Abstract
Background
A clinical trial of mometasone furoate/formoterol fumarate (MF/F) administered via a metered-dose inhaler in subjects with moderate to very severe chronic obstructive pulmonary disease (COPD) investigated the efficacy and safety of a fixed-dose combination of MF/F.
Methods
This multicenter, double-blind, placebo-controlled trial had a 26-week treatment period and a 26-week safety extension. Subjects (n = 1055; ≥40 years) were current or ex- smokers randomized to twice-daily treatment with inhaled MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, or placebo. The coprimary endpoints of the trial were mean changes from baseline in forced expiratory volume in 1 second (FEV1) over 0–12 hours (AUC0–12 FEV1) with MF/F versus MF, and in morning predose FEV1 with MF/F versus F. Key secondary endpoints were quality of life (Saint George’s Respiratory Questionnaire [SGRQ]), symptom-free nights, and partly stable COPD at 26 weeks, as well as time to first COPD exacerbation.
Results
Significant improvements in FEV1 AUC0–12 occurred at endpoint with MF/F 400/10 and MF/F 200/10 versus MF 400 (P ≤ 0.007). Significant bronchodilation occurred in 5 minutes with MF/F, and serial spirometry demonstrated sustained FEV1 improvements with MF/F over the treatment period. Significant improvements in morning predose FEV1 occurred with both MF/F doses, and these effects were further investigated by excluding results for subjects whose morning FEV1 data were collected >2 days after the last dose of study treatment. Improvements in SGRQ total scores surpassed the minimum clinically important difference of at least 4 units with MF/F 400/10. MF/F 400/10 significantly reduced the time-to-first COPD exacerbation. Similar proportions of subjects in all five treatment groups reported treatment-emergent adverse events. Rates of pneumonia were low (≤1.0%) across treatment groups.
Conclusion
MF/F 400/10 μg twice daily was shown to be an effective therapy for patients with moderate to very severe COPD, and both MF/F 400/10 μg twice daily and MF/F 200/10 μg twice daily were well tolerated.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by partially reversible airway obstruction, dyspnea, productive cough, and progressive decline in overall health.Citation1,Citation2 Although it may be preventable, COPD is debilitating for many patients afflicted with the illness. The classic course of COPD is a gradual downward spiral of lung function and activity level. As COPD worsens, patients are more likely to experience increasingly reduced functioning, acute exacerbations of COPD,Citation3 comorbid cardiovascular and metabolic disease, and possibly death.Citation1,Citation2,Citation4
Moderate COPD has an estimated worldwide prevalence of about 10%, based on data from 9425 patients in 12 cities around the world.Citation5 In recent years, COPD has surpassed stroke as the third leading cause of death in the US.Citation6,Citation7 Although COPD is a progressive disease, medications can alleviate symptoms, reduce the number and severity of exacerbations, and improve overall health status.Citation1,Citation2 In clinical practice, bronchodilators relax airway smooth muscle and improve lung emptying in patients with COPD.Citation1 Inhaled corticosteroids have also been found to provide some clinical improvements in patients with COPD due to their anti-inflammatory effects.Citation1 Although inhaled corticosteroids do not appear to affect the accelerated long-term decline in lung function typical of COPD,Citation1 they reduce the number of exacerbations and improve health status in patients with more advanced disease,Citation1 particularly when used concomitantly with an inhaled long-acting beta-agonist (LABA).Citation8 The additive clinical effects of inhaled corticosteroids when used with LABAsCitation8 may be due to synergistic effects via glucocorticoid and β2-adrenergic receptor regulation.Citation9,Citation10
The benefits of using concomitant therapy with inhaled corticosteroids plus inhaled LABA in the management of patients with exacerbations and persistent symptoms (eg, dyspnea, night-time awakenings) of COPD are cited by current COPD clinical guidelines.Citation1,Citation2,Citation11 The Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines recommend addition of an inhaled corticosteroid to long-acting bronchodilator therapy for patients with severe COPD (forced expiratory volume in 1 second [FEV1] <50% predicted) and repeated exacerbations.Citation2 In 2011 an official statement of the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society raised the FEV1 threshold from <50% to <60% predicted for symptomatic patients with stable COPD who may receive combination therapy.Citation12
Currently, three inhaled corticosteroid-LABA combinations are available for the treatment of COPD, ie, fluticasone propionate-salmeterol, budesonide-formoterol, and beclomethasone- formoterol. Although investigations of several inhaled corticosteroid-LABA combinations in the treatment of COPD have been undertaken,Citation13–Citation21 use of mometasone furoate with formoterol administered with a metered-dose inhaler is a novel combination in this setting. Individually, both mometasone furoate and formoterol have been shown to be effective in the treatment of COPD,Citation22–Citation24 and formoterol is approved for use in COPD. A fixed-dose combination of MF/F has been shown to be effective for the treatment of adolescents and adults with asthma.Citation25–Citation28 The combination product of mometasone furoate/formoterol fumarate (MF/F) offers both a potent inhaled corticosteroidCitation29 with relatively low systemic bioavailabilityCitation30 and a potent, selective LABA with fast onset of action.Citation31 These features of combined MF/F have the potential to be of benefit to patients with COPD. We undertook the present clinical trial, a 26-week randomized, placebo-controlled efficacy and safety study with a 26-week long-term safety extension, to evaluate two therapeutic doses of MF/F in patients with moderate to very severe COPD.
Materials and methods
Study design and treatments
This study was a randomized, placebo-controlled, parallel-group, multicenter, double-blind, double-dummy study (ClinicalTrials.gov identifier: NCT0383435) of MF/F in adults with moderate to very severe COPD conducted in 131 centers located in South America, Asia, Africa, Europe, and North America. The sponsor’s statistician produced a computer-generated randomization schedule with treatment codes in blocks using SAS. Randomization was stratified according to the subject’s smoking status at the time of randomization. Randomized treatment assignment was provided to the investigative site by means of an interactive voice response system at the time subjects were randomized. To place all prospective participants on the same type of treatment and to afford the same opportunity to demonstrate improvement in lung function, the study included an open-label run-in period in which long-acting bronchodilators and corticosteroids were discontinued and substituted with a short-acting β2-agonist (SABA)-anticholinergic fixed-dose combination. Patients were randomly assigned for 26 weeks (the treatment period) to one of five double-blind inhaled treatments given twice daily, ie, MF/F 400/10 μg, MF/F 200/10 μg, MF 400 μg, F 10 μg, or placebo (). For each treatment, patients were instructed to take two inhalations delivered via metered-dose inhaler in order to receive the full dose.
Figure 1 Study design schematic.
Abbreviations: bid, twice daily; F, formoterol; MDI, metered-dose inhaler; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination.
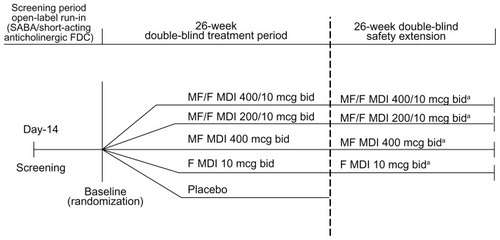
After 26 weeks of treatment, all patients randomized to placebo were discontinued, while 75% of those receiving active treatment were randomly selected to participate in a 26-week safety extension, continuing on the same treatment. We refer to weeks 1 through 52 of this study (the treatment period plus the safety extension) as the study period. All randomized subjects had study visits at screening, baseline (day 1), and weeks 1, 4, 13, and 26 in the treatment period. Subjects in the safety extension had additional visits at weeks 39 and 52.
Written informed consent was obtained from each patient prior to any study-related activity. Prior to study initiation at each study site, the clinical study protocol and the written informed consent form were reviewed and approved by an institutional review board/independent ethics committee.
Patients
We recruited adult males and females who were current or former smokers with a smoking history of ≥10 pack-years. Eligible patients were at least 40 years of age and had received a diagnosis of moderate to very severe COPD, based on a pre-bronchodilator FEV1/forced vital capacity (FVC) ratio of ≤0.70. They must have had symptoms of COPD (chronic cough and sputum production not attributable to another disease process) for at least 24 months, and a post-bronchodilator FEV1 ≤60% predicted normal and ≥25% predicted normal at screening. Females with childbearing potential were required to use a medically acceptable form of birth control. We excluded patients who experienced an increase in absolute volume of ≥400 mL at the screening visit or prior to the baseline visit within 30 minutes after administration of four inhalations of albuterol/salbutamol (total dose of 360 to 400 μg), or nebulized 2.5 mg albuterol-salbutamol. Patients requiring long-term administration of oxygen (>15 hours per day) or who experienced an exacerbation of COPD requiring medical intervention within four weeks prior to randomization, β-blocking agents, or treatment with additional excluded medication (other than SABA-short-acting anticholinergic to be used as rescue medication) were not enrolled. Patients with a history of significant medical illness or a disorder that might interfere with the study or that required treatment that might interfere with the study were also excluded. Other reasons for exclusion included pregnancy or breast-feeding, a diagnosis of asthma, lung cancer, or alpha-1-antitrypsin deficiency, or a history of lobectomy, pneumonectomy, lung volume reduction surgery, cataract extractions in both eyes, or other significant ocular problems (glaucoma, trauma, opacification).
Outcome measurements and assessments
Coprimary endpoints
In order to measure the contribution of each drug treatment to the combination, the study used two coprimary efficacy endpoints. The first coprimary endpoint was the mean change from baseline in FEV1 area under the curve from 0 to 12 hours postdose (AUC0–12 h) at the week 13 endpoint (to assess the contribution of F 10 to the combination). The second coprimary endpoint was the mean change from baseline in morning predose FEV1 at the week 13 endpoint (to assess the contribution of MF to the combination).
Key secondary endpoints
The study had four key secondary endpoints assessed for MF/F 400/10 and MF/F 200/10 at the end of the treatment period, ie, change in health status as assessed according to total scores on the St George’s Respiratory Questionnaire (SGRQ),Citation32 change in symptom-free nights, time-to-first mild, moderate, or severe COPD exacerbation, and the proportion of patients with partly stable COPD. The SGRQ consists of three component scores (symptoms, activity, impact) and a total score, each of which ranges from 0 to 100. The better SGRQ scores have a lower numeric value. The minimum clinically important difference (MCID) for SGRQ scores is defined as a 4-point difference from baseline or placebo.Citation33 A symptom-free night was defined as a combined score of 0 upon awakening, prior to the use of study drug or rescue medication, across three domains, ie, wheezing, cough, and difficulty breathing. Partly stable COPD was defined as a composite measure of the following outcomes: no use of oral steroid rescue medication; no morning or afternoon COPD weekly average symptom score >2 during at least 7 of 8 weeks; no moderate or severe COPD exacerbations; no unscheduled visits due to COPD worsening; and no study discontinuation due to treatment failure or a treatment-related adverse event.
Safety and e-diary assessments
Safety assessments at each study visit included monitoring of treatment-emergent adverse events, vital signs, oropharyngeal changes, and forearm bruising. Adverse events may have included the onset of new illness and the exacerbation of pre-existing conditions (eg, COPD). As directed by the study protocol, a medically qualified investigator at each site assessed the relationship of adverse events to study treatment, while treatment was blinded, as being unlikely, possibly, or probably related to treatment. Laboratory assessments, electrocardiography, and ophthalmologic examinations were conducted at screening and at the final visit. Chylack Incorporated (Duxbury, MA) provided guidance for ocular examinations and online training to ophthalmologists for Lens Opacities Classification System Version III (LOCS III) certification. Measurements of bone mineral density and 24-hour plasma cortisol were conducted at selected centers. Dual energy x-ray absorptiometry scans of the lumbar spine, left total femur, and femoral neck were obtained for a subgroup of subjects at selected centers. CCBR-Synarc (Portland, OR) provided centralized analysis of the scans, project management related to dual energy x-ray absorptiometry, and instrument quality control.
Each patient was given an e-diary (CareFusion Germany, 234 GmBH Services, Hoechberg, Germany) with a built-in spirometer to capture peak expiratory flow and a self-contained device to record information about medication use, nocturnal awakenings, COPD symptoms, and disease stability. COPD stability was evaluated with a 5-point or 6-point scale (0 = best, 4–5 = worst) measuring breathlessness, mucus production, chest tightness, cough, interference with personal care, and interference with outdoor activities.
Statistical analysis
The first coprimary efficacy endpoint (FEV1 AUC0–12) compared MF/F 400/10 versus MF 400, MF/F 400/10 versus placebo, and F 10 versus placebo. All of these comparisons had to be statistically significant at this dose level of MF/F to assess successfully the contribution of F 10 to the combination. The second coprimary efficacy endpoint (morning predose FEV1) compared MF/F 400/10 versus F 10, MF/F 400/10 versus placebo, and MF 400 versus placebo. All of these comparisons had to be statistically significant at this dose level of MF/F to assess successfully the contribution of MF 400 to the combination.
The target sample size was 1000 subjects (200 subjects per treatment group). This sample size was considered sufficient to detect a difference of 1.2 L × hour between MF/F 400/10 and MF 400 in change from baseline FEV1 AUC0–12 h with 91% power and a two-sided alpha level of 5% significance, assuming a pooled standard deviation of 3.6 L × hour. A 1.2 L × hour AUC converts to an average difference of 100 mL in FEV1 across 12 hours. A difference of this magnitude is considered clinically meaningful in subjects with this severity of COPD. For a morning predose FEV1 at the week 13 endpoint, the contribution of the MF 400 component was expected to be 80 mL for a target treatment difference of 160 mL between MF/F 400/10 and placebo. This treatment difference could be detected at 93% power with a two-sided alpha level of 4.9%, assuming a pooled standard deviation of 230 mL. The alpha level was adjusted to allow for a nominal penalty of 0.1%. The coprimary comparisons had to be statistically significant to proceed with additional efficacy analyses. The contribution of MF to the MF/F combination was evaluated by analyzing results in subjects whose morning predose FEV1 measurements were obtained in the protocol-defined time period, using values considered as actual trough FEV1 values. In a second analysis performed post database lock, FEV1 evaluations for each subject performed ≥2 days after the last dose of treatment were excluded, and the week 13 morning predose FEV1 endpoint was recalculated using the last remaining evaluation as specified in the study protocol.
To assess the coprimary endpoints, an analysis of covariance extracting sources of variation due to treatment, country, smoking status, and baseline as a covariate, was used. Pairwise comparisons were based on least square means from the model. An analysis of variance extracting sources of variation due to treatment, country, and smoking status, was performed also as a confirmatory analysis for these treatment comparisons.
Following testing of the coprimary endpoints at a given dose level, the key secondary endpoints were analyzed sequentially versus placebo. The first two key secondary variables (change from baseline in SGRQ total score and proportion of COPD symptom-free nights) were assessed using the same analysis of covariance as specified for the coprimary efficacy variables. The third key secondary variable (proportion of subjects with partly stable COPD) was analyzed using the Cochran-Mantel-Haenszel test controlling for smoking status. For the fourth key secondary variable (time-to-first mild, moderate, or very severe COPD exacerbation) the log-rank test for equality of survival curves was used. Kaplan-Meier curves were used to display these treatment responses. In addition, the effect of smoking status on the survival curves was examined. Assessments were repeated for MF/F 200/10.
Results
Subject disposition and demographics
Of 2313 subjects recruited, 1059 underwent randomization; however, four randomized subjects who had protocol violations were excluded from analyses, leaving 1055 randomized patients, two of whom did not receive the study drug. A total of 840 patients (80%) completed the treatment period, while 215 subjects (20%) discontinued from the study early, primarily for reasons unrelated to study treatment (). Treatment groups were well balanced regarding baseline demographic characteristics with respect to age, race, and sex (). The treatment groups had similar smoking histories.
Table 1 Disposition of patients following randomized treatment assignment: number (%) of patients during the treatment period
Table 2 Summary of demographic data and baseline characteristics (all randomized subjects)
A total of 529 patients on active treatment continued into the safety extension, 472 of whom (89%) completed the 26-week double-blind extension period. A total of 57 patients (11%) did not complete the safety extension. Of these patients, 17 (3%) discontinued due to adverse events, and 16 (3%) discontinued for reasons unrelated to study treatment.
Coprimary efficacy variables
Treatment with MF/F resulted in significant improvements in FEV1 which demonstrated the superiority of the combination versus the individual components. The contribution of F 10 to the MF/F 400/10 combination was demonstrated at the week 13 endpoint by the statistically significant effect of MF/F 400/10 over MF 400 in FEV1 AUC0–12 h (109 mL; ). An overall effect size of 163 mL was observed for MF/F 400/10 over placebo at this endpoint (P < 0.001). In addition, a statistically significant advantage of F 10 monotherapy over placebo was demonstrated, with an improvement of 74 mL (P = 0.003, ).
Table 3 Change from baseline in standardized FEV1 AUC0–12 h (mL)
Figure 2 FEV1 AUC0–12 h week 13 last observation carried forward results (all randomized subjects).
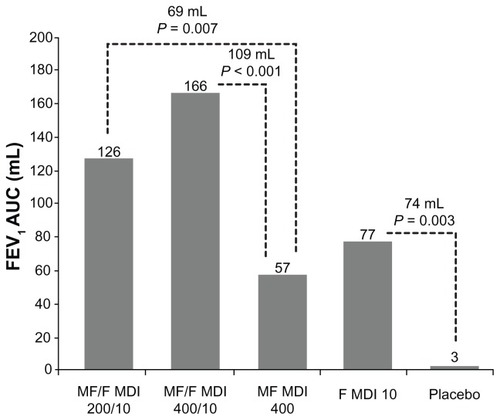
A comparison of MF/F 200/10 with MF 400 monotherapy at the week 13 endpoint also demonstrated the statistically significant effect of F 10, with an improvement of 69 mL (P = 0.007). The significant improvement of FEV1 AUC0–12 h with MF/F 400/10 versus F 10 (89 mL, P < 0.001) confirms the contribution of MF to the combination. The improvement of MF/F 200/10 over F 10 was shown to be 49 mL at this endpoint (P = 0.055). When compared with the MF 400 arm, MF/F 400/10 showed a 109 mL improvement at the week 13 endpoint and MF/F 200/10 showed a 69 mL improvement. Also at the week 13 endpoint, effect sizes of 123 mL for MF/F 200/10 and 163 mL for MF/F 400/10 over placebo were significant (P < 0.001) and demonstrated the overall benefit of MF/F. The additional benefit of 40 mL in the MF/F 400/10 group demonstrated a degree of dose-response compared with the MF/F 200/10 group. Serial spirometric assessment of FEV1 at the beginning (day 1) and end (week 26) of treatment identified the rapid onset and sustained duration of bronchodilator effects with MF/F ().
Figure 3 Serial FEV1 postdose at day 1 (A) and week 26 (B).
Abbreviations: bid, twice daily; FEV1, forced expiratory volume in 1 second; F, formoterol; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixed-dose combination.
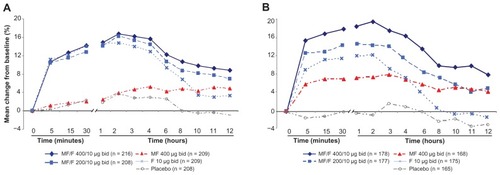
Substantial evidence indicated the contribution of the MF component to the combination, as demonstrated by the significant effects on morning predose FEV1 for MF/F 400/10 and MF/F 200/10 over F 10 alone (111 mL and 58 mL, respectively). An overall effect size of 128 mL was observed for MF/F 400/10 over placebo (P < 0.001). Although MF/F 400/10 demonstrated more efficacy than MF/F 200/10 (P = 0.045), both doses of MF/F demonstrated efficacy in this endpoint. Some subjects had FEV1 measurements long after they had stopped taking study treatment. In the second analysis of morning predose FEV1 which excluded subjects in whom FEV1 was measured >2 days after their last dose of treatment, a significant difference of 110 mL between MF/F 400/10 and F 10 (P < 0.001) confirmed the contribution of MF to the combination. Predose FEV1 data, collected from the daily diary recorded at the subject’s home support the effect observed from spirometry measured at the clinic visits.
Key secondary efficacy variables
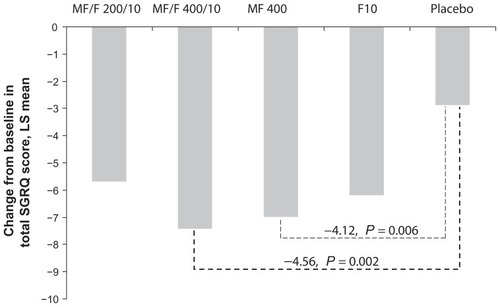
A 4-point increase over that of placebo (as well as over baseline) is considered the MCID for the SGRQ. MF/F 400/10 exceeded the MCID of 4 points compared with placebo, with a significant effect size of 4.56 points (P = 0.002) for SGRQ total score at the week 26 endpoint. In addition, a clinically meaningful difference of 4.12 points was observed for the MF 400 component, achieving significance compared with placebo (P = 0.006), while the F 10 component achieved significance with an effect size of 3.31 points compared with placebo (P = 0.026). Statistically significant improvements in SGRQ total score for MF/F 400/10 over placebo were demonstrated at weeks 4, 13, and 26 (P ≤ 0.040). MF/F 200/10 did not achieve the MCID, with a 2.82-point reduction compared with placebo ().
Figure 4 St George’s Respiratory Questionnaire total score change from baseline at week 26 endpoint.
Abbreviations: F, formoterol; LS, least squares; MCID, minimum clinically important difference; MF, mometasone furoate; MF/F, mometasone furoate/formoterol fixeddose combination; SGRQ, St George’s Respiratory Questionnaire.
The proportion of COPD symptom-free nights was improved with MF/F 400/10 (0.15) nights versus placebo (0.06) nights, achieving statistical significance (P = 0.001) over the 26-week treatment period. However, there was no treatment difference between MF/F 400/10 and placebo in the proportion of subjects with partly stable COPD at the 26-week endpoint (last 8 weeks on treatment for each subject). Percentages ranged from 37.8% to 45.9% across the treatment groups.
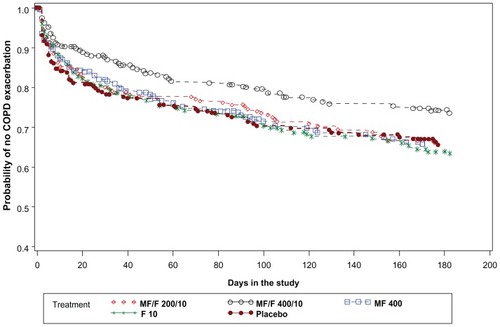
MF/F 400/10 demonstrated a significant improvement over F 10 (P = 0.015) for the time-to-first COPD exacerbation (). Subjects treated with MF/F 400/10 had the lowest rate of mild, moderate, or severe COPD exacerbations across treatments (25.8%), the relative reduction over placebo (20.6%) being nominally marginally significant (P = 0.079). MF/F 200/10 showed no reduction in mild, moderate, or severe exacerbations over placebo, and a 12.1% (P = 0.394) relative reduction in moderate or severe exacerbations. Since moderate or severe exacerbations are considered more clinically important, an additional analysis excluding mild exacerbations was performed. This analysis showed that subjects on placebo had a higher rate of moderate or severe COPD exacerbations (16.5%) than those on MF/F 400/10 (8.8%; P = 0.009), with a relative reduction for the MF/F 400/10 combination of 46.7% over placebo.
Figure 5 Time-to-first mild, moderate or severe COPD exacerbation over the 26-week treatment period: Kaplan-Meier survival curves by treatment (all randomized subjects).
Safety
Treatment-emergent adverse events
Treatment with MF/F 400/10 and MF/F 200/10 was well tolerated. During the 26-week treatment period, the percentage of subjects reporting treatment-emergent adverse events was similar between treatment groups, ranging from 26.3% in the MF/F 400/10 group to 33.5% in the F 10 group (). The most commonly reported treatment-emergent adverse events were headache, upper respiratory tract infection, cough, COPD, and hypertension (). The percentages of subjects reporting pneumonia during the 26-week treatment period were low, ranging from 0% to 1.0% across all treatment groups.
Table 4 Summary of treatment-emergent adverse events
Table 5 Summary of treatment-emergent adverse events ≥2% incidence (all randomized subjects) during the treatment period
During the study period (week 1 through week 52), the incidence of treatment-emergent adverse events in the active treatment groups ranged from 35.9% for MF/F 200/10 to 42.1% for F 10 (). The most commonly reported treatment- emergent adverse events among the four active-treatment groups were COPD, headache, nasopharyngitis, upper respiratory tract infection, and hypertension ().
Table 6 Summary of treatment-emergent adverse events ≥2% incidence (all randomized subjects) during the study period
During the treatment period, 68 subjects reported serious treatment-emergent adverse events. The most frequently reported adverse events were oral candidiasis, cough, and COPD. There were 16 all-cause mortality events across treatment groups during the 52 weeks of the study. Eleven of the deaths occurred during the treatment period (week 1 through week 26) and five occurred during the safety extension (week 27 through week 52). There were no abnormal, clinically relevant trends in laboratory values, vital signs, bone mineral density loss, plasma cortisol levels, ocular changes, or new findings of oral candidiasis or forearm bruising. Thirteen fractures were reported over the study, comprising one in the MF/F 400/10 group (vertebral), three in the MF/F 200/10 group (two lower limb, one wrist), three in the MF 400 group (one radius, one facial bone, one rib), five in the F 10 group (one each at the radius, ulna, and wrist, and two rib), and one in the placebo group (foot).
Rates of pneumonia were low across all treatment groups, with no notable differences between the treatment period and study period or between the MF-containing and non MF-containing treatment groups. During the 26-week treatment period, six subjects (0.6%) receiving active treatment or placebo reported pneumonia, and over the 52-week study period, 11 subjects (1.3%) across the active treatment groups reported pneumonia (). Most cases of pneumonia were of moderate severity.
Treatment-related adverse events
During the study, 62 of 843 subjects (7.4%) across the active treatment groups reported an adverse event considered by the investigator to be treatment-related. The most common adverse events considered possibly treatment-related were oral candidiasis, cough, and lenticular opacities. Only one case of pneumonia (in the placebo group) and one fracture (in the F 10 group) were considered possibly treatment-related.
Systemic and ocular effects
No clinically meaningful electrocardiographic changes were observed during the study period. Study treatments had minimal effects on the hypothalamic-pituitary-adrenal axis and bone mineral density, as measured at selected centers over the study period. Baseline 24-hour plasma cortisol levels ranged from 190.1 to 220.8 μg/dL · hour, and small, insignificant decreases in plasma cortisol were seen across all active treatment groups at week 26 and week 52. For bone mineral density in the lumbar spine, the region of greatest interest, decreases in bone mineral density were <1% for the MF/F 200/10 and MF 400 groups. The greatest loss of bone mineral density at lumbar spine was −0.6% in the MF 400 group at week 26. None of the between-treatment comparisons were statistically significant at weeks 26 or 52. Only five subjects had a bone mineral density loss at the lumbar spine >6% during the study period, comprising two subjects in the MF/F 200/10 group and three subjects in the MF 400 group.
Ophthalmologic examinations found that between 4.1% (MF/F 400/10) and 7.1% (MF 400) of subjects had LOCS III increases of ≥1 unit over the 52-week study period. One subject each in the MF/F 200/10 and MF/F 400/10 groups, two MF 400 subjects, and one placebo subject reported cataracts and were discontinued from the study, as per protocol. Additionally, intraocular pressure ≥22 mmHg was reported for four subjects (three on MF/F 400/10 and one on F 10) at week 26 and three subjects (one each on MF/F 200/10, MF 400, and F 10) at week 52.
Discussion
This clinical study demonstrated efficacy and safety for the combination of inhaled MF and F in the treatment of subjects with moderate to very severe COPD and showed that each component contributes to the combination. The F component demonstrated a significant and clinically meaningful contribution to the combination (109 mL) based on the comparison of MF/F 400/10 with MF 400 in change from baseline in the FEV1 AUC0–12 h at the week 13 endpoint. The MF component also demonstrated a significant contribution to the combination (111 mL) based on the comparison of MF/F 400/10 with F 10 in morning predose (trough) FEV1 at the week 13 endpoint. The results are significant and document a rapid onset of action of MF/F within 5 minutes, with significant bronchodilator effects that are sustained over the 12-hour dosing interval. In addition, the study demonstrates that both the rapidity of onset and magnitude of bronchodilation are maintained to the end of the 26-week treatment period without any evidence of tachyphylaxis. The study had a 12-hour period for serial spirometry, which was substantially longer than the 1-hour or 2-hour serial spirometry assessments in pivotal trials of other inhaled corticosteroids-LABA fixed-dose combinations,Citation15,Citation17,Citation18 although a recent trial of budesonide-F included serial spirometry data that extended for 12 hours.Citation34
A dose-related response for lung function was also observed, with MF/F 400/10 showing an additional benefit of 53 mL versus MF/F 200/10, when compared with F 10. At the week 13 endpoint, the MF contribution is also demonstrated by the 40 mL change in FEV1 AUC0–12 h between the MF/F 200/10 and MF/F 400/10 combinations, since the dose of the F component remains constant and only the dose of the MF component is increased. The effect seen in both FEV1 AUC0–12 h and morning predose FEV1 shows the additive effects of the inhaled corticosteroid and LABA components of the combination. At the week 13 endpoint, MF 400 showed a 27 mL improvement from baseline in morning predose FEV1 and F 10 had no improvement at the week 13 endpoint; however, MF/F 400/10 had a 111 mL improvement at the week 13 endpoint. In FEV1 AUC0–12 h, MF 400 had a 57 mL improvement from baseline and F 10 had a 77 mL improvement, whereas MF/F 400/10 had a 166 mL improvement. These findings are consistent with both preclinical and clinical reports of each component enhancing the effect of the other. The comparatively greater efficacy of MF/F 400/10 over MF/F 200/10 on lung function supports a dose-response effect of the MF component.
This study demonstrated a clinically relevant improvement in health-related quality of life compared with placebo for both the week 13 and week 26 endpoints, which is a clinically significant outcome rarely demonstrated in COPD trials. The MF/F 400/10 arm also demonstrated a significant improvement over placebo in the second key secondary endpoint of proportion of COPD symptom-free nights.
Comparison of the MF/F 400/10 combination with placebo showed a nominally significant relative reduction of 20.6% in the rate of mild, moderate, or severe exacerbations (P = 0.079). However, the relative reduction of 46.7% in moderate to severe exacerbations for the MF/F 400/10 combination compared with placebo is significant (P = 0.009) and clinically important. Statistically significant reductions were also shown for MF/F 400/10 compared with MF and F alone, despite a treatment period of only 6 months. Historically, it has been difficult to show significant effects on exacerbations with pharmacotherapy within this short timeframe.
Overall, treatment with MF/F was well tolerated in this study. The study demonstrated a low rate of pneumonia in all groups, including subjects who received treatments containing an inhaled corticosteroid. In contrast with findings of a meta-analysis reporting a dose-related increase in the risk of pneumonia with inhaled corticosteroid-containing treatments (inhaled corticosteroid monotherapy and inhaled corticosteroid-LABA combinations),Citation35 this study did not demonstrate any imbalance in the incidence of pneumonia across treatment groups. However, this study was not powered to have sufficient numbers or follow-up to detect a meaningful difference in pneumonia events, and the low event rate prevents any robust conclusions regarding any differences between treatment arms for this adverse event. In addition, the incidence of oral candidiasis was very low. There were no significant demonstrable adverse effects on the cardiovascular system, bone mineral density, lenticular opacities, or intraocular pressure. Effects on hypothalamicpituitary- adrenal axis suppression were quite modest. However, longer trials are needed to exclude any long-term effects of treatment.
In this study, MF/F was delivered using a metered-dose inhaler rather than a dry powder inhaler. This mode of administration, in our opinion, offers some advantages. Because a metered-dose inhaler requires a lower inspiratory flow rate compared with a dry powder inhaler, the amount of medication deposited in the oral cavity and the glottic area is reduced, which potentially accounts for the low incidence of oral candidiasis and dysphonia demonstrated during the study period. The generally finer particle sizes generated by metered-dose inhalers allow for greater lung and peripheral lung deposition. Additionally, patients with severe lung impairment may have difficulty generating adequate inspiratory force to allow for effective use of some dry powder inhalers. However, it is important for patients to be adequately instructed on appropriate use of both dry powder inhalers and metered-dose inhaler devices. Both devices have been associated with a similar percentage of errors.Citation36
In conclusion, the results of this study show MF/F 400/10 μg twice daily to be an effective therapy for patients with moderate to very severe COPD, based on improvements in lung function and quality of life as well as reduction in COPD exacerbations. Both MF/F 400/10 μg twice daily and MF/F 200/10 μg twice daily were well tolerated.
Disclosures
This study was sponsored by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Medical writing and editorial assistance was provided by Ken Kauffman and Genevieve Belfiglio, AdelphiEden Health Communications, New York, NY, and funded by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. Editorial assistance was also provided by Jorge Moreno-Cantu, Global Scientific and Medical Publications, Office of the Chief Medical Officer, Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ. DPT has served as a consultant for AstraZeneca, Boehringer-Ingelheim, Dey Laboratories, and Merck; received honoraria from AstraZeneca, Boehringer- Ingelheim and Dey Laboratories; and received grants from Almirall, AstraZeneca, Boehringer-Ingelheim, Dey Laboratories, Merck, Novartis, Pfizer, Sepracor, and Forest Laboratories. DED has served as a consultant for Forest, Ikaria, and Merck; has received via University of Kentucky research/grant support from AstraZeneca, Boehringer Ingelheim- Pfizer, Merck, and Novartis; and received honoraria from AstraZeneca, Boehringer Ingelheim, Forest, Merck, and Pfizer. EK has received consulting fees from Dey Laboratories, GlaxoSmithKline, MAP Pharma (AstraZeneca), and Sepracor (Sunovion) and speaking fees from AstraZeneca, GlaxoSmithKline, Merck, and Teva. TS, SB, BK, and HS are employees of Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc, Whitehouse Station, NJ.
References
- American Thoracic Society/European Respiratory Society Task ForceStandards for the Diagnosis and Management of Patients with COPD Available at: http://www.thoracic.org/clinical/copd-guidelines/resources/copddoc.pdfAccessed July 26, 2011
- Global Strategy for the Diagnosis, Management and Prevention of COPDGlobal Initiative for Chronic Obstructive Lung Disease (GOLD) Available at: http://www.goldcopd.org/Accessed July 21, 2011
- HurstJRVestboJAnzuetoASusceptibility to exacerbation in chronic obstructive pulmonary diseaseN Engl J Med20103631128113820843247
- DohertyDEA review of the role of FEV1 in the COPD paradigmCOPD2008531031818972280
- BuistSCOPD: a common disease that is preventable and treatablePrim Care Respir J2006157916701753
- AndersonRNSmithBLDeaths: leading causes for 2002Natl Vital Stat Rep20055318915786629
- KochanekKDZuJQMurphySLDeaths: Preliminary Data for 2009Hyattsville, MDNational Center for Health Statistics2011
- ManSFMcAlisterFAAnthonisenNRSinDDContemporary management of chronic obstructive pulmonary disease: clinical applicationsJAMA20032902313231614600190
- MakJCHisadaTSalmonMBarnesPJChungKFGlucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinasesBr J Pharmacol200213598799611861327
- MakJCNishikawaMShirasakiHMiyayasuKBarnesPJProtective effects of a glucocorticoid on downregulation of pulmonary beta 2-adrenergic receptors in vivoJ Clin Invest199596991067615841
- Chronic obstructive pulmonary disease. Management of chronic obstructive pulmonary disease in adults in primary and secondary careClinical Guideline 12National Institute for Clinical Excellence2010 Available at: guidance.nice.org.uk/CG101Accessed December 31, 2011
- QaseemAWiltTJWeinbergerSEDiagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians American College of Chest Physicians, American Thoracic Society, and European Respiratory SocietyAnn Intern Med201115517919121810710
- BourbeauJChristodoulopoulosPMaltaisFEffect of salmeterol/fluticasone propionate on airway inflammation in COPD: a randomised controlled trialThorax20076293894317557771
- CeylanEBudesonide-formoterol (inhalation powder) in the treatment of COPDInt J Chron Obstruct Pulmon Dis2006111512218046888
- HananiaNADarkenPHorstmanDThe efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the Diskus inhaler for the treatment of COPDChest200312483484312970006
- KoserAWestermanJSharmaSEmmettACraterGDSafety and efficacy of fluticasone propionate/salmeterol hydrofluoroalkane 134a metered-dose-inhaler compared with fluticasone propionate/salmeterol diskus in patients with chronic obstructive pulmonary diseaseOpen Respir Med J20104869121253451
- MahlerDAWirePHorstmanDEffectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20021661084109112379552
- RennardSITashkinDPMcElhattanJEfficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered-dose inhaler in patients with chronic obstructive pulmonary disease: results from a 1-year randomized controlled clinical trialDrugs20096954956519368417
- ShethKWassermanRLLincourtWRFluticasone propionate/salmeterol hydrofluoroalkane via metered-dose inhaler with integrated dose counter: Performance and patient satisfactionInt J Clin Pract2006601218122416981966
- TashkinDPRennardSIMartinPEfficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6-month randomized clinical trialDrugs2008681975200018778120
- VestboJPauwelsRAndersonJAJonesPCalverleyPEarly onset of effect of salmeterol and fluticasone propionate in chronic obstructive pulmonary diseaseThorax20056030130415790985
- AalbersRAyresJBackerVFormoterol in patients with chronic obstructive pulmonary disease: a randomized, controlled, 3-month trialEur Respir J20021993694312030736
- CalverleyPMRennardSNelsonHSOne-year treatment with mometasone furoate in chronic obstructive pulmonary diseaseRespir Res200897319014549
- CazzolaMCentanniSRegordaCOnset of action of single doses of formoterol administered via Turbuhaler in patients with stable COPDPulm Pharmacol Ther200114414511162418
- MeltzerEOKunaPNolteHNayakASLaForceCMometasone furoate/formoterol reduces asthma deteriorations and improves lung functionEur Respir JAugust 4, 2011 [Epub ahead of print.]
- NathanRANolteHPearlmanDSTwenty-six-week efficacy and safety study of mometasone furoate/formoterol 200/10 microg combination treatment in patients with persistent asthma previously receiving medium-dose inhaled corticosteroidsAllergy Asthma Proc20103126927920678306
- WeinsteinSFCorrenJMurphyKNolteHWhiteMTwelve- week efficacy and safety study of mometasone furoate/formoterol 200/10 microg and 400/10 microg combination treatments in patients with persistent asthma previously receiving high-dose inhaled corticosteroidsAllergy Asthma Proc20103128028920687982
- MasperoJFNolteHCherrez-OjedaILong-term safety of mometasone furoate/formoterol combination for treatment of patients with persistent asthmaJ Asthma2010471106111520874458
- SmithCKreutnerWIn vitro glucocorticoid receptor binding and transcriptional activation by topically active glucocorticoidsArzneimittel-forschung1998489569609793625
- AffrimeMBCussFPadhiDBioavailability and metabolism of mometasone furoate following administration by metered-dose and dry-powder inhalers in healthy human volunteersJ Clin Pharmacol2000401227123611075308
- AndersonGPLong acting inhaled beta-adrenoceptor agonists the comparative pharmacology of formoterol and salmeterolAgents Actions Suppl1993432532698103622
- JonesPWQuirkFHBaveystockCMLittlejohnsPA self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory QuestionnaireAm Rev Respir Dis1992145132113271595997
- JonesPWSt. George’s Respiratory Questionnaire: MCIDCOPD20052757917136966
- CelliBRTashkinDPRennardSIMcElhattanJMartinUJBronchodilator responsiveness and onset of effect with budesonide/formoterol pMDI in COPDRespir Med20111051176118821531124
- DrummondMBDasenbrookECPitzMWMurphyDJFanEInhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta-analysisJAMA20083002407241619033591
- MelaniASZanchettaDBarbatoNInhalation technique and variables associated with misuse of conventional metered-dose inhalers and newer dry powder inhalers in experienced adultsAnn Allergy Asthma Immunol20049343944615562882