
Abstract
Background
Addition of a second bronchodilator from a different pharmacological class may benefit patients with moderate-to-severe chronic obstructive pulmonary disease (COPD) whose symptoms are insufficiently controlled by bronchodilator monotherapy. GLOW6 evaluated the efficacy and safety of once-daily coadministration of the long-acting β2-agonist indacaterol (IND) and the long-acting muscarinic antagonist glycopyrronium (GLY) versus IND alone in patients with moderate-to-severe COPD.
Materials and methods
In this randomized, double-blind, parallel group, placebo-controlled, 12-week study, patients were randomized 1:1 to IND 150 μg and GLY 50 μg daily (IND + GLY) or IND 150 μg daily and placebo (IND + PBO) (all delivered via separate Breezhaler® devices). The primary objective was to demonstrate the superiority of IND + GLY versus IND + PBO for trough forced expiratory volume in 1 second (FEV1) at week 12. Other end points included trough FEV1 at day 1, FEV1 area under the curve from 30 minutes to 4 hours (AUC30min–4h), peak FEV1, inspiratory capacity and trough forced vital capacity (FVC) at day 1 and week 12, and transition dyspnea index (TDI) focal score, COPD symptoms, and rescue medication use over 12 weeks.
Results
A total of 449 patients were randomized (IND + GLY, 226; IND + PBO, 223); 94% completed the study. On day 1 and at week 12, IND + GLY significantly improved trough FEV1 versus IND + PBO, with treatment differences of 74 mL (95% CI 46–101 mL) and 64 mL (95% CI 28–99 mL), respectively (both P<0.001). IND + GLY significantly improved postdose peak FEV1, FEV1 AUC30min–4h, and trough FVC at day 1 and week 12 versus IND + PBO (all P<0.01). TDI focal score and COPD symptoms (percentage of days able to perform usual daily activities and change from baseline in mean daytime respiratory score) were significantly improved with IND + GLY versus IND + PBO (P<0.05). The incidence of adverse events was similar for the two treatment groups.
Conclusion
In patients with moderate-to-severe COPD, once-daily coadministration of IND and GLY provides significant and sustained improvement in bronchodilation versus IND alone from day 1, with significant improvements in patient-centered outcomes.
Introduction
Bronchodilators are central to the pharmacological management of chronic obstructive pulmonary disease (COPD).Citation1 Whereas short-acting bronchodilators are used for immediate relief from symptoms, one or more long-acting bronchodilators (long-acting β2-agonists [LABAs] and long-acting muscarinic antagonists [LAMAs]) are recommended for long-term maintenance therapy in patients with moderate-to-very severe COPD.Citation1,Citation2 Long-acting bronchodilators include well-established agents, such as the LAMA tiotropium (once-daily [od]) and the LABAs formoterol and salmeterol (both twice-daily [bid]), and the more recently introduced LAMAs glycopyrronium (NVA237; od)Citation3,Citation4 and aclidinium (bid),Citation5–Citation7 and the LABA indacaterol (od).
The efficacy and safety of glycopyrronium and indacaterol, given as long-acting bronchodilator monotherapies in patients with moderate-to-severe COPD, has been demonstrated in several Phase III studies.Citation8–Citation14 The effect of indacaterol on lung-function outcomes was shown to be superior to twice-daily LABAsCitation11,Citation14,Citation15 and comparable to tiotropium.Citation12,Citation16,Citation17 Clinical outcomes, such as dyspnea and health status, have also been shown to improve to a significantly greater extent with indacaterol compared with tiotropium.Citation16 Glycopyrronium was shown to have a comparable effect to tiotropium on lung function, symptoms, exacerbations, and rescue medication use, with a significantly more rapid onset of action on day 1 compared with tiotropium.Citation13 Glycopyrronium has also demonstrated an immediate and significant improvement in exercise tolerance over 3 weeks compared with placebo, beginning with the first dose; this was accompanied by sustained reductions in lung hyperinflation.Citation8
In patients whose symptoms are insufficiently controlled by bronchodilator monotherapy, the Global initiative for chronic Obstructive Lung Disease (GOLD) strategy for the management of COPD recommends the addition of a second bronchodilatorCitation1; this is supported by evidence showing that the addition of a second bronchodilator from a different pharmacological class improves lung function, symptoms, and health status compared with monotherapy, without significantly increasing the risk of side effects.Citation18–Citation20
Several studies have established the superior efficacy of free combinations of LABAs and LAMAs in bronchodilation, symptom control, and rescue medication use versus the LAMA monocomponentCitation21–Citation25 and versus the LABA monocomponent.Citation18,Citation20,Citation26 Recently, INTRUST-1 and INTRUST-2 study investigators reported that concurrent administration of a LABA (indacaterol) and a LAMA (tiotropium) provided superior bronchodilation and lung deflation compared with LAMA (tiotropium) monotherapy.Citation21 Furthermore, QVA149, a once-daily, fixed-dose combination of glycopyrronium 50 μg and indacaterol 150 μg (in development), has demonstrated superior efficacy compared with both monocomponents in a recent study.Citation27
In the present GLOW6 (GLycopyrronium bromide in COPD airWays clinical study 6), we aimed to evaluate the efficacy and safety of the coadministration of a LABA (indacaterol 150 μg od) and a LAMA (glycopyrronium 50 μg od) versus the LABA (indacaterol 150 μg od) alone in patients with moderate-to-severe COPD.
Materials and methods
Patients
The GLOW6 study enrolled men and women ≥40 years of age, with moderate-to-severe stable COPD (GOLD stage II or III according to the 2010 GOLD guidelines),Citation28 who were current or ex-smokers with a smoking history of at least 10 pack-years, and had a post-bronchodilator forced expiratory volume in 1 second (FEV1) ≥30% and <80% of the predicted normal and postbronchodilator FEV1/forced vital capacity (FVC) ratio of <0.70 at screening (GOLD stage II or III).Citation28 Postbronchodilator refers to 1 hour after sequential inhalation of 84 μg ipratropium bromide (or equivalent dose) and 400 μg salbutamol (or equivalent dose).
The main exclusion criteria included respiratory tract infection within 6 weeks prior to screening; COPD exacerbation requiring treatment with antibiotics and/or oral corticosteroids and/or hospitalization 6 weeks prior to screening; concomitant pulmonary disease (such as lung fibrosis, sarcoidosis, interstitial lung disease, pulmonary hypertension, clinically significant bronchiectasis, pulmonary tuberculosis); history of asthma, diabetes (with the exception of controlled type II diabetes), malignancy of any organ system, long QT syndrome or QTc >450 ms at screening, symptomatic prostatic hyperplasia, bladder-neck obstruction, moderate/severe renal impairment, urinary retention, narrow-angle glaucoma, a known history of α1-antitrypsin deficiency, or paroxysmal atrial fibrillation; clinically significant renal, cardiovascular (such as, but not limited to, unstable ischemic heart disease, New York Heart Association class III/IV left ventricular failure, myocardial infarction), neurological, immunological, psychiatric, gastrointestinal, hepatic, or hematological abnormality that could have interfered with the assessment of efficacy and safety of the study treatment; participation in the active phase of a supervised pulmonary rehabilitation program; and contraindications for tiotropium or ipratropium, or history of adverse reactions to inhaled anticholinergics.
All patients gave written, informed consent to participate in the study (NCT1604278).Citation29 The study protocol was reviewed and approved by institutional review boards and ethics committees at participating centers. The study was conducted in compliance with Good Clinical Practice (GCP). lists the study centers.
Study design and treatment
GLOW6 was a multicenter, randomized, double-blind, parallel-group, placebo-controlled, 12-week study. After a washout period (up to 7 days), followed by a 14-day run-in period, patients were randomized 1:1 to indacaterol 150 μg od and glycopyrronium 50 μg od (IND + GLY; 50 μg refers to the quantity of the glycopyrronium moiety present in the capsule, which corresponds to a delivered dose of 44 μg) or indacaterol 150 μg od and placebo (IND + PBO). All treatments were delivered via separate Breezhaler® (Novartis, Basel, Switzerland) devices, and were taken each morning between 8 and 11 (). The devices were not to be used interchangeably by the patients, and alternative inhalation devices were not permitted.
Figure 1 GLOW6 study design.
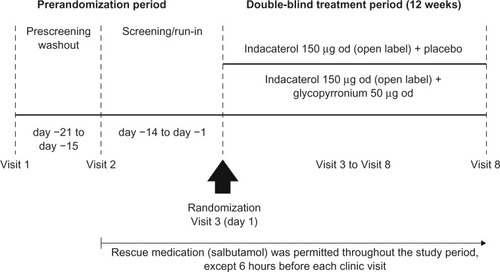
An automated, interactive, voice-response technology was used to assign randomization numbers to patients who met the study criteria. Randomization numbers were used to link patients to treatment groups and these were not communicated to the caller. Patients, investigators, site staff, persons performing the assessments and data analysts were blind to the identity of the treatment from the time of randomization. Randomization data were kept strictly confidential until the time of unblinding.
Patients were to discontinue taking long-acting bronchodilator therapy before starting the run-in period (for at least 7 days for LAMAs and the LABA indacaterol, and for 48 hours for other LABAs or LABA/inhaled corticosteroid [ICS] combinations). Those on fixed-dose LABA/ICS combinations were switched to ICS monotherapy at a dose equivalent to that contained in the fixed-dose combination. Patients were provided with a salbutamol (short-acting β2-agonist) inhaler to be used as rescue medication during the study. They were instructed to abstain from taking rescue medication within 6 hours of the start of each study visit. Further details are provided in .
Efficacy assessments
The primary efficacy variable was trough FEV1 (defined as the mean of the 23 hour 15 minute and 23 hour 45 minute postdose values, imputed with last observation carried forward) at week 12. Secondary variables included trough FEV1 at day 1, FEV1 area under the curve from 30 minutes to 4 hours (AUC30min–4h), peak FEV1, FEV1 and inspiratory capacity (IC) at individual time points, trough FVC at day 1 and week 12, transition dyspnea index (TDI) focal score, COPD symptoms collected via patient diaries, and mean change from baseline in daily number of puffs of rescue medication over the 12-week treatment period. Effects on the St George’s Respiratory Questionnaire – COPD (SGRQ-C) total score at week 12 versus baseline were assessed as an exploratory objective.
Spirometric measurements (recorded via centralized spirometry) were taken prior to the run-in period to determine study eligibility and to record postbronchodilator FEV1 1 hour after sequential inhalation of four 21 μg puffs of ipratropium bromide (or equivalent dose) and four 100 μg puffs of salbutamol. FEV1 and FVC were recorded at all clinic visits at the following time points relative to the morning dose: 45 and 15 minutes predose, and 30 minutes, 1 hour, 4 hours, and 24 hours postdose on day 1 and week 12. Dyspnea was assessed by an investigator-administered baseline dyspnea index and TDI, and health status by self-administered SGRQ-C at day 1 and week 12.
All patients were provided with an eDiary to record morning and evening daily clinical symptoms: cough, wheezing, shortness of breath, sputum volume, sputum purulence, cold, fever, sore throat, nighttime awakenings and rescue medication use, and time of study-drug administration. Patients were instructed to complete the eDiary routinely at the same time each morning (prior to taking the study drug) and again (approximately 12 hours later) each evening, considering events over the previous 12 hours. Patient diaries were reviewed at each clinic visit.
Safety assessments
Safety was assessed by recording all treatment-emergent adverse events (AEs) and serious AEs (SAEs), monitoring vital signs (pulse rate and systolic and diastolic blood pressure), and performing laboratory analyses (hematology, clinical chemistry, and urinalysis). AEs starting on or after the time of first inhalation of the study drug but not later than 7 days (30 days in the case of SAEs) after last inhalation of the study drug were classified as treatment-emergent AEs. AEs were coded using the Medical Dictionary for Regulatory ActivitiesCitation30 and summarized by primary system organ class, preferred term, maximum severity, and relationship to study drug. An independent adjudication committee classified the reported serious cardio- and cerebro-vascular (CCV) events.
Statistical analysis
Three populations were defined in the GLOW6 study for the purpose of analysis. The full analysis set (FAS) included all randomized patients who received at least one dose of the study drug; patients were analyzed according to the treatment they were assigned to at randomization. The per-protocol set (PPS) included all patients in the FAS who had no major protocol deviations; patients were analyzed according to the treatment to which they were randomized. The safety population included all patients who received at least one dose of the study medication, irrespective of randomization; patients were analyzed according to the treatment they received.
The primary analysis for trough FEV1 at week 12 was performed on the FAS using a mixed model. The mixed model contained treatment as a fixed effect, with the baseline FEV1 and FEV1 prior to and post inhalation of short-acting bronchodilator as covariates. The model also included smoking status at baseline (current/ex-smoker), history of baseline ICS use (yes/no) and region as fixed effects, and center (nested in the region) as a random effect. If any of the values contributing to trough FEV1 were collected within 6 hours of rescue medication or within 7 days of systemic corticosteroid use, then the individual FEV1 value was not included in the analysis. Superiority of IND + GLY versus IND + PBO was claimed if the difference in trough FEV1 was statistically significant at the 5% level and the 95% confidence interval (CI) was entirely to the right of (higher than) 0.0 L.
Other secondary variables were analyzed in the FAS using the same mixed model as the primary analysis, with the respective baseline values replacing baseline FEV1 as a covariate. Results are shown as least squares means (LSMs) with standard errors for group mean values and with 95% CIs for differences between treatments. The procedure for handling missing data is detailed in .
Additionally, exploratory subgroup analyses were performed for the primary end point to explore the treatment effect by age (<65 and ≥65 years), sex (male/female), smoking history (ex-smokers/current smokers), severity of disease, use of ICS at baseline (yes/no) and body mass index (BMI; >30 kg/m2 or ≤30 kg/m2) in the FAS. The analysis was performed with the appropriate interaction term in the model and the additional covariate as a fixed effect, if necessary. All safety end points were summarized for the safety population.
Sample-size calculation
To detect statistical significance in the primary end point (at α=0.05, with 80% power) for a treatment differential of 70 mL in trough FEV1 at week 12, and assuming a standard deviation of 250 mL and a 10% dropout rate, it was calculated that an estimated total sample size of 450 patients (225 per group) would be required (404 completers).
Results
Patient disposition and baseline characteristics
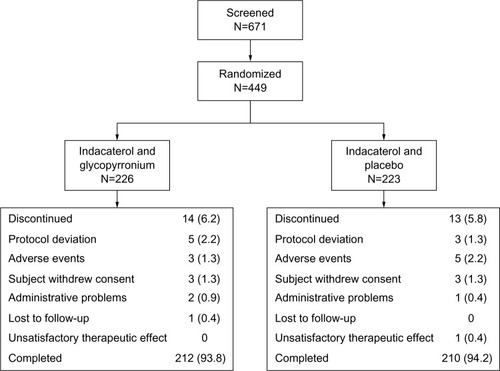
A total of 671 patients were screened, of whom 449 were randomized (IND + GLY, 226; IND + PBO, 223; ); 94% (422 patients) completed the study. The percentage of patients who discontinued was similar in both groups. The two most common reasons for discontinuing treatment were protocol deviation and AEs. Discontinuations were more frequent due to protocol deviation in the IND + GLY group (five of 14 patients; 2.2%) than in the IND + PBO group (three of 13 patients; 1.3%); they were more frequent due to AEs in the IND + PBO group (five of 13 patients; 2.2%) than in the IND + GLY group (three of 14 patients; 1.3%).
Figure 2 Patient disposition, n (%).
All baseline characteristics were similar between the treatment groups (). The mean age of the patients was 63.7 years, 82% of the patients were male and the majority (99%) were Caucasian. All patients had moderate (64%) or severe (36%) airflow limitation.Citation1 All patients were smokers (42%) or ex-smokers (58%), and the mean smoking history was 44.5 pack-years. The mean duration of COPD was 7.1 years; 30% of the patients had a documented history of at least one exacerbation in the previous year; 63% of the patients used ICS. Mean postbronchodilator FEV1 was 54.8% predicted, and mean postbronchodilator FEV1/FVC ratio was 48.5%.
Table 1 Baseline demographics, background characteristics, and spirometry (safety population)
Efficacy
Spirometry
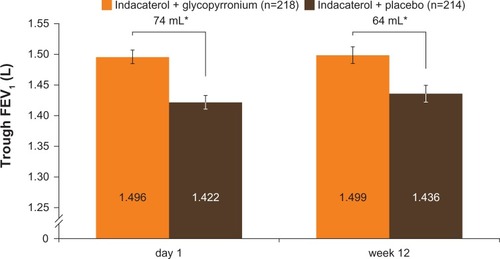
The primary end point in GLOW6 was met, as the LSM trough FEV1 at week 12 was significantly higher in the IND + GLY group than in the IND + PBO group, with a treatment difference of 64 mL (95% CI 28–99 mL, P<0.001; and ). Similar results were seen in the supportive analysis in the PPS, with a treatment difference of 64 mL (95% CI 28–101 mL, P<0.001). After first dose, the treatment difference for trough FEV1 (measured at the end of day 1 as the mean of the values at 23 hours 15 minutes and 23 hours 45 minutes) was 74 mL (95% CI 46–101 mL, P<0.001) in favor of IND + GLY versus IND + PBO ( and ). IND + GLY was also statistically significantly superior to IND + PBO for FEV1 AUC30min–4h postdose and peak FEV1 on day 1 and at week 12 (all P<0.001, ).
Table 2 Differences between treatments for primary and secondary efficacy outcomes on day 1 and at week 12 (FAS)
Figure 3 Trough FEV1 after first dose (end of day 1) and week 12 (FAS).
Abbreviations: FAS, full analysis set; FEV1, forced expiratory volume in 1 second.
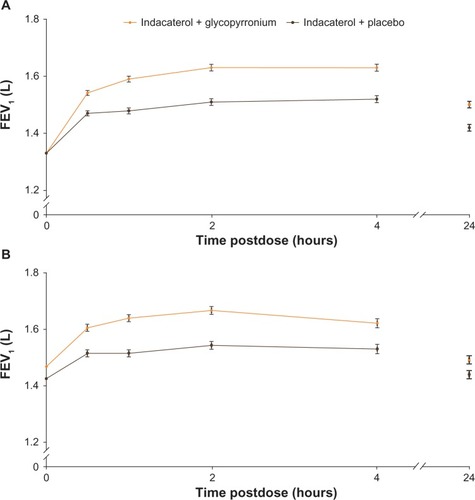
On day 1 and at week 12, FEV1 at all time points from 30 minutes to 4 hours and at 24 hours was significantly higher in the IND + GLY group compared with the IND + PBO group, with statistically significant treatment differences at each time point (all P<0.001 [except at 24 hours on week 12 P<0.01], ).
Figure 4 FEV1 from 30 minutes to 4 hours postdose and 24 hours postdose (A) on day 1 and (B) at week 12 (FAS).
Abbreviations: FEV1, forced expiratory volume in 1 second; FAS, full analysis set.
Trough FVC on day 1 and at week 12 was significantly higher in the IND + GLY treatment group than in the IND + PBO group (LSM treatment differences, 111 mL [P<0.001] and 93 mL [P=0.006], respectively; ). IC at 2 hours and 4 hours postdose on day 1 and at 30 minutes, 2 hours, and 4 hours postdose at week 12 was significantly higher in the ING + GLY treatment group compared with the IND + PBO treatment group ().
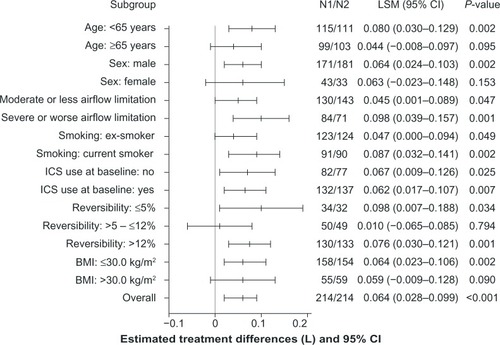
The exploratory subgroup analyses of trough FEV1 at week 12 demonstrated superiority of IND + GLY versus IND + PBO for most subgroups evaluated, consistent with the result for trough FEV1 in the overall FAS; younger patients (<65 years), patients with severe airflow limitation,Citation1 ICS users at baseline, and current smokers in the IND + GLY treatment group had a significantly higher improvement in trough FEV1 at week 12 versus patients in the IND + PBO group ().
Figure 5 Subgroup analyses of treatment differences in trough FEV1 at week 12 (FAS).
Abbreviations: BMI, body mass index; CI, confidence interval; FAS, full analysis set; FEV1, forced expiratory volume in 1 second; ICS, inhaled corticosteroid; LSM, least squares mean; N1, number of patients analyzed in the indacaterol + glycopyrronium treatment group; N2, number of patients analyzed in the indacaterol + placebo treatment group.
Symptoms, diary-card data, health status
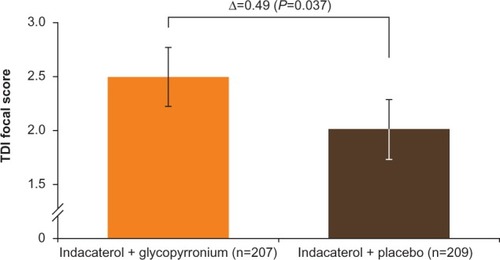
TDI focal score at week 12 showed a greater reduction in dyspnea with IND + GLY than with IND + PBO, with an LSM difference of 0.49 units (95% CI 0.03–0.96 units, P=0.037; and ). Patients receiving IND + GLY were significantly more likely to achieve a minimum clinically important improvement (MCID; ≥1 unit improvement) in dyspneaCitation31 than those taking IND + PBO (76.6% versus 62.2%, respectively; odds ratio [OR] 1.97, 95% CI 1.24–3.11; P=0.004).
Figure 6 TDI focal score at week 12 (FAS).
Abbreviations: FAS, full analysis set; TDI, transition dyspnea index.
Over the 12-week treatment period, diary-card data showed a significant improvement in the percentage of days able to perform usual activities in patients receiving IND + GLY, compared with patients receiving IND + PBO (LSM difference 6.2%, P=0.016; ). Significant differences were also observed in change from baseline in mean daytime respiratory symptom score with IND + GLY versus IND + PBO (LSM difference −0.1, 95% CI −0.1–0.0; P=0.025; ).
No significant differences were seen between the two treatment groups in the change from baseline in mean daily number of puffs of rescue medication (treatment difference −0.1, 95% CI −0.5–0.2; P=0.471), in the percentage of days with no rescue medication use (treatment difference 0.2, 95% CI −6.0–6.5; P=0.945), or in the change from baseline in mean daily total symptom score (treatment difference 0.0, 95% CI −0.3–0.3; P=0.810) over the 12-week treatment period ().
In the exploratory assessment of health status, there was a numerical difference in the SGRQ-C total score between IND + GLY versus IND + PBO at week 12; the LSM treatment difference was −1.47 points (95% CI −3.42–0.48 points; P=0.140; ). Raw mean (standard deviation) changes (improvements) from baseline were −6.22 (11.47) and −4.13 (10.38) with IND + GLY and IND + PBO, respectively. A higher percentage of patients taking IND + GLY achieved an MCID in SGRQ-C score (≥4-point reduction) versus patients taking IND + PBO, but the difference was not statistically significant (56.5% versus 46.8%, respectively; OR 1.43 95% CI 0.95–2.17; P=0.089).Citation32
Safety
The overall incidence of AEs was similar between the two treatment groups (IND + GLY 37.6%, IND + PBO 33.9%; ). The most frequently reported AE was COPD worsening, seen with similar frequency in both treatment groups (). Other frequently occurring AEs (at least three patients in either treatment group) included nasopharyngitis, lower respiratory tract infection, cough, back pain, upper respiratory tract infection and oropharyngeal pain. Lower respiratory tract infection occurred with higher frequency in the IND + GLY group, while the frequency of cough was higher in the IND + PBO group (). AEs leading to discontinuation occurred in a comparable number of patients in both groups ().
Table 3 Most frequent AEs (at least three patients in either treatment group) and discontinuations due to AEs (safety population), n (%)
SAEs occurred with similar frequency in both treatment groups (). The proportion of patients with newly occurring or worsening clinically notable QT interval with Fridericia’s correction (QTcF) values was similar in the IND + PBO group (3.8%) and the IND + GLY group (2.8%). The percentage of patients with an increase in QTcF of 30–60 ms from baseline was slightly higher in the IND + GLY group (6.5%) than in the IND + PBO group (4.2%). The incidence of CCV events was low in both groups: 1.3% patients in the IND + GLY group and 2.3% patients in the IND + PBO group. Only angina pectoris was experienced by more than one patient in either treatment group; two patients in the IND + PBO group reported these events, and only one of these events was classified as serious. No deaths were reported in the study.
Table 4 SAEs (safety population), n (%)
Discussion
In the GLOW6 study, once-daily coadministration of IND + GLY provided significantly greater improvement in trough FEV1 on day 1 and at week 12 versus indacaterol alone, with treatment differences of 74 mL and 64 mL, respectively (P<0.001). Peak FEV1 and FEV1 AUC30min–4h at day 1 and week 12 were also significantly superior with IND + GLY versus indacaterol alone. Coadministration of IND + GLY also provided statistically significant improvements in dyspnea (P=0.037) and COPD symptoms (percentage of days able to perform usual activities [P=0.026] and change from baseline in mean daytime respiratory symptom score [P=0.025]) over the 12-week treatment period versus monotherapy. Both treatments had an acceptable safety profile.
In the 2013 GOLD strategy document, the free combination of a LABA plus LAMA is a recommended treatment option for all patients except those with the mildest severity of the disease.Citation1 The additional benefit provided by combining bronchodilators from different pharmacological classes is a recognized phenomenon. This has been demonstrated in studies combining two short-acting agentsCitation33,Citation34 and also in studies investigating the combination of the LAMA tiotropium and twice-daily LABAs.Citation18,Citation20,Citation25,Citation26 More recently, the additional benefits of combining a once-daily LAMA with a once-daily LABA has been explored, and the results have demonstrated superior efficacy versus the respective monotherapies.Citation21,Citation27,Citation35
Results from the present GLOW6 study support these findings and also the GOLD recommendation. The treatment difference of 64 mL in trough FEV1 between IND + GLY versus IND + PBO observed at week 12 in the GLOW6 study was consistent with results from the SHINE study, where treatment with QVA149 (a fixed-dose combination of glycopyrronium and indacaterol in Phase III development) resulted in a treatment difference of 70 mL in trough FEV1 (P<0.001) versus indacaterol at week 26.Citation27 The magnitude of improvement in trough FEV1 at week 12 in the present study was also comparable to that reported in the INTRUST-1 and INTRUST-2 studies, where differences of 80 mL and 70 mL, respectively (both P<0.001), were observed with indacaterol plus tiotropium versus tiotropium plus placebo.Citation21
In GLOW6, coadministration of IND + GLY demonstrated a greater improvement in FEV1 AUC30min–4h compared with indacaterol alone on day 1 (106 mL) and at week 12 (111 mL). Furthermore, FEV1 at all time points from 30 minutes to 4 hours and at 24 hours on day 1 and at week 12 was significantly higher with IND + GLY versus indacaterol. These results indicate an early onset and sustained 24-hour bronchodilation with the coadministration of the two once-daily long-acting bronchodilators; this may have particular relevance for patients who struggle to undertake morning activities due to COPD symptoms.Citation36 It is noteworthy that in the GLOW2 study, which evaluated glycopyrronium 50 μg od versus open-label tiotropium 18 μg od, FEV1 AUC0–4h was significantly higher with glycopyrronium versus tiotropium on day 1 (treatment difference 56 mL, P<0.001) and at week 26 (treatment difference 50 mL; P<0.01).Citation13 Thus, the coadministration of IND + GLY appears to provide an additional improvement in FEV1 AUC0–4h. This improvement in FEV1 AUC30min–4h at week 12 with the coadministration in the GLOW6 study was similar in magnitude to that seen in the SHINE study, where a treatment difference of 110 mL was observed in FEV1 AUC0–4h between QVA149 and indacaterol at week 26 (P<0.001).Citation27 Furthermore, in GLOW6, the significantly greater improvement in IC observed with the coadministration of IND + GLY at almost all assessed time points on day 1 (except 30 min post dose) and at week 12 (except predose trough IC, ie 24 hours postdose) compared with indacaterol monotherapy indicates a superior reduction in hyperinflation.
In the present study, coadministration of IND + GLY also significantly improved dyspnea (measured by TDI) compared with indacaterol alone (treatment difference 0.49 units, P=0.037), suggesting that the coadministration results in additional improvement in dyspnea compared with monotherapy. Although the MCID of ≥1-unit improvement in TDI score is widely applied in COPD trials for comparisons versus placebo as well as between active treatments, it is not actually validated for comparison between different active treatments, only against placebo.Citation31 Indacaterol has been shown to achieve the MCID in TDI focal score versus placebo,Citation11,Citation12 as well as active treatment (tiotropium),Citation12,Citation16 and in GLOW6, the improvement in TDI focal score seen with coadministration of IND + GLY is an incremental benefit over indacaterol alone. Also, it is noteworthy that in the BLAZE study, QVA149 provided a clinically meaningful improvement in TDI focal score versus placebo (treatment difference 1.37 units, P<0.001), while the mean improvement over tiotropium was 0.49 units (P=0.021).Citation35
The percentage of patients in the current study who achieved a clinically meaningful improvement in dyspnea (≥ 1-unit improvement) was significantly higher in the IND + GLY treatment group than in the IND + PBO group (76.3% and 62.2%, respectively). In addition to the improvement in dyspnea, coadministration of IND + GLY also significantly improved patient-reported COPD symptoms versus indacaterol alone (the percentage of days able to perform usual activities and the change from baseline in mean daytime respiratory symptom score over 12 weeks), indicating better symptom control when using two long-acting bronchodilators from different classes.
Although the exploratory assessment of health status did not demonstrate a statistically significant difference between the two treatments, a numerical improvement was observed with the coadministration of IND + GLY versus indacaterol alone. The study duration of the GLOW6 study (12 weeks) might have impacted on this evaluation, and a longer-term study might potentially be able to demonstrate a greater and more significant separation between the two treatments for this end point. Studies of 6–12 months’ duration are proposed as the optimal length to capture data on patient-reported outcomes.Citation36
Overall, the results of the subgroup analyses were consistent with those of the overall analyses, and demonstrated the benefit of coadministration of two long-acting bronchodilators over monotherapy. Results of the subgroup analysis also provided an indication of the patient groups who could potentially derive immediate, greater benefit from the coadministration of IND + GLY compared with indacaterol alone, ie, patients with severe or worse airflow limitation, younger patients (<65 years), ICS users, and patients who are current smokers. However, it is not known whether these trends for the subgroups are maintained over the longer term.
The safety profile of the coadministration of IND + GLY was acceptable. A small imbalance was observed in the cardiac disorders AEs (IND + GLY, two patients [0.9%]; IND + PBO, seven patients [3.2%]), but was not considered clinically meaningful. This could potentially be attributed to the aging COPD population, and also to the multiple comorbidities that are usually prevalent in patients with COPD.Citation37
The study duration of GLOW6 was 12 weeks, which may be too short a period of time to detect improvements in outcomes for chronic diseases such as COPD, especially symptomatic end points. This could be a reason why significant separation was not seen on all secondary end points. Further, 63% of the patients in the study were on baseline ICS, and these patients continued on a stable daily dose of ICS throughout the study, which may have impacted on the magnitude of the treatment effects observed. Our study population did not enroll patients with all severities of COPD who may benefit from coadministration of two long-acting bronchodilators. It has been shown that it is patients with moderate COPD who show the greatest improvements in lung function,Citation38,Citation39 and the primary aim of our study was to assess the additional benefit in lung function from the coadministration of two long-acting bronchodilators versus one.
For the pharmacological management of COPD, long-acting bronchodilators given once-daily have been shown to be superior to short-acting bronchodilators and are also preferred.Citation1,Citation40 As demonstrated by the current study, once-daily combination therapy with a LABA plus a LAMA provides further benefits over LABA monotherapy, providing superior bronchodilation and symptom relief, with an acceptable safety profile. Hence, in COPD patients who are insufficiently controlled by long-acting bronchodilator monotherapy, a step up to long-acting bronchodilator combination therapy appears to be an effective and safe next step, potentially preferable over therapy with a combination of a LABA plus an ICS. Indeed, in non-frequent exacerbators, combination therapy with the fixed-dose, dual-bronchodilator QVA149 was more efficacious in terms of improvement in pulmonary function and symptom relief versus combination therapy with a LABA plus an ICS,Citation41 at the same time avoiding safety issues (such as pneumonia) encountered in COPD patients treated with ICS.Citation42
Conclusion
The results from the GLOW6 study demonstrate that in patients with moderate-to-severe COPD, coadministration of indacaterol 150 μg od and glycopyrronium 50 μg od via the Breezhaler® device was safe and well tolerated and provided a rapid onset of effect on day 1 and significant and sustained improvements in bronchodilation along with reduction in symptoms over 12 weeks, compared with indacaterol 150 μg od, demonstrating the benefits of adding a second long-acting bronchodilator for the treatment of patients with COPD.
Acknowledgments
The study was sponsored by Novartis Pharma AG. The authors were assisted in the preparation of the manuscript by Shilpa Mudgal, a professional medical writer contracted to CircleScience (Macclesfield, UK), and Mark J Fedele (Novartis). The authors would also like to thank Damon Jack (Novartis) for his contribution to the study conduct and manuscript preparation. Writing support was funded by the study sponsor. ClinicalTrials.gov identifier: NCT1604278.
Supplementary materials
Table S1 List of study centers
Table S2 Medications allowed in the GLOW6 study under certain conditions
Table S3 Procedure for handling missing data in the GLOW6 study
Disclosure
WV has acted as consultant for Boehringer Ingelheim/Pfizer, GlaxoSmithKline, AstraZeneca, Meda Pharmaceuticals, Chiesi, Mundipharma, Novartis, and MSD. JA has no conflicts of interest. HC, MH, DMcB, and PG are employees of the study sponsor and have no other conflicts to declare.
References
- Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD)2013 Available from: http://www.goldcopd.org/Accessed on January 8, 2014
- CazzolaMMateraMGLong-acting bronchodilators are the first-choice option for the treatment of stable COPDChest2004125191114718412
- European Medicines Agency (EMA)Seebri Breezhaler (glycopyrronium bromide: summary of product characteristics Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002430/WC500133769.pdfAccessed June 25, 2013
- Novartis GlobalNovartis receives European Commission approval for once-daily Seebri® Breezhaler® as maintenance COPD treatment in the EU [press release]Basel, SwitzerlandNovartis2012 [October 1]. Available from: http://www.novartis.com/newsroom/media-releases/en/2012/1645116.shtmlAccessed 22 November, 2012
- European Medicines Agency (EMA)Bretaris Genuair (aclidinium bromide): summary of product characteristics Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002706/WC500132732.pdfAccessed June 25, 2013
- European Medicines Agency (EMA)Eklira Genuair (aclidinium bromide): summary of product characteristics Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002211/WC500132661.pdfAccessed June 25, 2013
- US Food and Drug Administration (FDA)Tudorza Pressair (aclidinium bromide): US prescribing information Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202450s000lbl.pdfAccessed June 25, 2013
- BeehKMSinghDDiSLDrollmannAOnce-daily NVA237 improves exercise tolerance from the first dose in patients with COPD: the GLOW3 trialInt J Chron Obstruct Pulmon Dis2012750351322973092
- ChapmanKRRennardSIDograAOwenRLassenCKramerBLong-term safety and efficacy of indacaterol, a long-acting β2-agonist, in subjects with COPD: a randomized, placebo-controlled studyChest20111401687521349928
- D’UrzoAFergusonGTvan NoordJAEfficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trialRespir Res20111215622151296
- DahlRChungKFBuhlREfficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPDThorax201065647347920522841
- DonohueJFFogartyCLötvallJOnce-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropiumAm J Respir Crit Care Med2010182215516220463178
- KerwinEHébertJGallagherNEfficacy and safety of NVA237 versus placebo and tiotropium in patients with COPD: the GLOW2 studyEur Respir J20124051106111423060624
- KornmannODahlRCentanniSOnce-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparisonEur Respir J201137227327920693243
- KornSKerwinEAtisSAmosCOwenRLassenCIndacaterol once-daily provides superior efficacy to salmeterol twice-daily in COPD: a 12-week studyRespir Med2011105571972621367594
- BuhlRDunnLJDisdierCBlinded 12-week comparison of once-daily indacaterol and tiotropium in COPDEur Respir J201138479780321622587
- VogelmeierCRamos-BarbonDJackDIndacaterol provides 24-hour bronchodilation in COPD: a placebo-controlled blinded comparison with tiotropiumRespir Res20101113520920365
- van NoordJAAumannJLJanssensECombining tiotropium and salmeterol in COPD: effects on airflow obstruction and symptomsRespir Med20101047995100420303247
- VinckenWAn update of bronchodilator treatment of chronic obstructive pulmonary disease (COPD) [abstract]Ann Respir Med20101116
- VogelmeierCKardosPHarariSGansSJStengleinSThirlwellJFormoterol mono- and combination therapy with tiotropium in patients with COPD: a 6-month studyRespir Med2008102111511152018804362
- MahlerDAD’UrzoABatemanEDConcurrent use of indacaterol plus tiotropium in patients with COPD provides superior bronchodilation compared with tiotropium alone: a randomised, double-blind comparisonThorax201267978178822544891
- MaltaisFBeckEWebsterDFour weeks once daily treatment with tiotropium + olodaterol (BI 1744) fixed dose combination compared with tiotropium in COPD patients [abstract]Eur Respir J2010365557
- TashkinDPLittnerMAndrewsCPTomlinsonLRinehartMDenis-MizeKConcomitant treatment with nebulized formoterol and tiotropium in subjects with COPD: a placebo-controlled trialRespir Med2008102447948718258423
- TashkinDPPearleJIezzoniDVargheseSTFormoterol and tiotropium compared with tiotropium alone for treatment of COPDCOPD200961172519229704
- van NoordJAAumannJLJanssensEEffects of tiotropium with and without formoterol on airflow obstruction and resting hyperinflation in patients with COPDChest2006129350951716537846
- van NoordJAAumannJLJanssensEComparison of tiotropium once daily, formoterol twice daily and both combined once daily in patients with COPDEur Respir J200526221422216055868
- BatemanEDFergusonGTBarnesNDual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE studyEur Respir J2013124261484149423722616
- Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD)2010 Available from: http://www.goldcopd.org/Accessed November 23, 2013
- Clinicaltrials.gov identifier NCT1604278 http://clinicaltrials.gov/show/NCT1604278Accessed on 16th January 2014
- MozzicatoPMedDRAAn Overview of the Medical Dictionary for Regulatory ActivitiesPharm Med2009236575
- WitekTJJrMahlerDAMinimal important difference of the transition dyspnoea index in a multinational clinical trialEur Respir J200321226727212608440
- JonesPWSt George’s Respiratory Questionnaire: MCIDCOPD200521757917136966
- [No authors listed]In chronic obstructive pulmonary disease, a combination of ipratropium and albuterol is more effective than either agent alone. An 85-day multicenter trial. COMBIVENT Inhalation Aerosol Study GroupChest19941055141114198181328
- [No authors listed]Routine nebulized ipratropium and albuterol together are better than either alone in COPD. The COMBIVENT Inhalation Solution Study GroupChest19971126151415219404747
- MahlerDADecramerMD’UrzoADual bronchodilation with QVA149 reduces patient-reported dyspnoea in COPD: BLAZE studyEur Respir J10312013 [Epub ahead of print]
- CasaburiRMahlerDAJonesPWA long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary diseaseEur Respir J200219221722411866001
- YawnBPKaplanACo-morbidities in people with COPD: a result of multiple diseases, or multiple manifestations of smoking and reactive inflammation?Prim Care Respir J200817419920518338090
- CalverleyPMRennardSIWhat have we learned from large drug treatment trials in COPD?Lancet2007370958977478517765527
- DecramerMCelliBKestenSLystigTMehraSTashkinDPEffect of tiotropium on outcomes in patients with moderate chronic obstructive pulmonary disease (UPLIFT): a prespecified subgroup analysis of a randomised controlled trialLancet200937496961171117819716598
- VinckenWvan NoordJAGreefhorstAPImproved health outcomes in patients with COPD during 1 yr’s treatment with tiotropiumEur Respir J200219220921611871363
- VogelmeierCBatemanEPallanteJEfficacy and safety of once-daily QVA149 compared with twice-daily salmeterol – fluticasone in patients with chronic obstructive pulmonary disease (ILLUMINATE): a randomised, double-blind, parallel group studyLancet Respir Med201311516024321804
- PriceDYawnBBrusselleGRossiARisk-to-benefit ratio of inhaled corticosteroids in patients with COPDPrim Care Respir J20132219210023135217