
Abstract
Alpha 1 antitrypsin deficiency is a hereditary condition characterized by low alpha 1 proteinase inhibitor (also known as alpha 1 antitrypsin [AAT]) serum levels. Reduced levels of AAT allow abnormal degradation of lung tissue, which may ultimately lead to the development of early-onset emphysema. Intravenous infusion of AAT is the only therapeutic option that can be used to maintain levels above the protective threshold. Based on its biochemical efficacy, AAT replacement therapy was approved by the US Food and Drug administration in 1987. However, there remained considerable interest in selecting appropriate outcome measures that could confirm clinical efficacy in a randomized controlled trial setting. Using computed tomography as the primary measure of decline in lung density, the capacity for intravenously administered AAT replacement therapy to slow and modify the course of disease progression was demonstrated for the first time in the Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency (RAPID) trial. Following these results, an expert review forum was held at the European Respiratory Society to discuss the findings of the RAPID trial program and how they may change the landscape of alpha 1 antitrypsin emphysema treatment. This review summarizes the results of the RAPID program and the implications for clinical considerations with respect to diagnosis, treatment and management of emphysema due to alpha 1 antitrypsin deficiency.
Introduction
Alpha 1 antitrypsin deficiency (AATD) is a hereditary genetic disorder characterized by low serum levels of alpha 1 protease inhibitor (A1-PI; also known as alpha 1 antitrypsin [AAT]). In healthy individuals, AAT acts to inhibit nonspecific destruction by the serine protease neutrophil elastase (NE), an enzyme that can attack lung elastin and damage bronchial and alveolar wall integrity. The most widely recognized mechanism of action is associated with protease–antiprotease imbalance hypothesis of lung disease.Citation1 In this model, the pathogenesis of pulmonary emphysema occurs as a result of the imbalance between AAT and NE, driving excessive proteolysis that degrades alveolar and interstitial lung tissue. In patients with AATD, reduced levels of AAT result in destruction of lung tissue by NE, resulting in lung-related symptoms such as shortness of breath, wheezing, coughing and dyspnea.Citation2 In most cases, these symptoms appear between the ages of 20 and 40.Citation3,Citation4
Expression levels of AAT are determined in a codominant manner by a variety of mutations in the SERPINA1 gene.Citation5 The normal allele PI*M is associated with the genotype PI*MM in healthy individuals and is characterized by normal AAT levels (20–53 µM)Citation6 and a low risk of emphysema. Common pathogenic alleles include PI*S, PI*Z and the less-common Null alleles. Homozygosity for the PI*S allele results in moderately reduced serum levels, while the PI*ZZ and homozygous null genotypes are associated with very low or undetectable serum AAT, respectively, and a high risk of developing rapidly progressive emphysema.Citation7,Citation8 The majority of patients identified with an AAT deficiency have the PI*ZZ genotype (>90%),Citation9,Citation10 and these patients typically present with serum AAT levels well below the 11 µM protective threshold.Citation11 Individuals with a PI*SZ genotype may also have serum AAT concentrations below the protective threshold, whereas PI*MZ genotypes usually have concentrations within or just below the normal range. Intravenous AAT replacement therapy is the only available treatment that addresses the underlying cause of disease, aiming to raise serum levels above this protective threshold.Citation12 Symptomatic treatments such as bronchodilators and corticosteroids have been shown to relieve symptoms of dyspnea and improve exercise capacity, but have not been shown to alter the progression of emphysema.Citation13,Citation14
AATD is a progressive lung disease, and early diagnosis allows patients to implement lifestyle changes and begin treatment options that slow further loss of lung tissue. However, data suggest that AATD may be underdiagnosed; evidence from screening programs in the USA suggests that fewer than 10% of patients have been diagnosed.Citation15–Citation18 Patients often face delays or are misdiagnosed, for example, with COPD or asthma, due to the nonspecific nature of respiratory symptoms observed with AATD. An average delay of 5.6±8.3 years between the initial presentation of symptoms and diagnosis has been reported, and the mean age of first diagnosis is 43.9 years.Citation19 There is, therefore, a need to identify symptomatic patients who may benefit from treatment and those at risk who would benefit from counseling and increased monitoring. Physicians may lack awareness of the disease, available means of testing and available treatment options, further contributing to inaccurate or underdiagnosis.Citation20 AAT replacement therapy is the only available treatment known to affect the underlying cause of the disease; however, it is not currently available in all countries.
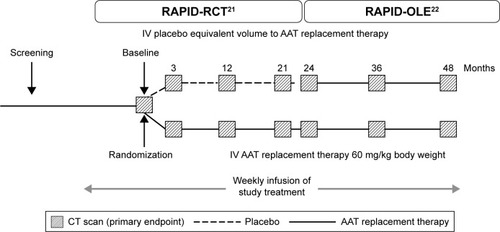
The clinical efficacy of AAT replacement therapy was assessed in the Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency (RAPID) program (), which consisted of an initial 2-year randomized, double-blind, placebo-controlled study (RAPID-RCT [randomized controlled trial]; N=180; 60 mg/kg/week A1-PI or placebo) followed by a 2-year open-label extension study (RAPID-OLE [open-label extension], N=140; 60 mg/kg/week A1-PI).Citation21,Citation22 In light of these data, and the challenges faced in the treatment of AATD, a symposium was held at the European Respiratory Society (ERS) Annual Conference 2016 to discuss these findings and recommendations for clinical practice. This review aims to explore the results of the RAPID program with respect to the use of computed tomography (CT) as a sensitive and specific measure of disease progression and its value when aiming to prove clinical efficacy. The specific features of the trial design are presented as a key component, allowing for the exploration of disease-modifying effects. The review focuses on the wider implications for the treatment of AATD and how new outcome measures and dose regimens, for example, 120 mg/kg weekly or every 2 weeks, can be incorporated into the current treatment landscape. Specific emphasis is placed on challenges associated with diagnosis and monitoring of patients and the timing of therapy. The review also discusses the various AAT therapy and lifestyle guidelines for AATD and how trial data may influence future treatment regimens.
Figure 1 Study design of the RAPID-RCT and RAPID-OLE trials employing lung density measures by CT scans at 0, 3, 12, 21, 24, 36 and 48 months.
Discussion
Evidence for clinical efficacy of AAT – update following completion of the RAPID clinical trial program
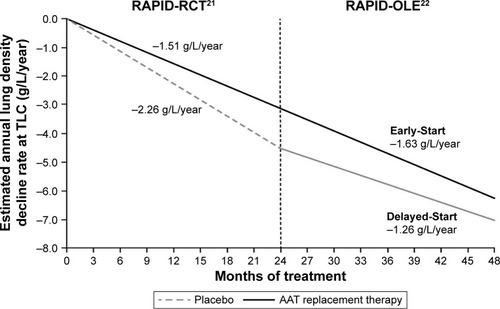
Early studies utilized FEV1 as a traditional surrogate marker for monitoring disease progression in COPD;Citation23–Citation25 however, changes in FEV1 occur slowly over time, and there are several limitations to its use.Citation23 The RAPID trial utilized CT densitometry as a more reliable, reproducible and sensitive tool for assessing lung function decline in patients with AATD.Citation23,Citation26,Citation27 CT densitometry has been shown to correlate with traditional outcome measures, for example, mortality and health status, and also with FEV1 decline.Citation28 During the RAPID-RCT, lung density decline at total lung capacity was significantly reduced in patients receiving AAT therapy compared with placebo (−1.51 versus −2.26 g/L/year, respectively, p=0.033; ).Citation21 After completion of the RAPID program, patients who received active therapy across all 4 years were referred to as the Early-Start group. Patients who initially received placebo during RAPID-RCT, who subsequently switched to active treatment in RAPID-OLE, were referred to as the Delayed-Start group. In RAPID-OLE, the beneficial effect of treatment over the first 2 years was maintained in the Early-Start subgroup of the patient population and remained statistically significant relative to the Delayed-Start group (−1.63 versus −1.26 g/L/year at total lung capacity, p=0.04). During RAPID-OLE, a statistically significant reduction in the rate of lung density decline was established in the Delayed-Start group temporal to the switch from placebo to active therapy at year two, reflecting a mean preservation of 0.52 g/L/year (p=0.001).Citation22 Despite this, patients in the Delayed-Start group were unable to regain lung tissue lost during the placebo treatment period and did not “catch up” to patients in the Early-Start group, demonstrating a disease-modifying effect of AAT therapy in patients with AATD.
Figure 2 Annualized rate of decline in physiologically adjusted P15 lung density (g/L) at TLC over 48 months.
Abbreviations: AAT, alpha 1 antitrypsin; adjusted P15, lung volume-adjusted 15th percentile of the lung density; OLE, open-label extension; RAPID, Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency; RCT, randomized controlled trial; TLC, total lung capacity.
The RAPID program was the first to demonstrate significant clinical efficacy, and the findings build on evidence from previous observational studies and randomized controlled trials (RCTs; summarized in ). Two previous RCTs utilized CT densitometry to investigate the efficacy of AAT therapy; in the Danish-Dutch trial, 56 ex-smokers possessing an AAT deficiency of the Pi*ZZ phenotype and moderate emphysema participated in a double-blind trial of AAT replacement therapy. CT lung density measurements were used to observe a supportive trend toward protection against the loss of lung tissue in these patients after monthly administration of 250 mg/kg of AAT.Citation29 A decade later, the EXAcerbations and Computed Tomography scan as Lung End-points (EXACTLE) trial aimed to study the use of CT densitometry as a means of ascertaining the therapeutic effect of AAT therapy using the previous trial as a basis for the study methodology. Patients with severe AATD (N=77) were randomized to 60 mg/kg weekly infusions of AAT therapy or placebo for at least 2 years. The loss of lung density in this study favored those receiving AAT therapy over the entire duration of the trial period.Citation30 While these trials alone did not reach unequivocal statistical significance, a pooled analysis combining data from both studies indicated a significant reduction in lung density decline following AAT treatment.Citation27
Table 1 Characteristics of clinical trials and observational studies using CT densitometric indices as outcome measures for emphysema progression
In addition to data from RCTs, there is a large body of evidence in support of clinical efficacy derived from observational studies. Several observational and registry studies have demonstrated an effect on FEV1 within specific FEV1 ranges.Citation35–Citation39 The largest of these studies, the AATD deficiency registry study (N=1,129) by the National Heart, Lung, and Blood Institute, demonstrated a significant treatment benefit in patients with severe impairment (35%–49% predicted) of lung function.Citation37 Similarly, the Danish-German comparison (N=295) showed the most pronounced difference in the group of patients with FEV1 31%–65% predicted.Citation36 In a study of patients with severe AATD (N=96), Wencker et al demonstrated that the greatest benefit in slowing the decline in lung function was observed in a subgroup of patients with mildly and moderately impaired lung function (baseline FEV1 >65% predicted), who had also been subject to a rapid decline in FEV1.Citation39. These findings have not been replicated in clinical studies by Dirksen et al, mainly due to smaller sample sizes and length of follow-up (N=77, 2–2.5 years of follow-up; N=56, 5 years of follow-up).Citation29,Citation30 When data from these clinical studies were pooled, the rate of FEV1 decline was demonstrated to be 23% slower in patients receiving AAT therapy, with the difference predominantly seen in patients with FEV1 30%–65% predicted.Citation40
In addition to FEV1, earlier studies also utilized quality of life (QoL), exacerbation and mortality as endpoints for clinical efficacy in RCTs. These parameters are less sensitive than other endpoints, and the trials utilizing them were not suitably powered to observe a reliable difference in the clinical outcomes. As a consequence, the effect of AAT replacement therapy on these measures was not confirmed in these studies.Citation29,Citation30 In contrast, the AATD registry has demonstrated a statistically lower mortality rate in patients receiving AAT replacement therapy compared with non-treated subjects, an effect predominantly observed in patients with an FEV1 <50% predicted.Citation37 Mortality in both mild and moderate lung disease is low; therefore, this apparent difference in mortality between patients with severe and mild lung disease is not surprising.Citation32 These findings have not been replicated in clinical trials; much larger sample sizes and longer duration placebo-controlled trials would be needed to show a significant difference. Given the rarity of AATD, such clinical trials would be impractical. It would be difficult to recruit sufficient patients in countries where AAT is already licensed. More importantly, given the significant body of evidence which now support the efficacy of AAT therapy, the extended duration of placebo treatment is unethical.
The impact and significance of disease modification in AATD
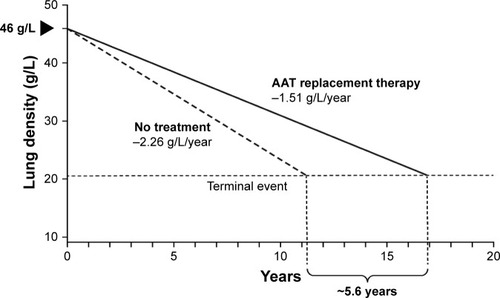
Disease modification can be defined as an improvement or stabilization of a disease state resulting from a reduction in the rate of disease progression that occurs following therapeutic intervention, which may persist after the intervention is discontinued.Citation41 It exerts its effects on the underlying pathology or pathophysiology of the disease, rather than the symptoms alone. The key hallmark of a disease-modifying treatment is the capacity to alter the course of the disease and have a beneficial effect on clinically significant trial endpoints (). RAPID-RCT and RAPID-OLE were the first trials to demonstrate the disease-modifying effect of AAT replacement therapy on emphysema progression. During RAPID-RCT, patients receiving active therapy achieved statistically significant reductions in the annual loss of lung tissue as compared with those receiving placebo. Continuous active treatment over 4 years favored the Early-Start group. Upon switching to active therapy, the Delayed-Start group demonstrated a statistically significant response to therapy, while lung tissue lost during the period of placebo treatment was never regained.Citation21,Citation22 This demonstrates that treatment with AAT replacement therapy is disease modifying, altering the course of disease progression, which has important implications for treatment. Early intervention, particularly in patients with fast lung density decline, would be beneficial to preserve functional lung tissue. Previous clinical studies failed to demonstrate this effect due to inadequate trial design or the use of less-sensitive clinical endpoints, such as lung function/spirometry (eg, FEV1).Citation23 Disease modification has significant implications for the design of future clinical trials. Following publication of data from the RAPID program, there is now a large body of evidence that confirms the efficacy of AAT replacement therapy. Although clinical studies have not demonstrated significant effects on mortality, given the large number of patients required and length of follow-up, it may not be ethical or feasible to conduct further placebo-controlled studies to assess this endpoint. Owing to the slow progression of AATD, Schluchter et al estimated that a trial of 684 patients with a baseline FEV1 of 35%–49%, studied over 5 years (recruited over the first 2 years and followed subsequently for a further 3 years) would be necessary to observe a 40% reduction in mortality.Citation42 Evidence from a post hoc analysis of the RAPID program suggests a mortality benefit following AAT treatment. During the program, the time required for progressive emphysema to develop into respiratory crisis was used to simulate the life-years gained as a result of AAT replacement therapy. Respiratory crisis was defined as death, lung transplant or a crippling respiratory condition. Seven patients withdrew with an average terminal lung density of 20 g/L. Using the average baseline lung density for all subjects (46 g/L) and the rate of decline in lung density in AAT versus placebo-treated patients, the projected time to terminal lung density was 16.9 years for those receiving AAT replacement therapy, compared with 11.3 years in the placebo group (). This indicates a gain in life-years of ~5.6 years with AAT treatment.Citation22 Although conducted in a small sample size, these data are supported by results from the National Heart, Lung, and Blood Institute observational study showing that patients receiving AAT replacement therapy had a greater survival than those not receiving treatment.Citation37
Figure 3 Change in clinical outcome measures after administration of a disease-modifying therapy.
Notes: Reproduced with permission from Taylor & Francis. The Version of Scholarly Record of this Article is published in COPD: Journal of Chronic Obstructive Pulmonary Disease (2016), available online at: http://www.tandfonline.com/10.1080/15412555.2016.1178224. This article was distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives license. Disease Modification in Emphysema Related to Alpha-1 Antitrypsin Deficiency, COPD: Journal of Chronic Obstructive Pulmonary Disease, Chorostowska-Wynimko J, Vol 13, pp. 807–815, published online: 12 May 2016, http://www.tandfonline.com reprinted by permission of the publisher.Citation23
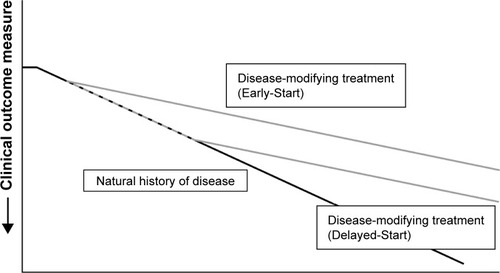
Figure 4 Extrapolation of the effect of AAT replacement therapy on the predicted time to reach terminal respiratory function in RAPID-RCT.
Abbreviations: AAT, alpha 1 antitrypsin; RAPID, Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency; RCT, randomized controlled trial.
These data also highlight the utility of CT as a clinical measure to monitor emphysema progression in AATD. It is now widely accepted that CT is the most sensitive measure for monitoring emphysema progression.Citation24 Furthermore, CT has been shown to correlate with other indices, such as pulmonary function, and is a better predictor of mortality than lung function.Citation27,Citation43 During the RAPID clinical trial program, CT was shown to correlate with several secondary endpoints, further validating the use of CT as a clinical endpoint. Significant correlations were observed for spirometry values, such as FEV1% predicted (r=0.338, p=0.0002).Citation22 Similar correlations between FEV1 and CT have been observed in other cross-sectional studies.Citation26,Citation44
Clinical considerations for the treatment of patients with AATD
The RAPID program, in conjunction with previous observational work and RCTs, has provided compelling evidence for the efficacy of AAT therapy. These data also highlight the importance of early detection and intervention in order to enable patients to receive appropriate treatment and preserve functional lung tissue. However, guidance for the treatment of AATD is limited; a previous statement from the American Thoracic Society (ATS)/European Respiratory Society (ERS)Citation45 precedes the RAPID program and there is a need for improved guidance on the practical aspects of AATD treatment. In view of this, during the ERS Expert Forum, the topics of monitoring AATD, identification of patients who would benefit from treatment and differences between treatment options were discussed. Considerations from these discussions are reviewed below.
Monitoring patients with AATD
There is considerable variability in the types and frequency of measures used to monitor disease progression in patients with AATD, and there is no clear consensus on baseline assessment and how and when patients should be monitored. Guidance published by Silverman and Sandhaus recommend full pulmonary and liver function tests in an initial consultation and further annual tests, with potential consideration for initial chest radiographs and baseline CT scans.Citation46 In contrast, the Spanish guidelines differ in the recommended frequency of spirometry and radiographic measures of lung function. Quarterly and biannual assessments are suggested, respectively, with additional annual testing of lung volume, transfer factor for carbon monoxide and liver function.Citation47
While many guidelines suggest regularly monitoring patients using spirometric measures of lung function, such as FEV1, these measures may not be the most sensitive. In AATD, FEV1 has been shown to change slowly over time and is subject to a considerable degree of inter- and intra-patient variability.Citation23,Citation48 Intra-patient variability can be attributed to technical factors, such as instrument performance, as well as observer and subject procedural errors. Additionally, intra-patient factors, such as the extent of airway obstruction, changes in bronchial tone and diurnal variations in FEV1, can contribute to further variability.Citation49–Citation51 Previous analysis has revealed a mean between-test difference in FEV1 of 0.1±0.1 L, which exceeds the volume change in FEV1 observed after intervention with AAT replacement therapy,Citation36 as well as the annual loss of lung function in patients classed as fast decliners.Citation52,Citation53 These factors can make it difficult to reliably detect changes in disease progression or treatment efficacy and create difficulties when attempting to set broadly applicable FEV1 ranges on which to base treatment recommendations.Citation54
Considering these shortcomings, other measures, including diffusing capacity of the lung for carbon monoxide (DLCO) or CT, may be more sensitive for monitoring patients with AATD. The US Food and Drug Administration has acknowledged the value of serial high-resolution CT lung densitometry as a more valuable outcome measure of the progression of emphysema and the effectiveness of AAT replacement therapy.Citation55 This is derived from the various studies showing that CT parameters correlate with anatomic pathology and various pulmonary function tests (). Despite these results, sequential quantitative CT scans of the lungs are not recommended as part of the routine assessment of emphysema progression in the current Global Initiative for Chronic Obstructive Lung Disease guidelines.Citation56 While quantitative CT scanning has been widely used in clinical trials, there remain a number of challenges to its wider adoption as part of standard clinical methods. These are predominantly associated with a lack of access to suitable CT scanners, the requirement for skilled operators to perform the acquisition and a lack of standardized methodologies among available CT imaging protocols. A number of studies have been dedicated to ensuring optimization and repeatability in lung densitometry measures with respect to choice of densitometric parameters,Citation57 data calibration,Citation57 standardization of CT acquisition protocol,Citation43 patient inspiration levelCitation58 and image processing.Citation59 A further concern regarding the wider implementation of CT scanning for determination of emphysema progression is the issue of subjecting patients to repeated doses of ionizing radiation. However, studies have already shown that low-dose CT measurements differ only marginally from those obtained with standard-dose scans.Citation60 Furthermore, the cumulative dose for patients experiencing multiple CT scans remains fairly low.Citation61 A typical two-view chest X-ray can expose a patient to 0.1 mSv of radiation. Effective low-dose protocols for quantitative CT are possible with exposures of ~0.5–1.5 mSv,Citation61 which is below the annual background radiation of 3 mSv/year observed in the USA.Citation62
In order to improve the monitoring of patients with AATD, clear consensus on the types and frequency of measurements is needed. In addition to highlighting measures that would be useful in the majority of patients (eg, FEV1, CT), it would also be useful to acknowledge the need to expand monitoring tests in some instances, for example, to include liver function tests or QoL questionnaires. For all patients, baseline measurements are essential to track disease progression and identify fast decliners who may obtain greater benefit from treatment.
Identifying the right patient and when to initiate treatment
Previous evidence from observational and registry studies indicated that AAT replacement treatment was most effective within certain FEV1 ranges, for example, between 30% and 65% predicted,Citation37,Citation40 and current guidance for treatment has been limited to patients who fall within these restrictions. This can lead to the neglect of patients outside of these parameters who may benefit from treatment. The more recently published Alpha-1 Global Foundation recommendations for the diagnosis and management of AATD in adult patientsCitation63 continue to support the treatment of patients with FEV1 between 30% and 65% and continuing therapy when the FEV1 falls below 30%, when lung transplantation becomes a viable treatment option.
Evidence is beginning to emerge demonstrating that there may be value in identifying and treating patients outside of established parameters. First, numerous testing initiatives are ongoing with the aim of improving the detection of broader genotypes containing new deficiency alleles currently being identified. Large-scale screenings in general populations, students, newborns and blood donors, as well as new case finding strategies that target patients with COPD and asthma, will aid in the discovery of new patients in need of treatment.Citation64 Furthermore, incorporation of SERPINA1 sequencing and/or next-generation sequencing methods in the diagnostic routine will also help recognize patients with rare and new pathologic alleles misdiagnosed by standard testing algorithms.Citation65,Citation66 Second, the evidence of disease modification and the potential mortality benefit with AAT replacement therapy from the RAPID program demonstrate the value of earlier treatment intervention where FEV1 is more likely to be outside of the recommended treatment bracket. This is reflected in updated recommendations from the Alpha-1 Foundation from the USA, which incorporate more inclusive parameters with broader treatment ranges.Citation63 Indeed, the labels of second-generation products are beginning to reflect these broader parameters in the treatment recommendations. The previous statement from the ATS/ERS is currently being updated and is likely to include recommended restrictions.
The Alpha-1 Foundation guidelines also address concerns relating to costs associated with testing and treating patients. The guidelines place a high value on complete baseline and follow-up pulmonary function testing to better identify patients with accelerated lung function decline and a low value on the costs associated with these tests. These can range from €13.40 to €33, and for sequencing can be up to €150 per test.Citation67 Furthermore, the guidelines indicate that a higher value (compared to the cost of treatment) can be placed on the potential for AAT replacement therapy to prolong survival in patients with FEV1 <30%–65% predicted.Citation63 The cost of augmentation therapy can range from $60,000 to $150,000 annually and depends on several factors including body weight, pricing and the cost of nursing care.Citation46 Importantly, reduced symptom severity and decreased hospitalizations can help to offset the costs associated with treatment.Citation68 Furthermore, the cost of AAT treatment is comparable or lower than that of other rare pulmonary diseases such as idiopathic pulmonary fibrosis (approximate annual drug cost for Nintedanib =$110,000) or cystic fibrosis (approximate annual drug cost for Ivacaftor =$325,000).Citation69 Due to the lower rates of lung density decline in individuals receiving intravenous AAT replacement therapy, as well as the potential to extend the time to terminal lung function as shown in the RAPID program, there is now renewed scope for the exploration of the overall cost–benefit of such a therapeutic intervention.
The disease-modifying implications of the RAPID program results based on CT densitometric parameters stress the importance of early intervention at the first signs of emphysema and the importance of targeting patients in the lower ranges of FEV1 deterioration. Updated guidance to reflect these recent findings is needed to emphasize the importance of early recognition, which allows clinicians to prevent, recognize and treat potential complications of emphysema such as hypoxemiaCitation70 and frequent exacerbations.Citation71 Furthermore, earlier recognition will allow for the recommendation of restraint from deleterious lifestyle habits, including cigarette smoking, which are known to accelerate the progression of emphysema. Importantly, not all patients with AATD have access to replacement therapy and not all patients experience lung function decline.Citation72 Whether AAT replacement therapy would be beneficial to all individuals with AATD is unclear. There is evidence that non-AATD patients with COPD can experience stabilization by simply giving up smoking,Citation48 and there is anecdotal evidence for a similar effect in AATD patients. As the rate of lung density decline differs between patients with AATD, personalized treatment approaches may be beneficial. These approaches aim to restrict AAT therapy to patients who are likely to gain the most benefit, for example, “rapid decliners”, who experience substantial annual FEV1 loss.Citation72 However, there is a lack of evidence for treatment within this subgroup and limited information on how to identify them.
Treatment of patients with intravenous AAT replacement and continuation of treatment
Current licensed treatment for AATD includes weekly infusions (60 mg/kg/week) of AAT; a variety of preparations are available, some of which may have advantages for patients. These second-generation products offer superior purity and, hence, higher specific activity,Citation73 which allows for faster infusion times, making treatment more convenient for patients. The differences between AAT preparations highlight a need for increased awareness of the available treatment options and the potential differences in individual patient tolerance. In addition, alternative dosing strategies, including bi-weekly dosing, higher doses or self-administration, may hold hope for improved efficacy of treatment in the future, as discussed below.
Intravenous AAT replacement therapy derived from purified pooled human plasma is currently recommended for 60 mg/kg weekly infusions by the ATS/ERS and updated US guidelines.Citation45,Citation63 This is based primarily on data from early biochemical studies demonstrating the efficacy of treatment in maintaining serum levels above the protective threshold of 11 µM.Citation74–Citation81 Data from the RAPID program reinforced these recommendations, demonstrating that 60 mg/kg weekly infusions maintained serum levels above the protective threshold and were able to significantly slow the decline in lung density. The Spanish guidelines suggest some value in alternative dosing intervals, such as 180 mg/kg every 3 weeks and 120 mg/kg every 2 weeks.Citation47
Alternative dosing regimens may provide a means to improve patient convenience. There is an increasing body of evidence for the safety and efficacy of alternative dosing intervals, such as bi-weekly dosing. During RAPID-RCT, patients were administered a higher bi-weekly dose of 120 mg/kg to account for periods where they may be unable to receive the normal weekly infusion (eg, during vacation). The 120 mg/kg bi-weekly dose achieved serum levels above the 11 µM protective threshold and was well tolerated, causing no serious adverse events.Citation82 In addition, regimens of 100/120 mg/kg every 2 weeks, 150/180 mg/kg every 3 weeks and 250 mg/kg every 4 weeks have also been studied in pharmacokinetic simulations to estimate the individual optimal dose for AAT replacement therapy. While infusions of 100/120 mg/kg every 2 weeks can sustain serum AAT levels above the protective threshold,Citation83 longer infusion intervals, such as monthly infusions of 250 mg/kg, failed to maintain the protective serum concentrations across the full 28 days of the dose window.Citation29 In order to prove the efficacy and safety of alternative dosing intervals, and of higher doses, further clinical trials are ongoing. For example, the upcoming SPARTA trial (NLM identifier: NCT1983241) will aim to further explore the efficacy of double dosing regimens (120 mg/kg) administered weekly over 3 years.Citation84
Finally, alternative means of treatment administration, such as home treatment or self-administration, may provide further options to improve convenience for patients. These methods have been employed with some degree of success; Wilke and Grohe performed a prospective study tracking seven patients over 3 years as they received nurse-based intravenous AAT replacement therapy at home. The results showed that home-based AAT replacement therapy was feasible, as there were few complications related to home infusion and less exacerbations and loss of lung function with respect to the historical cohort.Citation85 Self-administration is also an option for some patients; this has been successfully used in a number of indications, such as hereditary angioedemaCitation86 and hemophilia,Citation87,Citation88 and within the USA, it is provided for a number of patients with AATD. In order to implement these measures, training and awareness resources are required to support physicians, nurses and patients. Clinicians should be aware of these improved convenience options and of the differences in available products, so as to deliver a more personalized, patient-centric approach to AATD treatment.
Value of lifestyle modifications
Although AAT replacement therapy is the only available treatment that addresses the underlying cause of AATD, symptomatic treatment and lifestyle modifications may be of benefit to patients. However, managing the care of patients beyond the prescription of AAT therapy is often overlooked by physicians. Current guidelines, although broad, make few concrete recommendations on lifestyle modifications. The current ATS/ERS statement and US guidelines take a firm stance on smoking cessation, and patients are encouraged to maintain a level of physical activity while avoiding exercise that may lead to hyperventilation.Citation45,Citation63 Despite the recommendation of continued physical activity, unlike the guidelines for COPD, they do not provide comprehensive practical guidance on the types, duration and frequency of exercise that may be beneficial for AATD patients.
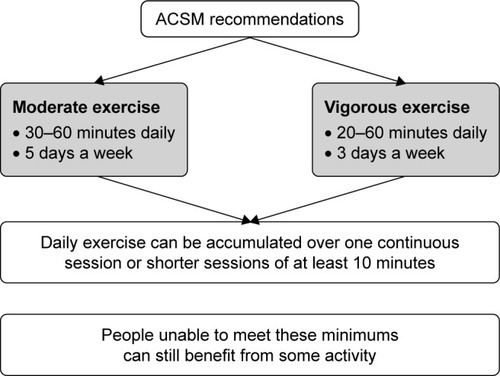
There may be potential to transfer the recommendations provided for COPD to AATD with respect to detailed physical activity and pulmonary rehabilitation guidelines. Guidelines from the Canadian Thoracic Society recognize the importance of nonpharmacologic methods for optimal disease management, such as pulmonary rehabilitation provided by specific exercise regimes.Citation89 Additionally, the American College of Sports Medicine recommends moderate or vigorous aerobic exercise and moderate intensity resistance training to improve the capacity to perform everyday tasks and enhance QoL ().Citation90 Pulmonary rehabilitation has been acknowledged as a valuable treatment for those suffering from lung disease, as it can improve the lung function, general exercise stamina and QoL and facilitate a reduced burden on health care resources.Citation91,Citation92 In particular, exercise capacity has been shown to be significantly related to the CT pixel index observed in patients with centrilobular emphysema.Citation93 Exercise capacity has also been shown to be a better predictor of health status than other parameters based on lung imaging or physiologic impairment.Citation94 There is, therefore, a need to provide greater guidance on pulmonary rehabilitation that is appropriate for patients with AATD. In addition, further guidance on additional measures may be warranted, including limiting the exposure to airborne pollutants (eg, passive smoking, dusts and fumes) that may aggravate the condition and ensuring the vaccination of patients against influenza, pneumococcal pneumonia and hepatitis.
Figure 5 ACSM exercise recommendations for pulmonary rehabilitation.Citation90
Future developments
The RAPID program is the first to confirm the clinical efficacy of AAT replacement therapy. The results indicate that the goal should be to focus on how to best incorporate disease-modifying AAT therapy into current clinical practice. New US treatment recommendations from the Alpha-1 Foundation have been developed,Citation63 which provide updated guidance on treatment of patients at earlier disease stages. Current ATS guidelines remain unchanged, and updates to the ERS statement are ongoing.
RAPID-RCT also suggested that alternative dosing such as bi-weekly infusions is well tolerated and plasma concentrations to a large extent exceed the 11 µM threshold during this regimen. This has been corroborated by additional studies suggesting value in a more personalized approach to therapy regimens with respect to dose, treatment interval and potential self-administration. Upcoming clinical studies such as the SPARTA trial aim to further explore the efficacy of double dosing regimens using regional lung density as measured by CT. This study will build on the results of the RAPID program that validated CT densitometry as a valuable outcome measure for the efficacy of AAT replacement therapy. Additionally, the heterogeneous distribution of emphysema has warranted the exploration of regional lung densitometry to elucidate the relationship between regional lung density decline and the decline in overall lung function. Current and future studies exploring the standardization of CT densitometry will help develop consensus on acquisition protocols and densitometric parameters to facilitate wider use of CT methods in this clinical setting.
Conclusion
The results of the RAPID program have demonstrated the efficacy and disease-modifying effect of AAT replacement therapy. In combination with data from earlier studies, there is now a large body of evidence that supports the use of AAT therapy. Despite this, AAT replacement therapy is not currently available in all countries, and this expert review has highlighted several key areas where improvements in the diagnosis and management of AATD patients are needed.
First, increased awareness of this rare lung condition and improved utilization of screening and diagnostic methods are needed to help reduce delays or misdiagnosis. Identification of patients earlier allows them to make lifestyle modifications (eg, avoidance of aggravating factors, smoking cessation and exercise/pulmonary rehabilitation) and allows timely access to disease-modifying therapy. Updated guidance on diagnosis, lifestyle modifications and pharmacotherapy is warranted.
Second, further guidance is required to facilitate the appropriate implementation of nonpharmacologic treatment measures such as exercise and dietary modifications to promote pulmonary rehabilitation. In addition, practical recommendations for the treatment of AATD are warranted that provide guidance on alternative dosing regimens and patient-centered convenience measures, such as home treatment or self-administration.
Finally, CT densitometry provides a new opportunity to change the landscape of how disease progression and treatment efficacy for AATD are assessed. The results of the RAPID program provide new insight into the utility of CT densitometry to detect the early signs of emphysema and the occurrence of complications such as hypoxemia. Further consideration of how different CT methodologies can be applied is necessary to facilitate implementation beyond trial settings and into routine clinical assessments.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Acknowledgments
Editorial assistance was provided by Meridian HealthComms, funded by CSL Behring.
Disclosure
Professor McElvaney reports grants and personal fees from CSL Behring, grants, personal fees and non-financial support from Grifols, outside the submitted work. Professor Chapman reports grants and personal fees from AstraZeneca, grants and personal fees from Boehringer Ingelheim, grants from Baxter, grants and personal fees from CSL Behring, grants and personal fees from Grifols, grants from GlaxoSmithKline, grants and personal fees from Sanofi, grants and personal fees from Genentech, grants and personal fees from Kamada, grants from Amgen, grants and personal fees from Roche, grants and personal fees from Novartis, personal fees from Merck and personal fees from CIHR-GSK Research Chair in Respiratory Health Care Delivery, UHN, during the conduct of the study. Professor Koczulla reports personal fees from CSL Behring, outside the submitted work. Dr Ferrarotti reports personal fees from CSL Behring, outside the submitted work. The authors report no other conflicts of interest in this work.
References
- SharafkhanehAHananiaNAKimVPathogenesis of emphysema: from the bench to the bedsideProc Am Thorac Soc20085447547718453358
- BernspångESvegerTPiitulainenERespiratory symptoms and lung function in 30-year-old individuals with alpha-1-antitrypsin deficiencyRespir Med200710191971197617532199
- National Human Genome Research InstituteLearning about alpha-1 antitrypsin deficiency (AATD)2012 Available from: https://www.genome.gov/19518992/learning-about-alpha1-antitrypsin-deficiency-aatd/Accessed October 23, 2017
- BrantlyMLPaulLDMillerBHFalkRTWuMCrystalRGClinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptomsAm Rev Respir Dis198813823273363264124
- AbboudRTNelsonTNJungBMattmanAAlpha(1)-antitrypsin deficiency: a clinical-genetic overviewAppl Clin Genet20114556523776367
- BrantlyMLWittesJTVogelmeierCFHubbardRCFellsGACrystalRGUse of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypesCHEST199110037037081889260
- FregoneseLStolkJHereditary alpha-1-antitrypsin deficiency and its clinical consequencesOrphanet J Rare Dis2008311618565211
- StockleyRAAlpha-1-antitrypsin deficiency: what next?Thorax200055761461810856323
- de SerresFJWorldwide racial and ethnic distribution of α1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveysChest200212251818182912426287
- BlancoIFernándezEBustilloEFAlpha-1-antitrypsin PI phenotypes S and Z in Europe: an analysis of the published surveysClin Genet2001601314111531967
- CraigTJSuspecting and testing for alpha-1 antitrypsin deficiency – an allergist’s and/or immunologist’s perspectiveJ Allergy Clin Immunol Pract20153450651126032475
- StollerJKAboussouanLSA review of alpha1-antitrypsin deficiencyAm J Respir Crit Care Med2012185324625921960536
- AlsaeediASinDDMcAlisterFAThe effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review of randomized placebo-controlled trialsAm J Med20021131596512106623
- AguilaniuBImpact of bronchodilator therapy on exercise tolerance in COPDInt J Chron Obstruct Pulmon Dis20105577120463947
- ColpCPappasJMoranDLiebermanJVariants of alpha 1-anti-trypsin in Puerto Rican children with asthmaChest199310338128158449073
- O’BrienMLBuistNRMurpheyWHNeonatal screening for alpha1-antitrypsin deficiencyJ Pediatr197892610061010307054
- SilvermanEKMiletichJPPierceJAAlpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screeningAm Rev Respir Dis198914049619662679271
- de SerresFJBlancoIPrevalence of alpha1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive reviewTher Adv Respir Dis20126527729522933512
- StollerJKSandhausRATurinoGDicksonRRodgersKStrangeCDelay in diagnosis of α1-antitrypsin deficiency: a continuing problemChest200512841989199416236846
- GreulichTVogelmeierCFAlpha-1-antitrypsin deficiency: increasing awareness and improving diagnosisTher Adv Respir Dis2016101728426341117
- ChapmanKRBurdonJGPiitulainenEIntravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trialLancet201538699136036826026936
- McElvaneyNGBurdonJHolmesMLong-term efficacy and safety of a-1 proteinase inhibitor treatment for emphysema caused by severe a-1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE)Lancet Respir Med201751516027916480
- Chorostowska-WynimkoJDisease modification in emphysema related to alpha-1 antitrypsin deficiencyCOPD201613680781527172295
- ParrDGDirksenAPiitulainenEDengCWenckerMStockleyRAExploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study of augmentation therapy in alpha 1-antitrypsin deficiencyRespir Res20091017519678952
- StolkJPutterHBakkerEMProgression parameters for emphysema: a clinical investigationRespir Med200710191924193017644366
- ParrDGStoelBCStolkJStockleyRAValidation of computed tomographic lung densitometry for monitoring emphysema in α1-antitrypsin deficiencyThorax200661648549016537666
- StockleyRAParrDGPiitulainenEStolkJStoelBCDirksenATherapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometryRespir Res201011113620920370
- RahaghiFFMiravitllesMLong-term clinical outcomes following treatment with alpha 1-proteinase inhibitor for COPD associated with alpha-1 antitrypsin deficiency: a look at the evidenceRespir Res201718110528558837
- DirksenADijkmanJMadsenFA Randomized Clinical Trial of α1-Antitrypsin Augmentation TherapyAm J Respir Crit Care Med199916051468147210556107
- DirksenAPiitulainenEParrDGExploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiencyEur Respir J20093361345135319196813
- DowsonLGuestPStockleyRLongitudinal changes in physiological, radiological, and health status measurements in α1-antitrypsin deficiency and factors associated with declineAm J Respir Crit Care Med2001164101805180911734427
- DawkinsPADowsonLJGuestPJStockleyRAPredictors of mortality in alpha1-antitrypsin deficiencyThorax200358121020102614645964
- StolkJNgWHBakkerMECorrelation between annual change in health status and computer tomography derived lung density in subjects with α1-antitrypsin deficiencyThorax200358121027103014645966
- ParrDGStoelBCStolkJStockleyRAPattern of emphysema distribution in α1-antitrypsin deficiency influences lung function impairmentAm J Respir Crit Care Med2004170111172117815306534
- ChapmanKRBradiACPatersonDNavickisRJWilkesMMSlower lung function decline during augmentation therapy in patients with alpha1-antitrypsin deficiency (A1ATD): results from the Canadian AIR registryProc Am Thorac Soc20052A808
- SeersholmNWenckerMBanikNDoes alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study groupEur Respir J19971010226022639387950
- The Alpha-1-Antitrypsin Deficiency Registry Study GroupSurvival and FEV1 decline in individuals with severe deficiency of α1-antitrypsinAm J Respir Crit Care Med1998158149599655706
- TonelliARRouhaniFLiNSchreckPBrantlyMLAlpha-1-antitrypsin augmentation therapy in deficient individuals enrolled in the alpha-1 foundation DNA and tissue bankInt J Chron Obstruct Pulmon Dis2009444345220054436
- WenckerMFuhrmannBBanikNKonietzkoNWissenschaftliche Arbeitsgemeinschaft zur Therapie vonLLongitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitorChest2001119373774411243951
- ChapmanKRStockleyRADawkinsCWilkesMMNavickisRJAugmentation therapy for alpha1 antitrypsin deficiency: a meta-analysisCOPD20096317718419811373
- HalpinDMGTashkinDPDefining disease modification in chronic obstructive pulmonary diseaseCOPD20096321122519811377
- SchluchterMDStollerJKBarkerAFFeasibility of a clinical trial of augmentation therapy for alpha(1)-antitrypsin deficiency. The Alpha 1-Antitrypsin Deficiency Registry Study GroupAm J Respir Crit Care Med20001613 Pt 179680110712324
- StoelBCStolkJOptimization and standardization of lung densitometry in the assessment of pulmonary emphysemaInvest Radiol2004391168168815486529
- GevenoisPADe VuystPde MaertelaerVComparison of computed density and microscopic morphometry in pulmonary emphysemaAm J Respir Crit Care Med199615411871928680679
- American Thoracic Society/European Respiratory SocietyStandards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiencyAm J Respir Crit Care Med2003168781890014522813
- SilvermanEKSandhausRAAlpha1-antitrypsin deficiencyN Engl J Med2009360262749275719553648
- VidalRBlancoICasasFJardiRMiravitllesMCommittee on the National Registry of Individuals with Alpha-1 Antitrypsin D. [Guidelines for the diagnosis and management of alpha-1 antitrypsin deficiency]Arch Bronconeumol2006421264565917178069
- AnthonisenNRConnettJEKileyJPEffects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of fev1: the lung health studyJAMA199427219149715057966841
- DonohueJFMinimal clinically important differences in COPD lung functionCOPD20052111112417136971
- HerpelLBKannerRELeeSMVariability of spirometry in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2006173101106111316497996
- EnrightPLConnettJEKannerREJohnsonLRLeeWWSpirometry in the lung health study: II. Determinants of short-term intraindividual variabilityAm J Respir Crit Care Med199515124064117842199
- WiseRAConnettJKurnowKSelection of spirometric measurements in a clinical trial, the Lung Health StudyAm J Respir Crit Care Med19951513 Pt 16756817881655
- WashkoGRRate of decline in FEV1: is emphysema the culprit?Am J Respir Crit Care Med201218512322210781
- CrapoROJensenRLStandards and interpretive issues in lung function testingRespir Care200348876477212890296
- U.S. Food and Drug Administration (FDA)Clinical and surrogate endpoints for evaluating efficacy of alpha1-proteinase inhibitor (human) augmentation therapyBlood products advisory committee 95th meetingJuly 20–21, 2009
- VogelmeierCFCrinerGJMartinezFJGlobal strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summaryAm J Respir Crit Care Med2017195555758228128970
- ParrDGStoelBCStolkJNightingalePGStockleyRAInfluence of calibration on densitometric studies of emphysema progression using computed tomographyAm J Respir Crit Care Med2004170888389015271692
- LamersRKemerinkGDrentMvan EngelshovenJReproducibility of spirometrically controlled CT lung densitometry in a clinical settingEur Respir J19981149429459623701
- FriedmanPJImaging studies in emphysemaProc Am Thorac Soc20085449450018453361
- GieradaDSPilgramTKWhitingBRComparison of standard-and low-radiation-dose CT for quantification of emphysemaAJR Am J Roentgenol20071881424717179344
- MetsOMde JongPAvan GinnekenBGietemaHALammersJWJQuantitative computed tomography in COPD: possibilities and limitationsLung2012190213314522179694
- BrennerDJDollRGoodheadDTCancer risks attributable to low doses of ionizing radiation: assessing what we really knowProc Natl Acad Sci U S A200310024137611376614610281
- SandhausRATurinoGBrantlyMLThe diagnosis and management of alpha-1 antitrypsin deficiency in the adultChronic Obstr Pulm Dis (Miami)20163366868228848891
- FerrarottiIPoplawska-WisniewskaBTrevisanMTHow can we improve the detection of alpha1-antitrypsin deficiency?PLoS One2015108e013531626270547
- FerrarottiIScabiniRCampoILaboratory diagnosis of alpha1-antitrypsin deficiencyTransl Res2007150526727417964515
- KueppersFSandersCState-of-the-art testing for alpha-1 antitrypsin deficiencyAllergy Asthma Proc201738210811428120746
- MiravitllesMHerrCFerrarottiILaboratory testing of individuals with severe alpha1-antitrypsin deficiency in three European centresEur Respir J201035596096820436173
- Barros-TizonJCTorresMLBlancoIMartinezMTInvestigators of the r EXAsgReduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-anti-trypsin augmentation therapyTher Adv Respir Dis201262677822354900
- GoodRX2017 Available from: https://www.goodrx.comAccessed October 26, 2017
- KentBDMitchellPDMcNicholasWTHypoxemia in patients with COPD: cause, effects, and disease progressionInt J Chron Obstruct Pulmon Dis2011619920821660297
- TeschlerHLong-term experience in the treatment of α1-antitrypsin deficiency: 25 years of augmentation therapyEur Respir Rev2015241354625726554
- StockleyRAEdgarRGPillaiATurnerAMIndividualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management?Int J Chron Obstruct Pulmon Dis2016111745175627536086
- BoeremaDJAnBGandhiRPBiochemical comparison of four commercially available human alpha1-proteinase inhibitors for treatment of alpha1-antitrypsin deficiencyBiologicals2017 pii:S1045-1056(17)30105-30107
- GadekJEKleinHGHollandPVCrystalRGReplacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjectsJ Clin Invest1981685115811657028785
- WewersMDCasolaroMASellersSEReplacement therapy for alpha 1-antitrypsin deficiency associated with emphysemaN Engl J Med198731617105510623494198
- BarkerAFIwata-MorganIOvesonLRousselRPharmacokinetic study of alpha1-antitrypsin infusion in alpha1-antitrypsin deficiencyChest199711236076139315791
- HubbardRCSellersSCzerskiDStephensLCrystalRGBiochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiencyJAMA19882609125912643261353
- StollerJKRouhaniFBrantlyMBiochemical efficacy and safety of a new pooled human plasma alpha(1)-antitrypsin, RespitinChest20021221667412114340
- StocksJMBrantlyMPollockDMulti-center study: the biochemical efficacy, safety and tolerability of a new alpha1-proteinase inhibitor, ZemairaCOPD200631172317175661
- CamposMAKueppersFStocksJMSafety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK)COPD201310668769523862647
- SandhausRAStocksJRouhaniFNBrantlyMStraussPBiochemical efficacy and safety of a new, ready-to-use, liquid alpha-1-proteinase inhibitor, GLASSIA (alpha1-proteinase inhibitor (human), intravenous)COPD2014111172523822603
- SeersholmNSandhausRChapmanKRSafety of bi-weekly infusion of A1-PI augmentation therapy in RAPIDEur Respir J20154659A999
- SoyDde la RozaCLaraBEsquinasCTorresAMiravitllesMAlpha-1-antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokineticsThorax200661121059106416928711
- SorrellsSCamprubiSGriffinRChenJAyguasanosaJSPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiencyRespir Med2015109449049925727857
- WilkeAGroheCProspective evaluation of clinical parameters of AAT patients with i. v. prolastin therapy in a homecare settingPneumologie2013671054555023999697
- CraigTJRecent advances in hereditary angioedema self-administration treatment: summary of an international hereditary angioedema expert meetingInt Arch Allergy Immunol2013161Suppl 1262723689242
- CarrMEJrFuture directions in hemostasis: normalizing the lives of patients with hemophiliaThromb Res2010125S78S8120163836
- OyesikuJOOHome treatment of haemophilia patients with inhibitorsHaemophilia201117217317821070497
- MarciniukDDHernandezPBalterMAlpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guidelineCan Respir J201219210911622536580
- GarberCEBlissmerBDeschenesMRQuantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exerciseMed Sci Sports Exerc20114371334135921694556
- GosselinkRTroostersTDecramerMPeripheral muscle weakness contributes to exercise limitation in COPDAm J Respir Crit Care Med199615339769808630582
- RiesALMakeBJReillyJJPulmonary rehabilitation in emphysemaProc Am Thorac Soc20085452452918453366
- JonesJHZeltJTHiraiDMEmphysema on thoracic CT and exercise ventilatory inefficiency in mild-to-moderate COPDCOPD201714221021827997255
- DowsonLNewallCGuestPHillSStockleyRExercise capacity predicts health status in α1-antitrypsin deficiencyAm J Respir Crit Care Med2001163493694111282769