
Abstract
Deficiency in the serine protease inhibitor, alpha-1 antitrypsin (AAT), is known to cause emphysema and liver disease. Other manifestations, including airway disease or skin disorders, have also been described. A 44-year-old woman presented to our emergency department with dyspnea and respiratory insufficiency. She had never smoked, and had been diagnosed with COPD 9 years earlier. Three months previously, she had suffered a pulmonary embolism. Chest computed tomography scan revealed severe cystic bronchiectasis with destruction of the lung parenchyma. The sweat test was normal and there was no evidence of the cystic fibrosis transmembrane conductance regulator (CFTR) mutation. Capillary zone electrophoresis showed a decrease of alpha-1 globin band and AAT levels were below the quantification limit (<25 mg/dL). No S or Z mutation was identified, but sequencing analysis found a homozygous cytosine and adenine (CA) insertion in exon 2 of the SERPINA-1 gene, probably leading to a dysfunctional protein (PI Null/Null). This mutation has not been previously identified. The atypical presentation of the patient, with severe cystic bronchiectasis, highlights AAT deficiency as a differential diagnosis in bronchiectasis. Further, awareness should be raised regarding a possible increased risk of thromboembolism associated with AAT deficiency.
Introduction
Alpha-1 antitrypsin (AAT) is an antiprotease, produced mainly by hepatocytes, that opposes the activity of human neutrophil elastase and several other proteases, including proteinase-3 and plasmin activator. Its normal plasma concentration is in the range of 120–200 mg/dL.Citation1,Citation2
AAT deficiency is an autosomal codominant disorder that predisposes to chronic obstructive pulmonary disease (COPD) and liver disease. A plasma concentration below 11 μM (50–80 mg/dL depending on the assay) is considered to represent a relevant deficiency. It is caused by inheritance of two severe deficiency alleles of the SERPINA-1 gene located on chromosome segment 14q31–32.3.Citation2 Variants of AAT alleles are conventionally classified as normal, deficient, and null alleles.
Isoelectric focusing has been the classical gold standard for phenotyping AAT deficiency alleles. It separates protein variants based on their migration in an electrophoretic field at alkaline pH, and is used for classification. Normal variants belong to the M alleles of AAT, whereas the A–L variants run faster and N–Z variants run slower than the M band in an isoelectric field.Citation2
The most common deficiency mutation leading to very low serum AAT levels (ie, 10%–15% of the normal level) is the Z mutation (p.Glu366Lys), whereas the S mutation (p.Glu288Val) is associated with mildly reduced AAT levels (ie, 30%–40% of normal). Several rare null variants, named Q0, associated with trace amounts (<1%) of plasma AAT have been described, resulting from a broad range of molecular mechanisms. Frame-shift or nonsense mutation leads to premature stop codons in SERPINA-1 with either unstable mRNA or truncated, unstable proteins (eg, Q0Granite Falls).Citation3 Other mechanisms include complete gene deletion (eg, Q0Isola di Procida)Citation4 or splicing mutations (eg, Q0Madrid).Citation5,Citation6
Emphysema is due to a protease-antiprotease imbalance causing destruction of the alveolar wall. Liver disease is caused by hepatocyte injury due to intracellular accumulation of polymerized AAT molecules occurring only in certain mutations, eg, the Z allele.Citation1,Citation2
Early-onset “pure” panacinar emphysema is the classical respiratory disease manifestation in adulthood. However, other pulmonary manifestations, such as airway hyperreactivity, wheezing, chronic cough, and expectoration of sputum, have been described. Additionally, extrapulmonary disease manifestations have been recognized, such as necrotizing panniculitis and other skin diseases,Citation7–Citation9 as well as an increased risk for vasculitisCitation10,Citation11 and thromboembolism.Citation7,Citation12,Citation13 We report a case of AAT deficiency presenting with severe bronchiectasis and absence of classical emphysema. Remarkably, the patient was homozygous for a novel mutation, ie, an insertion of cytosine and adenine (CA) in exon 2.
Case presentation
A 44-year-old woman of Turkish origin presented to the emergency department with severe dyspnea and inspiratory pleural chest pain that had developed over the previous few days. She had delivered healthy twins 6 weeks prior to presentation and had been diagnosed with a pulmonary embolism 3 months previously. She had never smoked. Her past medical history revealed a diagnosis of COPD that had been made 9 years earlier during her pregnancy with her first child, at which time she became symptomatic for the first time. There was no family history of respiratory or liver disease. She had no history of childhood asthma or allergies. Her current medication consisted of subcutaneous tinzaparin, a low molecular weight heparin, for pulmonary embolism, and inhaled tiotropium, formoterol, and budesonide.
Initial laboratory workup showed a leukocytosis of 12.6 g/L (normal range 6–11 g/L), mildly elevated C-reactive protein of 2.6 (normal range <0.5) and thrombocytosis of 485 g/L (normal range 150–400 g/L). Differential white blood cells showed an eosinophilia of 7% (normal <4%). Blood gas analysis showed severe respiratory insufficiency (paO2 54 mmHg, paCO2 38 mmHg).
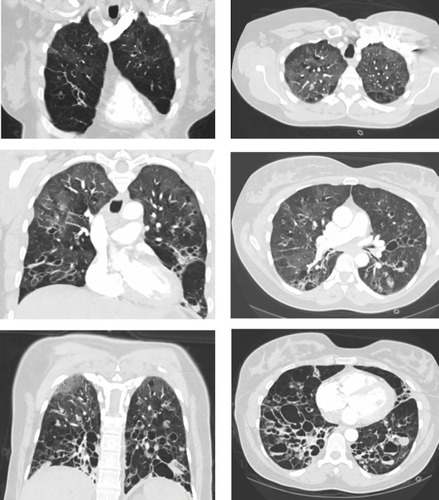
A chest computed tomography scan revealed very severe bronchiectasis with large cysts and mucus plugging, predominantly in the lower lung segments, suggestive of cystic fibrosis (). Additionally, ground glass opacities were noted in the upper lung segments and infiltrates in the right middle lobe. Marked emphysema was not present, and no residual thrombi were seen in the pulmonary arteries. Upon echocardiography, right and left heart function were normal and her brain natriuretic peptide level was normal.
Figure 1 Chest computed tomography scan. Coronal (left) and horizontal (right) images show severe bronchiectasis with large cysts and mucus plugging.
Lung function analysis showed severe obstruction (forced expiratory volume in 1 second [FEV1] 1,06 L/43%, FEV1/vital capacity 47%) and moderate relative hyperinflation (residual volume 2.2 L/140%, total lung capacity 4.37 L/99%). Sputum analysis revealed mucoid Pseudomonas aeruginosa and Aspergillus fumigatus. Mycobacterial infection was excluded by repeated sputum analysis, including polymerase chain reaction. An interferon-gamma release assay for tuberculosis antigen-specific T-cell response was negative. Bronchiectasis workup, including immunoglobulin (Ig)A, IgM, IgG, and subclasses, was normal. Total IgE levels were increased to 584 IU/mL (normal <100 IU/mL) and specific IgG and IgE were high for A. fumigatus (IgG 106 mgA/I; low range <14 mgA/I, intermediate range 14–39 mgA/I; high range >39 mgA/I; IgE 46 IU/mL, FEIA class 4), indicative of possible allergic bronchopulmonary aspergillosis (ABPA). Prick testing against common environmental antigens, such as pollen, house dust mite, and animal hair, was negative. An antinuclear antibody titer of 1:100 was noted, but extractable nuclear antigen screening (RNP/Sm, Sm, SS-A, SS-B, Scl70, CentB, Ho-1, Rib-P) was unremarkable. Antineutrophil cytoplasmic antibody (ANCA) testing was negative. A thrombophilia screen, including factor V Leiden, prothrombin gene mutation, antithrombin 3, proteins C and S, and lupus anticoagulant had been performed at the time of diagnosis and had revealed no abnormalities.
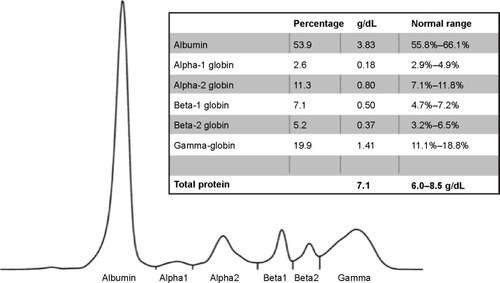
Sweat chloride testing was normal (<25 mM) and no CFTR gene mutation was found. Capillary zone electrophoresis indicated a marked decrease in alpha-1 globin band 0.18 g/dL, 2.6% (). The AAT level in serum was below 25 mg/dL (measured by the Cobas C702 nephelometry kit, Roche, Basel, Switzerland; quantification range 25–600 mg/dL, detection limit 10 mg/dL).
Figure 2 Capillary zone electrophoresis. A decrease of the alpha-1 globin peak is notable.
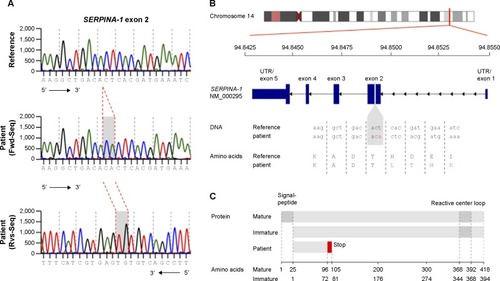
Upon genetic testing, no S or Z mutations were found in the sequencing of exon 3 or exon 5 of the SERPINA-1 gene. Further sequencing analysis of all exons of the SERPINA-1 gene revealed a homozygous CA insertion in exon 2, leading to a frame shift starting at codon 73. This mutation probably leads to a dysfunctional truncated protein lacking the reactive center loop (). There was no suggestion of liver disease on sonography and liver enzymes (alanine transaminase, aspartate transaminase, gamma-glutamyl transpeptidase, alkaline phosphatase) were normal. There was no apparent skin disease.
Figure 3 Sequence of a regular and mutated SERPINA-1 gene. (A) DNA sequencing chromatogram of reference sequence (upper) and patient sequence (middle and lower). (B) Overview of SERPINA-1 gene location, structure, and patient’s mutation with DNA and amino acid sequence. (C) Overview of mature, immature, and patient SERPINA-1 protein and domains.
The patient was treated with oxygen therapy, oral ciprofloxacin, and inhaled colistin twice daily for P. aeruginosa infection. Later, itraconazole and prednisolone were added for treatment of probable ABPA.
Discussion
AAT deficiency is a clinically underdiagnosed genetic disease classically presenting with panacinar pulmonary emphysema. Although less common, an association between AAT deficiency and bronchiectasis has also been described,Citation14,Citation15 and testing for AAT deficiency is recommended in the workup of bronchiectasis.Citation2
Mechanistically, the protease-antiprotease imbalance responsible for the development of emphysema is also thought to cause bronchiectasis. Unopposed human neutrophil elastase from polymorphonuclear influx in the airways leads to injury of bronchial connective tissue, since normal microbial and native airway cellular interaction may be amplified.Citation16
It has been suggested that AAT deficiency may be associated with an increased risk of venous thromboembolism.Citation7,Citation12,Citation13 Our patient experienced thromboembolism during pregnancy, which in itself is a sufficiently strong risk factor. A larger study would be necessary to establish if such a correlation can be confirmed.
There are two possible mechanisms for increased thrombophilia in AAT deficiency. An unopposed proteolytic activity of plasminogen activator could cause activation of the coagulation cascade. Additionally, unopposed proteinase-3 activity may damage and/or activate endothelial cells, resulting in a prothrombotic state. An increased risk for ANCA-associated vasculitis (Wegener’s granulomatosis) has been established,Citation10,Citation11 but signs of vasculitis or ANCAs were not found in our patient. AAT plays an important role as an inhibitor of proteinase-3, a neutrophil elastase-like serine protease located in the primary granules of the neutrophil. AAT deficiency could increase unchecked proteinase-3, which in turn exerts a potent tissue-destructive capacity, triggering an autoimmune response by allowing increased extracellular exposure to proteinase-3.Citation10
Diagnosis of ABPA was probable in our patient because she had A. fumigatus in sputum, increased total IgE, and increased specific IgE and IgG titers, as well as exacerbation of her disease with a pulmonary infiltrate. ABPA classically occurs in patients with asthma or cystic fibrosis, but cases in patients with COPD or non-cystic fibrosis bronchiectasis have also been described. APBA may also be the cause of bronchiectasis in patients with asthma. Our patient did not have a history of allergies or asthma. Hence, we assume ABPA might have aggravated the disease, but was not the primary cause of bronchiectasis.
Via sequencing analysis, a novel SERPINA-1 mutation was identified, consisting of a homozygous CA insertion in the exon 2 that leads to a frameshift mutation with a premature stop codon. The resulting protein is truncated at one-third of the amino acid sequence and lacks the reactive center loop (). Hence, there is no antiproteolytic activity, and a null phenotype (PI Q0/Q0) must be assumed. Classically, different AAT alleles have been characterized by phenotyping using isoelectric focusing, and this method is considered the gold standard for phenotypic analysis. However, this method is reserved for experienced reference laboratories. Moreover, it may miss relevant mutations because mutated proteins may exhibit similar migration characteristics as the M allele, as seen in MMalton and MDuarte alleles, or in the event of heterozygosity for the M allele and a null allele.Citation17 Since genotyping by whole gene sequencing is now widely available, we used a combination of AAT quantification in plasma by nephelometry and whole gene sequencing as suggested by a current diagnostic algorithm.Citation18 A phenotypic analysis of the mutant protein by isoelectric focusing as well as RNA expression analysis by reverse transcription polymerase chain reaction would help to characterize the novel mutation in more detail, but was not available at our center. Generally, null mutations are considered to be very rare; however, their prevalence may be underestimated by a lack of general awareness of AAT deficiency as well as diagnostic strategies omitting whole gene sequencing.
Interestingly, in the present case, the mutation was found in the homozygous form, pointing toward a hereditary mechanism. It is possible that this mutation has a local prevalence in the patients’ region of origin (Kayseri, Turkey), but this has not been investigated. A new spontaneous mutation involving the same insertion in both alleles seems unlikely.
Similar mutations leading to stop codons in exon 2 are the Q0soest, Q0amersfoort,Citation19 and Q0granite falls,Citation3 resulting in undetectable AAT messenger RNA and severe AAT deficiency in homozygotes or compound heterozygotes. Patients with Q0soest and Q0amersfoort had severe COPD in their fortiesCitation19 and carriers of Q0granite falls had severe emphysema.Citation3
The possibility of AAT augmentation with intravenous AAT therapy for long-term treatment was discussed with the patient. The indication for therapy in Germany is severe AAT deficiency (<80 mg/dL) and FEV1 between 35% and 60%, but this may vary in different countries. Augmentation therapy has been shown to reduce the progression of emphysema, as assessed by computed tomographic densitometry.Citation20 Effectiveness in slowing the decline in lung function has been suggested by a meta-analysis including controlled and uncontrolled trial data.Citation21 A few reports have suggested positive effects of augmentation therapy on airway inflammation,Citation22,Citation23 but the effects on bronchiectasis have not been conclusively studied. Given the limited evidence for augmentation treatment in cases such as the one described, we favor treatment only within the setting of a clinical study. Furthermore, genetic testing was advised for the patient’s children.
Conclusion
We describe a novel SERPINA-1 mutation associated with severe AAT deficiency. The atypical presentation of the patient with severe cystic bronchiectasis highlights AAT deficiency as a differential diagnosis in bronchiectasis. Awareness should be raised as to a possible increased risk of thromboembolism associated with AAT deficiency. Further studies are necessary to investigate the risk of thrombosis as well as the effectiveness of augmentation therapy in AAT deficiency-associated bronchiectasis.
Acknowledgments
The authors thank Sandhya Matthes for proofreading and English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
- SilvermanEKSandhausRAClinical practice. Alpha1-antitrypsin deficiencyN Engl J Med2009360262749275719553648
- No authors listedAmerican Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiencyAm J Respir Crit Care Med2003168781890014522813
- NukiwaTTakahashiHBrantlyMCourtneyMCrystalRGalpha 1-Antitrypsin nullGranite Falls, a nonexpressing alpha 1-antitrypsin gene associated with a frameshift to stop mutation in a coding exonJ Biol Chem19872622511999120043040726
- TakahashiHCrystalRGAlpha 1-antitrypsin Null(isola di procida): an alpha 1-antitrypsin deficiency allele caused by deletion of all alpha 1-antitrypsin coding exonsAm J Hum Genet19904734034131975477
- BrantlyMAlpha1-antitrypsin: not just an antiprotease: extending the half-life of a natural anti-inflammatory molecule by conjugation with polyethylene glycolAm J Respir Cell Mol Biol200227665265412444023
- LaraBMartínezMTBlancoISevere alpha-1 antitrypsin deficiency in composite heterozygotes inheriting a new splicing mutation QOMadridRespir Res20141512525287719
- ElsensohnANCurtisJASecrestAMAlpha-1-antitrypsin deficiency panniculitis presenting with severe anasarca, pulmonary embolus, and hypogammaglobulinemiaBr J Dermatol12152014 Epub ahead of print
- GrossBGrebeMWenckerMStollerJKBjurstenLMJanciauskieneSNew findings in PiZZ alpha1-antitrypsin deficiency-related panniculitis: Demonstration of skin polymers and high dosing requirements of intravenous augmentation therapyDermatology200921837037519218787
- WarterJStorckDGrosshansEMetaisPKuntzJLKlumppTWeber-Christian syndrome associated with an alpha-1 antitrypsin deficiency. Familial investigationAnn Med Interne (Paris)197212310877882 French4659249
- EsnaultVLAudrainMASesboüéRAlpha-1-antitrypsin phenotyping in ANCA-associated diseases: one of several arguments for protease/antiprotease imbalance in systemic vasculitisExp Clin Immunogenet19971432062139493789
- MahrADEdbergJCStoneJHAlpha1-antitrypsin deficiency-related alleles Z and S and the risk of Wegener’s granulomatosisArthritis Rheum201062123760376720827781
- GuptaRSridharaSWoodJAA rare case of alpha 1-antitrypsin deficiency associated with hypogammaglobulinemia and recurrent pulmonary thrombosisAnn Thorac Med201491394124551017
- FiechtnerJJMagninGEMultiple pulmonary thromboemboli and pneumothorax. Complications of exogenous estrogen in an alpha-1-antitrypsin deficient womanWis Med J19787710S103S106309692
- ParrDGGuestPGReynoldsJHDowsonLJStockleyRAPrevalence and impact of bronchiectasis in alpha1-antitrypsin deficiencyAm J Respir Crit Care Med2007176121215122117872489
- ShinMSBronchiectasis in patients with alpha 1-antitrypsin deficiency. A rare occurrence?Chest19931045138413868222792
- StrangeCAirway disease in alpha-1 antitrypsin deficiencyCOPD201310Suppl 1687323528021
- TaylorNJShawcrossDLAlpha1-antitrypsin deficiencyN Engl J Med200936121210219938318
- FerrarottiIScabiniRCampoILaboratory diagnosis of alpha1-antitrypsin deficiencyTransl Res2007150526727417964515
- PrinsJVan Der MeijdenBBKraaijenhagenRJWieldersJPInherited chronic obstructive pulmonary disease: new selective-sequencing workup for alpha1-antitrypsin deficiency identifies 2 previously unidentified null allelesClin Chem200854110110718024524
- StockleyRAParrDGPiitulainenEStolkJStoelBCDirksenATherapeutic efficacy of α-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometryRespir Res20101113620920370
- ChapmanKRStockleyRADawkinsCWilkesMMNavickisRJAugmentation therapy for alpha1 antitrypsin deficiency: a meta-analysisCOPD20096317718419811373
- StockleyRABayleyDLUnsalIDowsonLJThe effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiencyAm J Respir Crit Care Med2002165111494149812045122
- LiebermanJAugmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting dataChest200011851480148511083705