi KC, Khochbin S, Fässler R, Massoumi R. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010;29(1):131–144. doi:10.1038/emboj.2009.317
- Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi:10.1038/417455a
- Lee Y-S, Lim K-H, Guo X, et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68(18):7561–7569. doi:10.1158/0008-5472.CAN-08-0188
- Rodgers MA, Bowman JW, Fujita H, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med. 2014;211(7):1333–1347. doi:10.1084/jem.20132486
- Rébé C, Ghiringhelli F. Interleukin-1β and Cancer. Cancers. 2020;12(7):1791. doi:10.3390/cancers12071791
- Chariot A, Leonardi A, Müller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the IκB Kinase (IKK) regulator NEMO connects IKK complexes with IKKε and TBK1 kinases. J Biol Chem. 2002;277(40):37029–37036. doi:10.1074/jbc.M205069200
- Tu D, Zhu Z, Zhou AY, et al. Structure and ubiquitination-dependent activation of TANK-binding Kinase 1. Cell Rep. 2013;3(3):747–758. doi:10.1016/j.celrep.2013.01.033
- Durand J, Zhang Q, Baldwin A. Roles for the IKK-related kinases TBK1 and IKKε in cancer. Cells. 2018;7(9):139. doi:10.3390/cells7090139
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi:10.1016/j.cell.2007.06.009
- Xie X, Zhang D, Zhao B, et al. IκB kinase ε and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci. 2011;108(16):6474–6479. doi:10.1073/pnas.1016132108
- Goktuna Sİ. IKBKE inhibits TSC1 to activate the mTOR/S6K pathway for oncogenic transformation. TURKISH J Biol. 2018;42(4):268–278. doi:10.3906/biy-1801-57
- Bodur C, Kazyken D, Huang K, et al. The IKK‐related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists. EMBO J. 2018;37(1):19–38. doi:10.15252/embj.201696164
- Tooley AS, Kazyken D, Bodur C, Gonzalez IE, Fingar DC. The innate immune kinase TBK1 directly increases mTORC2 activity and downstream signaling to Akt. J Biol Chem. 2021;297(2):100942. doi:10.1016/j.jbc.2021.100942
- Blagosklonny MV. Answering the ultimate question “What is the Proximal Cause of Aging?”. Aging. 2012;4(12):861–877. doi:10.18632/aging.100525
- Blagosklonny MV. The hyperfunction theory of aging: three common misconceptions. Oncoscience. 2021;8:103–107. doi:10.18632/oncoscience.545
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi:10.1038/nrm3025
- Page A, Navarro M, Suárez-Cabrera C, Bravo A, Ramirez A. Context-dependent role of IKKβ in cancer. Genes. 2017;8(12):376. doi:10.3390/genes8120376
- Wei Y, Zhao Q, Gao Z, et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J Clin Invest. 2019;129(8):3347–3360. doi:10.1172/JCI127726
- Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.584626
- Belgnaoui SM, Paz S, Samuel S, et al. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe. 2012;12(2):211–222. doi:10.1016/j.chom.2012.06.009
- Musella M, Galassi C, Manduca N, Sistigu A. The Yin and Yang of Type I IFNs in cancer promotion and immune activation. Biology. 2021;10(9):856. doi:10.3390/biology10090856
- Baydoun HH, Bai XT, Shelton S, Nicot C. HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS One. 2012;7(8):e42226. doi:10.1371/journal.pone.0042226
- Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13(7):443–454. doi:10.1038/nrc3537
- Pannem RR, Dorn C, Ahlqvist K, Bosserhoff AK, Hellerbrand C, Massoumi R. CYLD controls c-MYC expression through the JNK-dependent signaling pathway in hepatocellular carcinoma. Carcinogenesis. 2014;35(2):461–468. doi:10.1093/carcin/bgt335
- Poli V, Fagnocchi L, Fasciani A, et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat Commun. 2018;9(1):1024. doi:10.1038/s41467-018-03264-2
- Alameda JP, Ramírez Á, García-Fernández RA, et al. Premature aging and cancer development in transgenic mice lacking functional CYLD. Aging. 2019;11(1):127–159. doi:10.18632/aging.101732
- Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129(1):9–16. doi:10.1242/jcs.175521
- Strobelt R, Adler J, Paran N, et al. Imatinib inhibits SARS-CoV-2 infection by an off-target-mechanism. Sci Rep. 2022;12(1):5758. doi:10.1038/s41598-022-09664-1
- Hantschel O, Wiesner S, Güttler T, et al. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell. 2005;19(4):461–473. doi:10.1016/j.molcel.2005.06.030
- Yuan Z-M, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399(6738):814–817. doi:10.1038/21704