
Abstract
Alpha-1 antitrypsin deficiency (AATD) is a genetic condition characterised by low circulating levels of alpha-1 antitrypsin (AAT), a serine proteinase inhibitor. The most common deficiency variants are the S and Z mutations, which cause the accumulation of misfolded AAT in hepatocytes resulting in endoplasmic reticular stress and insufficient release of AAT into the circulation (<11μmol/L). This leads to liver disease, as well as an increased risk of emphysema due to unopposed proteolytic activity of neutrophil-derived serine proteinases in the lungs. AATD has been traditionally viewed as an inflammatory disorder caused directly by a proteinase-antiproteinase imbalance in the lung, but increasing evidence suggests that low AAT levels may affect other cellular functions. Recently, AAT polymers have been identified in both monocytes and macrophages from AATD patients and evidence is building that these cells may also play a role in the development of AATD lung disease. Alveolar macrophages are phagocytic cells that are important in the lung immune response but are also implicated in driving inflammation. This review explores the potential implications of monocyte and macrophage involvement in non-liver AAT synthesis and the pathophysiology of AATD lung disease.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is an autosomal co-dominant genetic condition that is characterised by low circulating levels of alpha-1 antitrypsin (AAT) protein, a serine proteinase inhibitor synthesised and secreted mainly by hepatocytes, but also by immune and other cells.Citation1 AAT circulates in the blood and enters tissues including the lungs by diffusion, where it binds and inhibits proteinases including neutrophil elastase (NE), cathepsin G and proteinase 3, as well as potentially exhibiting anti-inflammatory properties.Citation2 Low levels of AAT lead to an increased likelihood of developing emphysema, especially at an early age (30–40 years) and the accumulated protein in hepatocytes may lead to clinically overt liver disease.Citation1 AAT is released as a single polypeptide chain that binds neutrophil elastase via a methionine residue at position 358 on the reactive centre loop of the protein. This then translocates to the opposite side of the protein and inserts the reactive loop into a β sheet, forming a covalently linked complex inactivating both the enzyme and inhibitor and is cleared from the circulation.Citation3 In the lung, neutrophil elastase and other proteinases are released from immune cells, primarily neutrophils where they aid extravasation from the blood. AAT inactivates the neutrophil serine proteinases in order to limit any bystander lung damage and thus maintain a physiological proteinase-anti-proteinase balance important for maintaining lung integrity.Citation4 AAT is an acute phase protein and plasma levels normally increase within hours of inflammation or infection adding extra protection. However, in AATD, little or no AAT is released as part of this acute phase response disturbing the physiological balance further.Citation5
AATD is caused by a genetic mutation on the SERPINA1 gene that encodes the AAT protein and results in low circulating levels of the AAT protein in the ZZ variant (2–8µmol/L). An estimated 3.4 million people worldwide are affected; however, under diagnosis means many individuals remain undetected so this number is likely to be higher, highlighting the need for screening programmes in at risk individuals.Citation6,Citation7 There are over 120 reported mutations in this gene, with the most common being the S and Z mutations, which affect 1.4 million and 250,000 people worldwide, respectively.Citation8 Both mutations result in misfolding of the protein, with the S variant resulting in a proportion of the AAT protein (~40%) being retained within hepatocytes, which reduces plasma levels but may not affect susceptibility to either liver or lung disease.Citation9–Citation11 The Z mutation occurs in over 95% of diagnosed AATD patients and causes greater misfolding and polymerisation of the protein. These polymers have no anti-neutrophil elastase activity and accumulate in the endoplasmic reticulum (ER) of cells. Eighty-five percent of the misfolded protein is retained in the hepatocytes and partially removed by ER degradation pathways, while the remaining 15% is secreted into the serum largely as monomers but still retains the potential to polymerise. The retention of polymers in the ER causes cell damage through ER stress, ER overload response, mitochondrial dysfunction and autophagy which damages cells and leads to liver disease with AAT protein overload, and lung disease due to circulating protein insufficiency.Citation1,Citation12
Although AAT is produced mainly by hepatocytes,Citation13 it is also produced by lung epithelial cells,Citation14 neutrophils,Citation15,Citation16 intestinal epithelial cells,Citation17 and pancreatic islet cells.Citation18 AATD polymers have been found in the lungCitation19 (including the bronchoalveolar lavage), skin and kidneys,Citation20 around capillaries and in epithelial cells.Citation21 The lack of systemic AAT leads to a reduced ability to control serine proteinases in the lungs. This results over time in parenchymal lung destruction due to increased connective tissue breakdown, and the development of panlobular emphysema, especially in the presence of smoking.Citation22 Z-AAT polymers also co-localise with neutrophils in the lung tissue, which increases the local proteinase release thereby contributing to further interstitial damage, as previously reviewed.Citation23,Citation24 Recently, AAT polymers have been identified in both monocytesCitation25 and macrophagesCitation26 from AATD patients and evidence is building that these cells may also play a role in the development of AATD lung disease. The purpose of this review is to explore the potential implications of the local lung cells involved in non-liver AAT synthesis.
Monocytes
Human monocytes account for 5–10% of circulating blood leukocytes. Monocytes are equipped with chemokine and adhesion receptors which mediate their migration from the blood to sites of infection or injury. Once migrated, activated monocytes release pro-inflammatory cytokines such as IL-1β, IL-6 and TNFα as well as other inflammatory mediators which induce the recruitment of further immune cell types, and moderates inflammation (for review seeCitation27,Citation28). There are three main monocyte phenotypes – classical CD14++ CD16−, intermediate CD14+ CD16+ and non-classical CD14−, CD16+.Citation29 Classical monocytes are primed for phagocytosis, migration and immune sensing. Intermediate monocytes express CCR5 and play a role in antigen presentation and cytokine secretion. Non-classical monocytes are involved in complement and Fcγ mediated phagocytosis and adhesion.Citation30 However, these functions overlap and the spectrum of activity of these subsets is not exclusive.Citation28 Once migrated to peripheral tissues, monocytes differentiate into macrophages or dendritic cells depending on the local milieu. This increases the phagocytic or antigen presentation capacity of the local environment as required, and aids in clearing the infectious agent/s and modulating the immune response. Monocytes are thus a vital component of the secondary immune response to infection.
Monocytes and Alpha-1 Antitrypsin
Monocytes express the AAT geneCitation31,Citation32 at a level that is 200 times less than hepatocytes, indicating that they are a minor source of overall AAT but may contribute to the localised pool.Citation33 Monocytes secrete AAT and also maintain this property when differentiated into macrophages.Citation31 Alongside the main function of AAT to inhibit the activity of free neutrophil elastase locally, AAT also has some anti-inflammatory properties that may be enhanced by monocyte secretion locally. In monocytes, AAT inhibits lipopolysaccharide-stimulated release of pro-inflammatory cytokines TNFα and IL-1β, increases anti-inflammatory IL-10 release,Citation34 and reduces CD14 expressionCitation35 via increased cAMP.Citation36 Together this may prevent the over-activation of monocytes and other immune cells and would curtail the immune response, promoting the resolution of inflammation. This secondary role of AAT is independent of its anti-proteinase activity, as it occurs with both oxidised and heat-inactivated protein.Citation34 This may important in the management of AATD, as it suggests that targeting the neutrophil elastase inhibitory function independently of AAT (for instance with chemical elastase inhibitors) may only partly restore normal homeostasis in the AATD lung, as the effect of AAT on other cell functions may be an important component for the development and treatment of lung disease.
Monocytes in Alpha-1 Antitrypsin Deficiency
Monocyte subset analysis has shown that PiZZ patients have similar levels of HLA-DR+ monocyte subsets, but fewer non-classical HLA-DR− cells. These latter cells protect the vascular endothelium during inflammation through the removal of damaged cells and secretion of anti-inflammatory mediators,Citation37,Citation38 and alteration in this subset variation has been suggested as a contributing factor to the early disease inflammation in AATD, although this is based on a single study of low numbers of patients that requires validation at least in vitro.Citation39
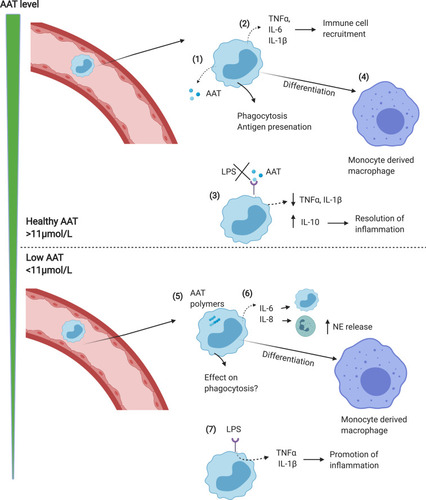
In response to neutrophil elastase, monocytes increase AAT mRNA; however, PiZZ monocytes (as with hepatocytes) secrete little AAT due to the accumulation of polymers within the cell.Citation32 This leads to an “unfolded protein response” in the monocytes, with accumulation of AAT polymers within the ER, activation of NFĸB and increased expression of IL-6 and CXCL-8.Citation25 Increased release of CXCL-8 would result in further recruitment of neutrophil to the lungs, increasing the elastase burden and further amplifying tissue degradation in the AAT deficient lung. IL-6 recruits’ further monocytes to the lungs perpetuating chronic neutrophil and monocyte induced inflammation (see ). This monocyte-driven loop may in some instances be the precipitating step in the initiation of a progressive destructive process in AATD especially in the 30% of never smokers who develop lung disease.Citation40 The exact mechanism remains speculative but is worthy of further study. This also suggests that augmentation therapy in such individuals may work through both its anti-proteinase and anti-inflammatory effects.
Figure 1 Monocyte function in normal vs AATD lungs. (1) In response to infection/inflammation, circulating monocytes enter the lungs where they release AAT which acts to neutralise NE. (2) Monocytes also release cytokines including TNFα, CXCL-8, LTB4 and IL-6 to recruit immune cells including neutrophils and monocytes. (3) AAT blocks LPS induced pro-inflammatory cytokine release, thus limiting inflammation. (4) Monocytes differentiate into monocyte-derived macrophages, adding to the alveolar macrophage pool. (5) In AATD, AAT polymers form in monocytes, leading to elevated release of pro-inflammatory cytokines including IL-6 and CXCL-8 which increases neutrophil recruitment (6), thus increasing NE levels and activity in the lungs. (7) Levels of AAT below the putative protective threshold (<11μmol/L) causes LPS induced pro-inflammatory cytokine release to increase, driving inflammation.
Macrophages
Alveolar macrophages line the alveoli and the airways of the lungs where they provide an important part of innate immunity, including phagocytosis of inhaled pathogens and releasing mediators to modulate the physiological immune response if required. The macrophage pool is long lived but also supplemented through the recruitment of monocytes, especially during acute and chronic episodes of inflammation, generating monocyte-derived macrophages with similar activity. Macrophages exhibit plasticity and are able to alter their phenotype and function in response to the local environmental requirements – from pro-inflammatory during infection, to anti-inflammatory during resolution of inflammation.Citation41,Citation42
Macrophages and Alpha-1 Antitrypsin
Both peripheral lung macrophages isolated from bronchoalveolar lavage and monocyte-derived macrophages (MDM) transcribe the AAT gene, and secrete the protein.Citation32 Monocyte differentiation to macrophages increases the level of gene expression 3-fold, though still 70 times less than hepatocytes, suggesting that macrophages contribute a greater component of local AAT in the lungs than monocytes. Furthermore, MDM differentiated into a pro-inflammatory phenotype using GM-CSF secrete even higher levels of AAT than MDM differentiated into an anti-inflammatory phenotype using M-CSF.Citation43 LPS further increases gene expression of AAT in MDM, suggesting that exposure to gram negative bacteria in the lungs may drive AAT release from macrophages, which will contribute to the anti-proteinase activity during infection. How this relates to both the peripheral acute phase response and increased protein transudation from plasma during such episodes remains unknown.
AAT has several other effects on macrophages unrelated to its direct anti-proteinase activity. Neutrophil elastase binding to macrophages induces the release of LTB4,Citation44 a major neutrophil chemoattractant in AATD,Citation45,Citation46 but this pathway is inhibited by AAT leading to reduced LTB4 release in vitroCitation44 and in vivo.Citation47 This reduces neutrophil recruitment to the airways, limiting the potential proteinase damage.
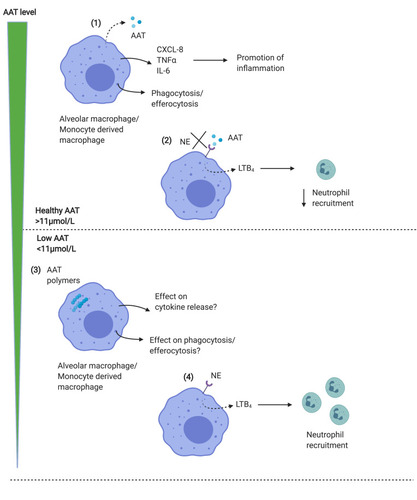
Exogenous AAT has also been shown to restore efferocytosis and phagocytosis by macrophages exposed to cigarette smoke, due to a reduction in shedding of CD206 and phosphatidylserine receptors.Citation48 This suggests that the increased proteinase levels in the lungs of AATD patients may strip scavenger receptors from the macrophage surface in a similar manner to that shown in cystic fibrosis, where similar or even higher levels of proteinases are present,Citation49 resulting in a reduced ability to bind bacteria as part of phagocytosis. However, this has yet to be studied in AATD. AAT is also able to suppress infection of MDM with Mycobacterium abscessus suggesting a potential to prevent bacterial infection in the lungs by acting directly on the macrophage.Citation50 Further work is needed to elucidate the full effects of exogenous AAT on macrophage function and particularly the role in clearance of bacteria relevant to COPD (see ).
Figure 2 Macrophage function in normal vs AATD lungs. (1) Alveolar macrophages or MDM release AAT in response to inflammatory stimuli, together with proinflammatory cytokines including CXCL-8 and IL-6 which promote inflammation. Macrophages also phagocytose bacteria, and efferocytose apoptotic cells. (2) AAT acts to block the binding of neutrophil elastase to macrophage receptors, limiting the release of LTB4 resulting in reduced neutrophil recruitment to the airways, which limits inflammation. AAT is thus anti-inflammatory in the lungs. (3) In AATD, both MDM and alveolar macrophages contain AAT polymers, which may limit phagocytic function. (4) Levels of AAT below the putative protective threshold (<11μmol/L) results in the ability of NE to bind to receptors on macrophage surface, increasing LTB4 release and recruiting neutrophils, which further contribute to NE levels in the lungs, driving inflammation.
Macrophages in Alpha-1 Antitrypsin Deficiency
Macrophages from PiZZ patients express normal levels of the AAT gene but release 10 times less AAT than normalCitation33 resulting potentially in greater overall proteolytic activity associated with concomitant increased MMP14 expression.Citation51 This is postulated to be due to the accumulation of AAT polymers within the cell, reducing the release of protein. However, a further study showed polymer accumulation did not occur in all PiZZ macrophages but only in 27% of cells observed in lung slices imaged by immunohistochemistry.Citation26 Interestingly, the authors also found AAT polymers in lung slices from non-deficient smokers with and without COPD, but not in non-smokers which may reflect the effect of oxidants on polymerisation even with normal AAT. The authors also showed that the percentage of alveolar macrophage containing AAT polymers correlated with pack year smoking history, FEV1/FVC ratio, small airways dysfunction, CD8 T cell and neutrophil numbers in the alveolar walls, suggesting that polymer accumulation could be important in both AATD and non-deficient COPD. This observation is important as it is the first to show polymer accumulation within the macrophage, and it would be important to determine whether this relates to all macrophages within the slices and that the antibody used was specific to polymeric AAT.
In non-AATD COPD, macrophages are believed to be the key drivers of processes leading to disease initiation and progression.Citation52 COPD macrophages are predominantly pro-inflammatory, display dysfunctional phagocytosisCitation53–Citation55 and efferocytosis,Citation56 and have altered mitochondrial function.Citation57 This likely contributes to the chronic colonisation of the respiratory tract, due to an inability to clear invading bacteria and apoptotic cells, despite the 20 fold increase in macrophage number within the lungs.Citation52 Targeting macrophage function in COPD is a potential treatment strategy being explored. Whether this is also of importance in AATD macrophages requires exploration. The function of AATD macrophages is currently unknown, but the accumulation of polymers within the cell has implications for mechanisms such as phagocytosis and cytokine release.
Macrophage-Neutrophil Crosstalk in AATD
The role of the neutrophil in AATD has been extensively reviewed;Citation23,Citation24 however, the impact of altered neutrophil function on macrophage biology has not been studied. Macrophages and neutrophils not only share similar functions, including phagocytosis and inflammatory mediator release, they also work in concert to coordinate the response to invading pathogens. This process is tightly regulated to modulate non-physiological damage to the host.
Macrophages recruit neutrophils to the lungs via the release of LTB4 and CXCL-8. Neutrophils are equally capable of recruiting monocytes to the lungs via the release of cathepsin G and azurocidin, which upregulates E-selectin and VCAM-1 on the vascular endothelium, promoting monocyte rolling, sticking and recruitment into the parenchyma.Citation58,Citation59 This results in increased numbers of both neutrophils and macrophages in the lungs during inflammation, especially to infectious organisms, to facilitate effective clearance of the invading organism.
Neutrophils are able to modify macrophage phenotype, and thereby modulate their function, through the release of reactive oxygen species, which activates macrophage AMPK, suppresses NFkB and induces an anti-inflammatory phenotype.Citation60,Citation61 This helps to limit the damage caused by these pro-inflammatory cells, and, on clearance of the invading organism induces the resolution of inflammation. During this, macrophages efferocytose apoptotic neutrophils in order to remove them from the tissue safely, and avoid excess proteinase release during necrosis. This process induces an anti-inflammatory macrophage phenotype which further promotes resolution of inflammation in the lungs once the initial infection is cleared. Efferocytosis occurs by binding of neutrophil apoptosis receptors, such as phosphatidylserine,Citation62 annexin 1 and calreticulinCitation63 to macrophage receptors including T cell immunoglobulin and mucin receptors (TIM) 1 and 4Citation64 and integrins.Citation65
In AATD, polymers have been shown to increased degranulation in airway neutrophil, resulting in increased apoptosis, possibly due to high levels of endoplasmic reticulum stress, and activation of the internal death pathways induced by polymers.Citation66 This would lead to an increase in clearance requirements by alveolar macrophages, which may overwhelm the normal macrophage capacity, and may drive the macrophages into an anti-inflammatory phenotype, thus impairing the normal clearance of bacteria in the AATD lung. Clearly studies to examine this potential physiological failure should be undertaken as it may have a central role in AATD particularly during exacerbations when excess proteinase activity is generated. It is known that in non-AATD COPD, efferocytosis by alveolar macrophages is reduced,Citation56 and since the inflammatory and neutrophilic response in AATD is enhanced this is likely to be a pathologically important pathway.Citation24 Further, supplementation of macrophages in vitro with exogenous AAT restores cigarette smoke induced decrease in efferocytosis, suggesting AAT is protective to macrophages, although the mechanism is currently unknown.Citation48
Augmentation Therapy
Augmentation therapy is currently the only therapy specific for AATD approved in the US and Europe (though not in all countries including the UK). However, there are issues of cost-effectiveness and benefits to the patient which currently questions its use, particularly in the UK. Although augmentation can modulate (though not prevent) the progression of emphysema,Citation67–Citation69 likely due to the reduction in the effects of neutrophil elastase in the lungs and improvement of the serine proteinase:anti-proteinase imbalance, it cannot affect the formation of polymers within hepatocytes, monocytes or macrophages, and so these cells may remain in a pro-inflammatory state, and therefore continue causing damage through alternative pathways. However, studies are emerging which suggest potential beneficial effects of augmentation therapy on these cells themselves.
Augmentation restores AAT, which in turn reduces LTB4 production in airways due to the prevention of neutrophil elastase binding to macrophage surfaces.Citation44 This will reduce neutrophil recruitment, and thereby reduce inflammation in the lungs. In mice, inhaled AAT restores cigarette smoke induced decrease in efferocytosis and also reduces neutrophil apoptosis,Citation48 suggesting that AAT is cytoprotective although the mechanism is unclear. In human augmentation patients, MDMs isolated after an infusion of AAT are better able to control infection to Mycobacterium intracellulare due to greater phagosome-lysosome fusion and increased autophagosome formation/maturation.Citation70 Whether this is also an effect with organisms more likely to be relevant to COPD and exacerbations needs to be confirmed however this suggests that augmentation therapy may potentially prevent bacterial exacerbation in AATD patients due to improved macrophage function, although this requires further and more relevant research. While augmentation therapy may slow progression of emphysema, the effect on lung function is less clear due to a lack of longitudinal studies, although observational studies suggest this to be the case.Citation71 However, the impact of restoring lung AAT levels to normal on the whole immune response requires further research in order to determine whether this additional benefit aids in the treatment of this disease.
Alternative Therapies
Alternative approaches to treating AATD are being developed, which target the polymeric AAT protein itself. These include chemical chaperones, which reverse cellular mislocalisation and misfolding of the protein, such as 4-phenylbutyric acid (PBA). PBA which has been shown to increase the secretion of functional Z-AAT in human skin fibroblasts expressing Z-AAT, and produced a 20–50% increase in serum AAT level in PiZZ mice.Citation72 In human trials, PBA improves release of AAT from hepatocytes, and increased serum levels, but was associated with severe side effects.Citation73 Further trials on the benefits of these chaperones on cellular AAT polymers, in hepatocytes and immune cells may show benefit in improving macrophage function.
Alternatively, small molecule correctors facilitate proper folding of the AAT protein within affected cells, and thus may increase systemic (via hepatocytes) and local release of AAT (by macrophage/monocytes). These include small peptides that fit into the open beta sheet of abnormal AAT, to block insertion of the inhibitory site loop of one AAT into the sheet of the next,Citation74 thus preventing polymerisation. However, although this may prevent polymerisation it also inactivates the protein as an inhibitor and thus the main benefit may relate to protection of the liver and not the lung. To date, there is no published data on the effectiveness of these drugs in humans.Citation75
Conclusion
AATD is typically considered a disease driven by the proteolytic nature of neutrophil enzymes, but evidence is increasing that other cells of the innate and secondary immune system are also implicated. Fully understanding the interactive role and function of monocytes and macrophages in AATD may help explain the partial response to augmentation therapy and identify important alternative targets for treatment, and determine whether drugs that remove polymers from within the cells is essential for effective disease treatment.
Disclosure
Kylie BR Belchamber reports grants from Alpha-1 foundation, during the conduct of the study. Robert A Stockley reports personal fees from Vertex, grants and personal fees from CSL Behring, grants from Mereo biopharma and Takeda, Advisory Board for Z factor, and Chair DSMB for Kamada, during the conduct of the study. Elizabeth Sapey reports grants from Medical Research Council, grant from Wellcome Trust, grants from NIHR, grants from British Lung Foundation, grants from Alpha 1 Foundation, and grants from HDR-UK, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
- StockleyRA. Alpha1-antitrypsin review. Clin Chest Med. 2014;35(1):39–50. doi:10.1016/j.ccm.2013.10.00124507836
- BerginDA, HurleyK, McElvaneyNG. Alpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp. 2012;60:81–97. doi:10.1007/s00005-012-0162-5
- DickensJA, LomasDA. Why has it been so difficult to prove the efficacy of alpha-1-antitrypsin replacement therapy? Insights from the study of disease pathogenesis. Drug Des Devel Ther. 2011;5:391–405.
- StockleyRA. Neutrophils and Protease/Antiprotease Imbalance. Am J Respir Crit Care Med. 1999;160(1):S49–S52. doi:10.1164/ajrccm.160.supplement_1.1310556170
- HillAT, BayleyDL, CampbellEJ, HillSL, StockleyRA. Airways inflammation in chronic bronchitis: the effects of smoking and alpha1-antitrypsin deficiency. Eur Respir J. 2000;15(5):886–890. doi:10.1034/j.1399-3003.2000.15e12.x10853853
- StollerJK, AboussouanLS. A review of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246–259. doi:10.1164/rccm.201108-1428CI21960536
- BlancoI, DiegoI, BuenoP, Perez-HolandaS, Cases-MaldonadoF, MiravitllesM. Prevalence of α1-antitrypsin PiZZ genotypes in patients with COPD in Europe: a systematic review. Eur Respir Rev. 2020;29(157):200014. doi:10.1183/16000617.0014-202032699024
- Society AT, Society ER. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency; 2003.
- TanL, DickensJA, DeMeoDL, et al. Circulating polymers in α1-antitrypsin deficiency. Eur Respir J. 2014;43:1501–1504. doi:10.1183/09031936.0011121324603821
- LiebermanJ, WinterB, SastreA. Alpha-1 antritrypsin Pi-types in 965 COPD patients. Chest. 1986;89(3):370–373. doi:10.1378/chest.89.3.3703485034
- FranciosiAN, HobbsBD, McElvaneyOJ, et al. Clarifying the risk of lung disease in SZ Alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2020;202(1):73–82. doi:10.1164/rccm.202002-0262OC32197047
- HaqI, IrvingJA, SalehAD, et al. Deficiency mutations of Alpha-1 antitrypsin. Effects on folding, function, and polymerization. Am J Respir Crit Care Med. 2016;54(1).
- RogersJ, KalshekerN, WallisS, et al. The isolation of a clone for human alpha1-antitrypsin and the detection of alpha 1-antitrypsin in mRNA from liver and leukocytes. Biochem Biophys Res Commun. 1983;116(2):375–382. doi:10.1016/0006-291X(83)90532-66606425
- CichyJ, PotempaJ, TravisJ. Biosynthesis of alpha 1 proteinase inhibitor by human lung-derived epithelial cells. J Biol Chem. 1997;272(13):8250–8255. doi:10.1074/jbc.272.13.82509079644
- BerginDA, ReevesEP, MeleadyP, et al. Alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120(12):4236–4250. doi:10.1172/JCI4119621060150
- Du BoisRM, BernaudinJF, PaakkoP, et al. Human neutrophils express the alpha 1-antitrypsin gene and produce alpha 1-antitrypsin. Blood. 1991;77(12):2724–2730. doi:10.1182/blood.V77.12.2724.27242043769
- MolmentiEP, PerlmutterDH, RubinDC. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J Clin Invest. 1993;92(4):2022–2034. doi:10.1172/JCI1167978408656
- BoscoD, MedaP, MorelP, et al. Expression and secretion of alpha1- proteinase inhibitor are regulated by proinflammatory cytokines in human pancreatic islet cells. Diabetologia. 2005;48(8):1523–1533. doi:10.1007/s00125-005-1816-116001235
- MulgrewAT, TaggartCC, LawlessMW, et al. Z alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest. 2004;125(5):1952–1957. doi:10.1378/chest.125.5.195215136414
- MahadevaR, AtkinsonC, LiZ, et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166(2):377–386. doi:10.1016/S0002-9440(10)62261-415681822
- ElliottPR, BiltonD, LomasDA. Lung polymers in Z alpha1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol. 1998;18(5):670–674. doi:10.1165/ajrcmb.18.5.30659569237
- ParrDG, StoelBC, StolkJ, StockleyRA. Pattern of emphysema distribution in α1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004;170(11):1172–1178. doi:10.1164/rccm.200406-761OC15306534
- JanciauskieneS, WrengerS, ImmenschuhS, et al. The multifaceted effects of Alpha1-antitrypsin on neutrophil functions. Front Pharmacol. 2018;9:341. doi:10.3389/fphar.2018.0034129719508
- McCarthyC, ReevesEP, McElvaneyNG. The role of neutrophils in Alpha-1 antitrypsin deficiency. Ann Am Thorac Soc. 2015;13(4):297–304. doi:10.1513/AnnalsATS.201509-634KV
- CarrollTP, GreeneCM, O’ConnorCA, NolanAM, O’NeillSJ, McElvaneyNG. Evidence for unfolded protein response activation in monocytes from individuals with a-1 antitrypsin deficiency. J Immunol. 2010;184:4538–4546. doi:10.4049/jimmunol.080286420228200
- BazzanE, TineM, BiondiniD, et al. α1-Antitrypsin Polymerizes in Alveolar Macrophages of Smokers With and Without α1-Antitrypsin Deficiency. Chest. 2018;154(3):607–616. doi:10.1016/j.chest.2018.04.03929763589
- KarlmarkKR, TackeF, DunayIR. Monocytes in health and disease – mini review. Eur J Microbiol Immunol (Bp). 2012;2(2):97–102. doi:10.1556/EuJMI.2.2012.2.124672677
- KapellosTS, BonaguroL, GemundI, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.02035
- PasslickB, FliegerD, Ziegler-HeitbrockL. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74(7):2527–2534. doi:10.1182/blood.V74.7.2527.25272478233
- WongKL, TaiJ, WongWC, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):16–31. doi:10.1182/blood-2010-12-326355
- PerlmutterDH, ColeFS, KilbridgeP, RossingTH, ColtenHR. Expression of the a1-proteinase inhibitor gene in human monocytes and macrophages. Proc Natl Acad Sci. 1985;82:798–799. doi:10.1073/pnas.82.3.795
- PerlmutterDH, TravisJ, PunsalPI. Elastase regulates the synthesis of its inhibitor, alpha 1-proteinase inhibitor, and exaggerates the defect in homozygous PiZZ alpha 1 PI deficiency. J Clin Invest. 1988;81(6):1774–1780. doi:10.1172/JCI1135193260245
- MornexJF, Chytil-WeirA, MartinetY, CourtneyM, LeCocqJP, CrystalRG. Expression of the alpha-1-antitrypsin gene in mononuclear phagocytes of normal and alpha-1-antitrypsin-deficient individuals. J Clin Invest. 1986;77(6):1952–1961. doi:10.1172/JCI1125243486887
- JanciauskieneS, LarssonS, LarssonP, VirtalaR, JanssonL, StevensT. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by a1-antitrypsin. Biochem Biophys Res Commun. 2004;321:592–600. doi:10.1016/j.bbrc.2004.06.12315358147
- NitaIM, SerapinasD, JanciauskieneS. alpa1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int J Biochem Cell Biol. 2007;39:1165–1176. doi:10.1016/j.biocel.2007.02.01717448722
- JanciauskieneS, NitaIM, StevensT. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282(12):8573–8582. doi:10.1074/jbc.M60797620017261591
- OlingyCE, San EmeterioCL, OgleME, et al. Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci Rep. 2017;7:447. doi:10.1038/s41598-017-00477-128348370
- ThomasG, TackeR, HedrickCC, HannaRN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. 2015;35(6):1306–1316. doi:10.1161/ATVBAHA.114.30465025838429
- StolkJ, AggarwalAN, HochnadelI, et al. Blood monocyte profiles in COPD patients with PiMM and PiZZ α1-antitrypsin. Respir Med. 2019;148:60–62. doi:10.1016/j.rmed.2019.02.00130827477
- StockleyRA, EdgarRG, PillaiA, TurnerAM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obstruct Pulmon Dis. 2016;11:1745–1756. doi:10.2147/COPD.S11150827536086
- MurrayPJ, AllenJE, BiswasSK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.00825035950
- MantovaniA, BiswalS, GaldieroMR, SicaA, LocatiM. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. doi:10.1002/path.413323096265
- Van’t WoutEFA, van SchadewijkA, SavageNDL, StolkJ, HiemstraPS. Alpha-1 antitrypsin production by proinflammatory and antiinflammatory macrophages and dendritic cells. Am J Respir Cell Mol Biol. 2012;46(5):607–613. doi:10.1165/rcmb.2011-0231OC22162908
- HubbardRC, FellsG, GadekJ, PacholokS, HumesJ, CrystalRG. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest. 1991;88(3):891–897. doi:10.1172/JCI1153911653278
- CrooksSW, BayleyDL, HillSL, StockleyRA. Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. Eur Respir J. 2000;15(2):274–280. doi:10.1034/j.1399-3003.2000.15b09.x10706491
- WoolhouseIS, BayleyDL, StockleyRA. Sputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha(1)-antitrypsin deficiency and the role of leukotriene B(4) and interleukin 8. Thorax. 2002;57(8):709–714. doi:10.1136/thorax.57.8.70912149532
- StockleyRA, BayleyDL, UnsalI, DowsonLJ. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165(11):1494–1498. doi:10.1164/rccm.210901312045122
- SerbanKA, PetruscaDN, MikoszA, et al. Alpha-1 antitrypsin supplementation improves alveolar macrophages efferocytosis and phagocytosis following cigarette smoke exposure. PLoS One. 2017;12(4):e0176073. doi:10.1371/journal.pone.017607328448535
- VandivierRW, FadokVA, HoffmannPR, et al. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002;109(5):661–670. doi:10.1172/JCI021357211877474
- ChanED, KaminskaAM, GillW, et al. Alpha-1-antitrypsin (AAT) anomalies are associated with lung disease due to rapidly growing mycobacteria and AAT inhibits Mycobacterium abscessus infection of macrophages. Scand J Infect Dis. 2007;39:690–696. doi:10.1080/0036554070122574417654345
- KrotovaK, MarekGW, WangRL, et al. Alpha-1 antitrypsin-deficient macrophages have increased matriptase-mediated proteolytic activity. Am J Respir Cell Mol Biol. 2017;57(2):238–247. doi:10.1165/rcmb.2016-0366OC28362108
- BelchamberKBR, DonnellyLE. Macrophage function in respiratory disease In: KlocM, editor. Macrophages: Origin, Functions and Biointervention. Results and Problems in Cell Differentiation. Vol. 62 Springer; 2017:299–313.
- TaylorAE, Finney-HaywardTK, QuintJK, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35(5):1039–1047. doi:10.1183/09031936.0003670919897561
- BerensonCS, GarlippMA, GroveLJ, MaloneyJ, SethiS. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194(10):1375–1384. doi:10.1086/50842817054066
- WrenchC, BelchamberKBR, BercussonA, ShahA, BarnesPJ, DonnellyLE. Reduced clearance of fungal spores by COPD GM-CSF and M-CSF derived macrophages. Am J Respir Cell Mol Biol. 2018;58(2):271–273. doi:10.1165/rcmb.2017-0351LE29388829
- HodgeS, HodgeG, ScicchitanoR, ReynoldsPN, HolmesM. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–296. doi:10.1046/j.1440-1711.2003.t01-1-01170.x12848850
- BelchamberKBR, SinghR, BatistaCM, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD. Eur Respir J. 2019;54(3). doi:10.1183/13993003.02244-2018.
- ChertovO, UedaH, XuL, et al. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med. 1997;186(5):739–747. doi:10.1084/jem.186.5.7399271589
- SoehnleinO, LindbornL. Neutrophil‐derived azurocidin alarms the immune system. J Leukoc Biol. 2008;85(3).
- YangW, TaoY, WuY, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun. 2019;10.
- MarwickJA, MillsR, KayO, et al. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-κB activation. Cell Death Dis. 2018;9. doi:10.1038/s41419-018-0710-y
- MartinSJ, ReutelingspergerCP, McGahonAJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;185(5):1454–1456.
- ScannellM, FlanaganMB, deStephaniA, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178(7):4595–4605. doi:10.4049/jimmunol.178.7.459517372018
- KobayashiN, KarisolaP, Pena-CruzV, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–940. doi:10.1016/j.immuni.2007.11.01118082433
- FadokVA, BrattonDL, KonowalA, FreedPW, WestcottJY, HensonPM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–898. doi:10.1172/JCI11129466984
- HurleyK, LaceyN, O’DwyerCA, et al. Alpha-1 antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individuals. J Immunol. 2014;193:3979–3991. doi:10.4049/jimmunol.1400132
- EdgarRG, PatelM, BaylissS, CrossleyD, SapeyE, TurnerAM. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308. doi:10.2147/COPD.S13044028496314
- ChapmanKR, StockleyRA, DawkinsC, WilkesMN, NavickisRJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6(3):177–184. doi:10.1080/1541255090290596119811373
- TonelliAR, BrantlyML. Augmentation therapy in alpha-1 antitrypsin deficiency: advances and controversies. Ther Adv Respir Dis. 2010;4(5):289–312. doi:10.1177/175346581037391120650978
- BaiX, BaiA, HondaJR, et al. Alpha-1-antitrypsin enhances primary human macrophage immunity against non-tuberculous mycobacteria. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.01417
- FerrarottiI, OttavianiS, De SilvestriA, CorsicoAG. Update on α 1 -antitrypsin deficiency. Breathe. 2018;14:17–24. doi:10.1183/20734735.015018
- BurrowsJA, WillisLK, PerlmutterDH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci. 2000;97(4):1796–1801. doi:10.1073/pnas.97.4.179610677536
- LoffingJ, MoyerBD, ReynoldsD. PBA increases CFTR expression but at high doses inhibits Cl(-) secretion in Calu-3 airway epithelial cells. Am J Physiol. 1999;227(4):700–708.
- MahadevaR, DaffronTR, CarrellRW, LomasDA. 6-mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerization. J Biol Chem. 2001;277:6771–6774. doi:10.1074/jbc.C10072220011773044
- PyeA, TurnerAM. Experimental and investigational drugs for the treatment of alpha-1 antitrypsin deficiency. Expert Opin Investig Drugs. 2019;28(10):891–902. doi:10.1080/13543784.2019.1672656