
Abstract
The prevalence rate of thalassemia, which is endemic in Southeast Asia, the Middle East, and the Mediterranean, exceeds 100,000 live births per year. There are many genetic variants in thalassemia with different pathological severity, ranging from a mild and asymptomatic anemia to life-threatening clinical effects, requiring lifelong treatment, such as regular transfusions in thalassemia major (TM). Some of the thalassemias are non-transfusion-dependent, including many thalassemia intermedia (TI) variants, where iron overload is caused by chronic increase in iron absorption due to ineffective erythropoiesis. Many TI patients receive occasional transfusions. The rate of iron overloading in TI is much slower in comparison to TM patients. Iron toxicity in TI is usually manifested by the age of 30–40 years, and in TM by the age of 10 years. Subcutaneous deferoxamine (DFO), oral deferiprone (L1), and DFO–L1 combinations have been effectively used for more than 20 years for the treatment of iron overload in TM and TI patients, causing a significant reduction in morbidity and mortality. Selected protocols using DFO, L1, and their combination can be designed for personalized chelation therapy in TI, which can effectively and safely remove all the excess toxic iron and prevent cardiac, liver, and other organ damage. Both L1 and DF could also prevent iron absorption. The new oral chelator deferasirox (DFX) increases iron excretion and decreases liver iron in TM and TI. There are drawbacks in the use of DFX in TI, such as limitations related to dose, toxicity, and cost, iron load of the patients, and ineffective removal of excess iron from the heart. Furthermore, DFX appears to increase iron and other toxic metal absorption. Future treatments of TI and related iron-loading conditions could involve the use of the iron-chelating drugs and other drug combinations not only for increasing iron excretion but also for preventing iron absorption.
Introduction
Iron is an essential metal found in all living organisms, and is involved in many physiological processes, such as oxygen transport and energy transduction.Citation1,Citation2 It is estimated that more than a quarter of the human population is affected at some stage of their life by abnormalities of iron metabolism, in particular iron-deficiency anemia. Similarly, many millions of people suffer from iron overload, including hereditary hemochromatosis and thalassemia intermedia (TI), which are caused by increased iron absorption.Citation3–Citation6 In these and other iron-loading conditions, excess iron is toxic, causing increased morbidity and mortality as a result of tissue damage. Iron plays an important catalytic role in free radical pathology and oxidative stress damage, which is not only observed in iron-overload diseases but also in non-iron-loaded diseases of focal or labile iron toxicity, such as cardiovascular, neurodegenerative, hepatic, and renal diseases, as well as in cancer and aging.Citation7,Citation8
A major group of diseases related to iron abnormalities involving millions of people are the inherited hemoglobinopathies, especially thalassemia and sickle-cell disease. It is estimated that the prevalence of these two inherited hemoglobinopathies exceeds 200,000 live births per year, with approximately 100,000 for each disease.Citation3,Citation4 Sickle-cell disease is endemic in Africa, Central and South America, and the Afro-American population of North America, and is also distributed with lower frequency in Middle East and the Mediterranean countries. Thalassemia is endemic mainly in Southeast Asia, the Middle East, and Mediterranean countries.
There are many genetic variants of thalassemia related to the hemoglobin (Hb) chains (α, β, γ, δ, and ε). Many of the thalassemia variants can cause pathological effects of different severity, ranging from a mild and asymptomatic anemia to death, eg, in hydrops fetalis in α-thalassemia (4/4 α-globin gene deletion).Citation9 A major group of thalassemia patients with life-threatening clinical effects requiring life long treatments, such as regular red blood cell transfusions within a few years of life, is β thalassemia major (TM) (2/2 β-globin gene deletion).Citation3,Citation4 Iron overload in TM is caused mainly from chronic transfusions and to a lesser extent by increased iron absorption. Another major group of thalassemias of milder severity than TM are the iron-loaded non-transfusion-dependent thalassemias (IL-NTDTs), which include many TI variants where iron overload is caused by chronic increase in iron absorption due to ineffective erythropoiesis.Citation3,Citation4,Citation9–Citation12 The same mechanism, but with a higher rate of iron absorption, is also observed in nontransfused TM patients in developing countries, where no transfusion or chelation therapy is available.
Many patients with IL-NTDT may have a number of red blood-cell transfusions because of low Hb levels, infections, and other causes. The rate of transfusion increases with age in most cases of IL-NTDT, eg, many TI patients at the age of 30–50 years may be regularly transfused at a rate similar to that of TM patients.Citation9–Citation12 The rate of iron overload from increased iron absorption in IL-NTDT is generally much slower in comparison to iron accumulated from transfusions in regularly transfused TM patients.Citation12,Citation13 However, the rate of iron absorption in IL-NTDT is generally similar to that observed in hereditary hemochromatosis, and in both conditions iron overload is manifested in many cases very early in life, even from early childhood.Citation9–Citation15
Genetic, dietary, immunological, and other factors influence the rate of iron-overload and related toxicity in both IL-NTDT and TM. Iron-overload toxicity is manifested in many organs, including the heart, liver, spleen, and pancreas.Citation11,Citation16–Citation19 Iron overload in IL-NTDT can lead progressively to cardiomyopathy, liver fibrosis, cirrhosis, hepatocellular carcinoma, and diabetes.Citation11,Citation16–Citation21 In addition to iron overload-related toxicity, ineffective erythropoiesis, and chronic anemia, there are also many other abnormalities related to the underlying condition in TI and other similar IL-NTDTs, such as hypothyroidism, hypogonadism, hypersplenism, pulmonary hypertension, and osteoporosis.Citation11,Citation16–Citation21
Different clinical strategies are generally used for the treatment of TI and other similar IL-NTDT patients, which mainly include red blood-cell transfusions, increase in HbF production using hydroxycarbamide (hydroxyurea), splenectomy, and iron-chelation therapy.Citation9–Citation11
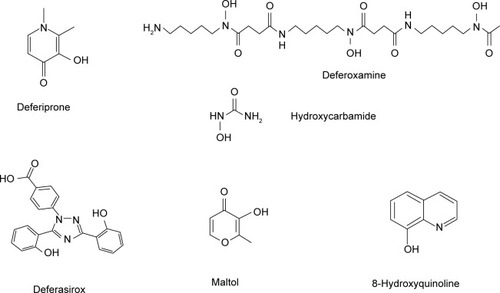
Iron-chelation therapy can decrease and in many cases eliminate the morbidity and mortality rates associated with iron toxicity in IL-NTDT syndromes. Iron-chelation strategies in patients with this syndrome are similar to those in TM, and are generally based on the early diagnosis and prevention of iron overload and toxicity and also the rapid normalization of the iron stores.Citation14,Citation15 The chelating drugs and chelation protocols used in IL-NTDT are similar to those in TM. Most of these protocols involve the administration of subcutaneous deferoxamine (DFO), oral deferiprone (L1), and DFO–L1 combinations, which have been effectively used for more than 20 years for the treatment of iron overload in TM and TI patients ().Citation14,Citation15 In addition, the recent introduction of oral deferasirox (DFX) has increased the options for the treatment of iron overload in TM and other conditions, including IL-NTDT (). Several studies sponsored by the manufacturers of DFX have been recently published on the effect of the drug on IL-NTDT.Citation22
Figure 1 Structure of the chelating drugs.
Personalized medicine is designed and appropriately used in each case for optimizing chelation therapy in patients with IL-NTDT and TM. This approach is generally based on the risk–benefit assessment of each chelator and chelation protocol, and also the idiosyncratic clinical profile of the patients, as determined by drug absorption, distribution, metabolism, excretion, and toxicity effects and other relevant parameters, such as organ function.Citation23 However, other factors are also considered in the selection criteria of chelating drugs, including costs, influence of the pharmaceutical companies, and drug policies of the regulatory authorities.Citation24,Citation25
Epidemiology and severity of iron-loaded non-transfusion-dependent thalassemia syndromes
IL-NTDT is a syndrome characterized by excess iron deposition in organs similar to hereditary hemochromatosis, which is caused by increased body-iron intake as a result of an increase in net body-iron intake from gastrointestinal absorption in comparison to total body-iron excretion and other losses.Citation12–Citation15,Citation26,Citation27 The level of iron overload and its severity in this syndrome is related primarily to hereditary abnormalities, but also a number of metabolic, environmental, dietary, aging, and other factors influencing the ferrokinetics of iron metabolism through different mechanisms.Citation12–Citation15,Citation26,Citation27
In relation to the hereditary factors related to IL-NTDT syndromes, β-TI is the best-characterized genetic disorder, which is associated with a variety of gene mutations involving the β-globin chain of Hb and is predominantly found in Southeast Asia, Mediterranean, and Middle Eastern countries, similar to that of TM patients ().Citation3,Citation4 In TI patients, the normal production of Hb is affected to a variable extent depending on the genotype and age of the patient. Thousands of TI patients diagnosed with Hb variants of the α-, β-, γ, δ-, and ε-globin genes exhibit milder forms of anemia than TM patients ().Citation3,Citation4
Table 1 Examples of thalassemia intermedia and other conditions where iron overload can be caused from the increased gastrointestinal absorption of ironTable Footnote*
There are several categories of TI patients where red blood-cell transfusions may be initiated to compensate for the decreasing level of Hb production. In some cases, the rate of transfusions may increase to levels similar to those observed in TM patients. There are also other groups of TI patients who may receive less frequent transfusions and some where transfusions are not necessary. In the latter group, TI patients usually maintain Hb levels at 6–10 g/dL, which in many cases is sufficient in younger patients to be maintained without complications, and transfusion can be avoided.Citation3,Citation4 For example, increased production of HbF is associated with milder TI disease. In general, Hb production in TI decreases with age, and the general health of TI patients also deteriorates as they grow older.Citation10,Citation11,Citation15
Similar variations are observed in HbE β-thalassemia patients, most of whom live in developing countries of Southeast Asia, and also in immigrants from this region.Citation3,Citation4,Citation17,Citation27,Citation28 It is estimated that the HbE β-thalassemia genotype is responsible for approximately 50% of all severe β-thalassemias worldwide (). The disorder is characterized by marked clinical variability and Hb production, ranging from a mild and asymptomatic anemia to a life-threatening disorder requiring transfusions from infancy. For example, no treatment is required for mild HbE β-thalassemia that is characterized by normal Hb levels (9–12 g/dL). Patients in this category do not usually develop clinically significant symptoms. However, more severe forms are characterized by decreased Hb levels (6–8 g/dL) and the clinical manifestations are similar to those of TI, where transfusions are not required unless infections or other complications increase the anemia further. Most patients of HbE β-thalassemia (~50%) are thought to be included in this category of IL-NTDT, where iron overload from progressive increases in iron absorption may occur. It is estimated that about a third of the cases of HbE β-thalassemia are present with the severe form of the disease, which is characterized by very low Hb levels (4–5 g/dL), and the clinical symptoms are similar to those of TM patients. Regular red blood-cell transfusions and iron-chelation therapy are required for the survival of this category of HbE β-thalassemia patients similar to TM patients.Citation3,Citation4,Citation17,Citation27,Citation28
There are many other categories of patients with similar rates of body-iron loading to that of IL-NTDT that are due to increased iron absorption and require chelation therapy (). These include hereditary hemochromatosis patients who cannot be treated with venesection because of complications, such as severe congestive heart failure, cardiomyopathy, severe anemia, advanced liver cirrhosis, or poor peripheral venous access, and also patients with porphyria cutanea tarda ().Citation15 Additionally, there are other categories of patients, such as those with sickle-cell anemia or sickle-cell anemia with β-thalassemia, with similar rates of body-iron loading to that of IL-NTDT, which are caused by occasional red blood-cell transfusions ().Citation10,Citation11,Citation15 In all these cases, the body-iron intake from increased gastrointestinal absorption and occasional red blood-cell transfusions is much lower and iron accumulation is at a slower rate in comparison to regularly transfused TM patients. Accordingly, the toxic side effects of iron overload are manifested at different ages in IL-NTDT, and less intensive chelation therapy may be used at younger ages for the treatment of iron overload and toxicity in comparison to TM.
Molecular mechanisms of increased iron absorption and storage in iron-loaded non-transfusion-dependent thalassemias
Under normal conditions, the regulatory uptake and utilization of iron appear to involve a number of pathways and proteins that control the rate of iron transfer from the gastrointestinal tract to the bloodstream such as ferrireductase(s), DMT1, ferroportin, hepcidin, transferrin, and ferritin ().Citation1,Citation2 Different changes in the regulatory mechanisms of dietary iron uptake from the gastrointestinal tract are suggested for causing increased iron absorption in hereditary hemochromatosis, TI, and other similar conditions.Citation1,Citation2,Citation15
Figure 2 Iron-absorption and iron-overload mechanisms in non-transfusion-dependent thalassemias: the role of chelators and chelating drugs.
Abbreviations: DMT1, divalent metal transporter 1; EDTA, ethylenediaminetetraacetic acid; DTPA, diethylenetriaminepentaacetic acid.
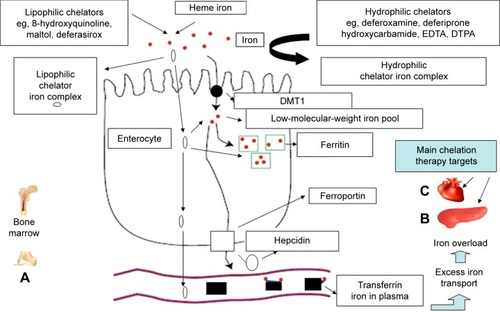
The mechanism and the regulatory pathway of iron absorption is thought to involve initially the conversion of dietary ferric iron to ferrous iron by a ferrireductase present at the cell surface of the enterocyte and then its intracellular transport by DMT1 (). Once inside the cell, iron is partly incorporated into ferritin or transferred into the low-molecular-weight intracellular iron pool. Some of the iron is then transported to ferroportin at the basolateral membrane of the enterocyte, which exports iron into the plasma. The critical role of iron absorption is thought to be played by hepcidin, a peptide hormone produced in the liver. The mode of action of hepcidin is the prevention of iron release into plasma by binding ferroportin, causing its internalization and degradation within the enterocyte. Following the binding of ferroportin by hepcidin, iron is not released into plasma, but remains within the enterocytes and returns in the gut lumen, since enterocytes are shed every 2–3 days. Within this context, a decrease in plasma hepcidin concentration will increase iron absorption.Citation1,Citation2,Citation15
Iron released into plasma from the basolateral membrane of the enterocyte by ferroportin is mobilized by plasma transferrin and transferred to all the cells of the body. The uptake of transferrin iron by cells is mediated by transferrin receptors that are present at the cell surface. Increased transferrin receptors are present especially in hepatocytes for iron storage and the erythroid cells for Hb synthesis. Intracellular release of iron is mediated by receptor-mediated endocytosis, through the internalization in a vesicle and acidification of the transferrin receptor–transferrin–iron complex.Citation1,Citation2
The expression of hepcidin, DMT1, and ferroportin appears to be affected by a number of factors. In particular, the signal to the intestine to increase iron absorption appears to be influenced by regulatory molecules (iron-responsive elements) which sense the iron stores.Citation1,Citation2,Citation29,Citation30 Different iron-responsive element regulatory molecules are also present, which appear to sense the production of Hb or anemia. In both cases, the influence of the regulatory molecules cause an increase in the expression of DMT1 and a corresponding increase in the uptake of iron. In general, it would appear that in TI and other IL-NTDT, the signal arriving at the enterocytes is a combination of ineffective erythropoiesis, anemia, and hypoxia, which leads to the initiation of compensatory mechanisms, including an increase in plasma levels of erythropoietin, as well as a decrease in plasma levels of hepcidin. The decrease in hepcidin increases the rate of transfer of iron by ferroportin on the intestinal epithelium into the bloodstream, followed by an increase of transferrin iron uptake, transferrin iron delivery, and intracellular storage, mainly in the liver hepatocytes but also in other organs, including the heart and the pancreas.Citation12,Citation13,Citation16–Citation19
The level of iron overload in IL-NTDT patients is mainly characterized by increases in serum ferritin, plasma iron, and transferrin iron saturation, as well as by increases in iron concentration in the liver, heart, and other organs, which can be measured by magnetic resonance imaging (MRI) T2 and T2* relaxation-time values.Citation10–Citation12,Citation16,Citation17,Citation31,Citation32 The same methods are also used for monitoring the progress of chelation therapy, as well as other treatments used for the reduction of iron absorption or the elimination of excess iron in organs of patients with IL-NTDT.
Iron overload has important clinical consequences in both patients with TI and TM. However, unlike TM, where iron overload is mostly caused from transfused red blood cells and is initially stored in the Kupffer cells of the liver, the iron overload caused in IL-NTDT originates from the absorption and intake of excess iron present in the gastrointestinal tract, which is mainly delivered and stored in the hepatocytes. Since iron accumulation primarily occurs in the hepatocytes, iron overload can predispose patients to liver fibrosis and cirrhosis, hepatocellular carcinoma, and other toxic side effects similar to hereditary hemochromatosis patients.Citation20,Citation21,Citation30,Citation31 Cardiac complications, including cardiac iron overload, are also very common in TI and other IL-NTDT patients, and in both cases appropriate therapeutic interventions are needed.Citation16–Citation20,Citation30,Citation31
Nongenetic factors influencing the rate of iron absorption and body-iron intake in iron-loaded non-transfusion-dependent thalassemias
The genotype of thalassemia is the major factor affecting the rate of gastrointestinal iron absorption and body-iron loading in IL-NTDT. However, there are many other additional factors that also appear to influence this process, and this is reflected by the wide variation in iron-loading in IL-NTDT patients with the same genotype. Some of these factors may be influenced by changes in relation to the iron-metabolism pathways and related proteins and regulatory molecules, such as hepcidin and ferroportin. Infection, inflammation, and pathological conditions may also influence normal iron metabolism and the rate of iron absorption in IL-NTDT ().Citation1,Citation2,Citation5,Citation6,Citation10,Citation11
Table 2 Examples of nonregulatory factors affecting iron absorptionTable Footnote*
Dietary habits are also important factors affecting the rate of iron absorption in IL-NTDT patients, but also in normal individuals. Within this context, the quantity and quality (ferrous, ferric, heme) of iron in food, as well as other dietary factors or constituents, such as ascorbic acid, polyphenols, inorganic phosphates, and tannins, can also influence the rate of iron absorption.Citation15,Citation26 Increased iron absorption is anticipated when dietary iron is in the form of heme, in comparison to ferrous iron and to a lesser extent ferric iron, which is the least soluble and absorbable form.Citation15,Citation26 The presence of reducing agents, such as ascorbic acid, could increase the absorption of iron by reducing ferric to ferrous iron. Low pH and dietary factors that increase the solubility of iron can also increase the absorption of iron ().Citation15,Citation26 In contrast, decrease in iron intake and iron deficiency is generally observed in vegetarians, where absorbable dietary iron is low in concentration. Consequently, vegetarian meals could decrease the rate of iron overload in IL-NTDT in comparison to meals with increased and more absorbable forms of iron from animal food products, such as red meat.Citation15,Citation26
Drugs may also influence the rate of iron absorption. Within this context, increased absorption of iron can be facilitated by natural or synthetic lipophilic chelators, such as maltol, 8-hydroxyquinolone, and DFX, all of which form lipophilic iron complexes.Citation14,Citation15,Citation26,Citation33 In contrast, decreased absorption of iron could be caused using hydrophilic chelators, such as DFO and L1, which form hydrophilic and/or charged iron complexes, as well as by chelators forming insoluble iron complexes (, ).Citation14,Citation15,Citation26,Citation33
The ferrokinetic, tissue-distribution, and toxicity profiles vary among the different categories of iron-loading conditions, including TM, IL-NTDT, and hereditary hemochromatosis. In normal individuals and under normal conditions, the amount of iron absorbed from the diet is equivalent to the amount of iron excreted (1–4 mg).Citation12,Citation15,Citation26 Different levels of increased iron absorption are observed (2.6–8.6 mg/day) in nontransfused TI patients, which in most cases can progressively over many years lead to iron overload and subsequently to tissue and organ damage.Citation12–Citation15 This pattern of increased rate of iron absorption from the diet, which is estimated to be up to approximately 6 mg/day, is similar to other IL-NTDT and also in hereditary hemochromatosis patients.Citation12–Citation15 In comparison, it is estimated that the daily body-iron intake in TM from red blood-cell transfusions for maintaining Hb levels at approximately 9–10 g/dL is approximately 20 mg/day and from increased gastrointestinal absorption 1–6 mg/day.Citation12–Citation15,Citation26
In TM patients, organ damage due to iron overload toxicity in the absence of chelation therapy appears within a few years of initiating red blood-cell transfusions or when approximately 50 units of packed red blood cells (equivalent to 10.0–12.5 g of iron) have been transfused.Citation14,Citation15 Similar levels of iron overload appear to cause organ toxicity in TI and other related IL-NTDT conditions, but at an older age.Citation14,Citation15
Alternative treatments to red blood-cell transfusions are generally used in early diagnosed TI and other related IL-NTDT conditions for increasing Hb levels. Within this context, early diagnosed nontransfused TI patients with low Hb levels are currently treated with hydroxyurea, which appears to be effective in increasing HbF levels in some TI genotypes.Citation28,Citation34,Citation35 The response to hydroxyurea is variable, and iron-loading, pharmacogenomic, and other factors appear to influence its efficacy.Citation28,Citation34–Citation36 However, in many cases, and especially in older TI patients, transfusions may be initiated because of ineffective hydroxyurea treatment and increasing anemia. Erythropoietin has also been tested for elevating Hb levels, but the results were not satisfactory and further development in this area was not pursued.Citation37
In undiagnosed and untreated IL-NTDT patients, excess toxic iron is accumulated over the years, leading to organ damage, including mainly liver cirrhosis, hepatocellular carcinoma, cardiomyopathy, and diabetes. During the progress of the disease, excess iron gradually accumulates initially in the liver and later to other organs, leading to an increase in serum iron, transferrin iron saturation, and serum ferritin and hepatic iron concentration, usually before the age of 30 years. In many cases, tissue damage can be diagnosed by the age of 30 years and liver cirrhosis by the age of 40 years. However, there is great variability in the progress and development of iron overload and organ damage among IL-NTDT patients and other related conditions.Citation38 For example, Hb levels, dietary habits, hydroxyurea treatment, and many other factors can lead to different rates of iron absorption and iron overload-related tissue damage, which overall can influence the progress of the disease in IL-NTDT.
Within this context, the early diagnosis and treatment of TI and related IL-NTDT could prevent iron-overload toxicity, related tissue and organ damage, and overall decrease the morbidity and mortality in these conditions. Normal life expectancy is expected for early diagnosed and treated IL-NTDT patients who have not developed permanent organ damage, such as liver cirrhosis, cardiomyopathy, and diabetes.Citation24
Reduction of body-iron load in IL-NTDT and other related conditions of iron overload due to increased gastrointestinal iron absorption, as well as transfusional iron overload in TM, can only be accomplished using chelation therapy and specifically the iron-chelating drugs DFO, L1, and DFX ().Citation14,Citation24
Properties and effects of chelators and chelating drugs in relation to iron absorption
Chelating drugs and chelators in general have different properties that can affect iron excretion as well as absorption, and can play a major role in the management of many iron-metabolism disorders, including TM, TI, and other IL-NTDT. In each case, a risk–benefit assessment is required for the selected therapies, based on individual variations and the long-term efficacy, safety, and cost of the chelating drug.
The pharmacological, toxicological, biochemical, chemical, and iron-binding properties of different chelators and the chelating drugs DFO, L1, and DFX, including their effects on iron absorption and excretion, have been previously reviewed ().Citation14,Citation26,Citation39 L1 is a bidentate chelator forming a 3L1–1Fe complex, DFX is a tridentate chelator forming a 2DFX–1Fe complex, and DFO is a hexadentate chelator forming a 1DFO–1Fe complex at physiological pH.
Table 3 Mode of action and property differences of the chelating drugs deferoxamine, deferiprone, and deferasirox and other chelatorsTable Footnote*
The mode of action and effects of chelators are variable in relation to different metabolic pathways, which is a reflection of the differences in their physicochemical and other properties (). This variability has been shown in many in vitro, in vivo, and clinical studies. For example, the lipid–water partition coefficient of the chelators and their iron complexes has been shown to affect the transfer of iron across the cell membrane in different cell types, as well as to cause variable effects related to the intracellular iron-metabolism pathways in these cells.Citation40–Citation42
In studies on the effect of chelators on iron absorption, it was observed that the long-term oral administration of lipophilic chelators, such as 8-hydroxyquinoline, appears to cause iron overload in rats, by gradually increasing the absorption of iron present in the diet.Citation43 An increase in iron absorption in mice has also been shown by other lipophilic chelators, such as maltol and 2-hydroxy-4-methoxypyridine-1-oxide.Citation33 The maltol–iron complex is currently undergoing clinical development for general use in iron deficiency and especially in patients with inflammatory bowel disease.Citation44–Citation46 The same effect is anticipated by the chronic administration of other lipophilic oral iron chelators and especially the chelating drug DFX.Citation47,Citation48 DFX is also anticipated to increase aluminum and other toxic metal absorption (, ).Citation47,Citation48
In contrast to maltol, DFX, and other lipophilic chelators, many hydrophilic chelators, including DFO and L1, appear to cause a decrease in iron absorption under similar conditions.Citation33 Because of these differences in properties, DFO and L1 could also be used in the treatment of acute oral iron poisoning.Citation49,Citation50 In contrast, the use of DFX in the treatment of acute iron poisoning is inappropriate and not recommended, because it may increase the absorption of iron and increase further iron toxicity in this condition.Citation47,Citation48
Inhibition of iron absorption has also been observed by such chelators as 2-hydroxypyridine-1-oxide, which form complexes with iron that precipitate and become insoluble.Citation33 Increase or inhibition of iron absorption can also be observed with commonly used drugs with chelating properties, such as tetracycline and hydroxyurea.Citation51,Citation52 It is anticipated that similar chelators could be used to decrease the rate of gastrointestinal iron absorption in TI, other IL-NTDT, hereditary hemochromatosis, and TM.
Clinical use and efficacy of deferoxamine, deferiprone, and deferasirox in iron overload
The primary use of DFO, L1, and DFX is the treatment of transfusional iron overload in TM, other transfusional iron-loaded conditions, and IL-NTDT. Iron-overload toxicity from chronic transfusions and/or increased gastrointestinal iron absorption leads to progressive multiorgan damage and associated increase in morbidity and mortality, which in most cases is directly related to the level of excess iron load. For example, in the absence of chelation therapy, regularly transfused TM patients die by the age of 20 years, mainly from congestive cardiac failure caused by cardiac iron-overload toxicity.Citation53,Citation54 In contrast, the mean survival of TM patients in the UK treated with DFO was estimated to be 35 years.Citation54 However, the use of specific chelation-therapy protocols with L1 and the L1–DFO combination appear to increase life expectancy and reduce the morbidity of iron-loaded patients to levels approaching those of normal individuals.Citation24,Citation55
DFO has been used parenterally for the treatment of iron-overloading conditions, including TI, other IL-NTDT, and especially TM in the last 50 years. Similarly, oral L1, which is a generic drug like DFO, has been approved and used in India since 1995 and in Europe and other regions since 1999, but in the US since only recently – in 2011. DFX is a relatively new oral but very expensive patented drug that has been used worldwide since 2005, and the patent expires in 2017.
The three chelating drugs have major differences in the efficacy, tolerance, site of action, toxicity profile, and cost, all of which affect the morbidity and mortality of iron-overloaded patients in both developed and developing countries ().Citation14,Citation24,Citation25 Because of the high cost of the transfusions and chelating drugs, most of the TM and IL-NTDT patients in developing countries are left to die untreated. The local manufacturing of cheaper formulations for L1 and DFX in developing countries, such as India, Thailand, and Iran, has substantially reduced the cost of chelating drugs and increased the number of patients receiving chelation therapy in these countries.Citation14,Citation24,Citation25,Citation56,Citation57
There are general variations among patients not only related to the level of iron overload and organ distribution but also in the response and pharmacology to each of the chelating drugs. These responses are also related to individual profile differences in the absorption, distribution, metabolism, excretion, and toxicity of the drugs.Citation24,Citation25,Citation58,Citation59 For example, L1 causes an increase in iron excretion exclusively in the urine, DFX exclusively in the feces, and DFO mostly in the urine and also the feces ().Citation14,Citation24,Citation25,Citation59
The general recommended doses of the chelating drugs in TM and IL-NTDT are 40–60 mg/kg/day for subcutaneous DF, 75–100 mg/kg/day for oral L1, and 20–40 mg/kg/day for oral DFX. However, variations in iron overload and targeted therapies can result in the application of a range of therapeutic protocols from an intensive chelation, eg, combinations of DF (40–60 mg/kg/day) and L1 (75–100 mg/kg/day) in heavily transfused iron-loaded patients, to much lower doses or intermittent withdrawal of chelation in TM and IL-NTDT patients whose normal iron-store levels have been achieved and further chelation may cause iron deficiency and other complications.Citation24,Citation60
In general, the selection of the chelation-therapy protocol in each iron-loaded patient depends on several other factors in addition to the iron load, such as drug efficacy, organ targeting, toxicity, cost, and compliance. However, there are many controversies in the selection and use of chelating drugs in TM and IL-NTDT that arise mainly from commercial influence, the lack of consensus in the ultimate goal or aim of the chelation therapy, and the selective use of each of the chelating drugs for optimal therapy.Citation23,Citation25 Furthermore, there is no consensus in the evaluation criteria and risk–benefit assessment for the use of each of the chelating drugs in personalized medicine.Citation23,Citation25 In most Western and other developed countries, the selection of the chelating drug for the treatment of TM and IL-NTDT depends on the choice of the physician and the commercial influence of pharmaceutical companies.Citation24,Citation25 In developing countries, where most patients live, the main issue is the cost of the chelating drug and its availability and not necessarily risk–benefit assessment issues. These issues have been recently highlighted within the broader context of the use of orphan drugs, which include DFO, L1, and DFX, and also of orphan and rare diseases, which include TM and IL-NTDT.Citation24,Citation25
The monitoring of efficacy of different chelation-therapy protocols on the gross body as well as individual organ iron-load levels has been recently achieved following the introduction of new diagnostic techniques, such as MRI T2 and T2*. These methods have increased our understanding of iron-metabolism and chelation pathways of iron removal and resulted in improved drug-targeting therapies of the gross body, as well as focal iron deposits in organs, which are the cause of toxic side effects.Citation61–Citation64 Furthermore, these developments have increased the prospects of the introduction of personalized medicine in TM, IL-NTDT, and other iron-metabolism disorders.Citation23 The use of these new diagnostic methods in combination with serum ferritin monitoring made it possible to introduce chelation-therapy protocols of L1 or the L1–DFO combination that can achieve the complete elimination of iron overload in the heart and liver of almost all categories of iron-loaded patients.Citation65–Citation67
The clearance of excess iron in both organs can be achieved in most TM cases in 0.5–1.5 years using the International Committee on Chelation protocol on L1 (75–100 mg/kg/day) and DFO (40–60 mg/kg/day at least 3 days per week). The clearance of excess iron from these organs is faster when using higher overall doses of L1 and DFO and in patients who are less heavily iron-loaded.Citation65–Citation69 The removal of excess toxic iron in TM by L1 and the L1–DFO combination is accompanied by improvement in organ function, such as elevation of left ventricular ejection fraction (LVEF) and other cardiac and endocrinological benefits.Citation24,Citation65,Citation66,Citation69 In contrast, excess iron removal from the heart by DFX is not effective and in most cases very slow, with no improvement in LVEF, especially in heavily iron-loaded TM patients.Citation70 It should be noted that even after 5 years of treatment with DFX (30–40 mg/ kg/day), excess toxic amounts of iron remain in the heart of heavily iron-loaded TM patients and with no improvement in LVEF.Citation70,Citation71
Excess iron removal from the liver by DFX appears to be effective usually at high doses and equivalent to that caused by DFO, L1, and their combination at effective doses.Citation65–Citation72 The most effective and rapid clearance of excess liver iron, which is also used for intensive chelation in heavily iron-loaded patients, is intravenous DFO in combination with L1. Following the clearance of excess iron from the liver and heart and the normalization of the iron stores, which is characterized by normal levels of MRI T2 and T2* and serum ferritin, normal levels of iron are usually maintained using much lower overall chelation doses and in particular L1 monotherapy.Citation65–Citation67
Toxicity aspects and limitations in the use of chelating drugs in iron-loaded non-transfusion-dependent thalassemias and other patient categories
Patients with IL-NTDT and TM are regularly monitored for iron toxicity and other effects caused by their underlying condition.Citation9–Citation11,Citation24,Citation73 Clinical examination and biochemical tests are regularly carried out for organ function, including liver-enzyme levels, echocardiography, serum ferritin, and serum Zn levels.Citation9–Citation11,Citation16,Citation17,Citation24,Citation73
The task of treating iron overload in IL-NTDT is generally easier in comparison to TM, since IL-NTDT patients are not as heavily iron-loaded as TM patients and also the rate of iron loading in IL-NTDT in general is much slower than that in TM. However, the prospect of chelator toxicity is a major issue, and increases especially in patients with low iron stores.Citation24,Citation60 Regular monitoring and prophylactic measures for chelating drug toxicity are generally recommended for these categories of patients.Citation24,Citation74
The treatment of patients with TM, IL-NTDT, and other categories of iron-loading conditions with normal iron stores or not heavily iron-loaded is restricted for DFX, due to toxicity implications, as underlined by the labeling information for its use by the manufacturers of the drug.Citation75,Citation76 In particular, the use of DFX is not recommended for iron-loaded patients with serum ferritin lower than 500 μg/L.Citation75,Citation76 This limitation is important for the safety of many patients and in particular the vast majority of IL-NTDT, who are not heavily iron-loaded and have serum ferritin lower than 500 μg/L.Citation75–Citation76
High morbidity and mortality have been reported in different categories of patients, and especially non-iron-loaded categories of patients who have been treated with DFX.Citation77,Citation78 Many toxic side effects, such as fatal renal, liver, and bone marrow failure, including agranulocytosis, as well as renal toxicity, skin rashes, and gastric intolerance, have also been reported in iron-loaded patients treated with DFX.Citation74–Citation78
Patients treated with DFX are regularly monitored for kidney function, and withdrawal of the drug is recommended for patients with persistent increase in serum creatinine levels. The same limitations and restrictions that apply in the use of DFX in patients with low iron stores also apply for the use of DFO, despite the incidence of serious toxic side effects being much lower in the latter. Within this context, the use of DFO in IL-NTDT and other similar categories of not heavily iron-loaded patients, as well as other categories of patients with normal iron stores, is restricted, due to toxicity implications. Fatal cases of mucormycosis, acute respiratory distress syndrome, and Yersinia enterocolitica was reported in different categories of patients treated with DFO.Citation39,Citation79 Furthermore, ocular and auditory toxicity has also been reported in non-heavily iron-loaded TM patients using DFO.Citation39,Citation80,Citation81
In contrast, the safety of L1 in TM, IL-NTDT, and other categories of patients with low or normal iron stores has been shown during the regular use of the drug in thousands of patients in the last 20 years and also in studies in TM patients with normal iron stores exceeding 100 patient-years.Citation24,Citation65,Citation67,Citation74 The prospect of wider clinical use of L1 as a universal antioxidant in non-iron-overloading diseases, such as neuro-degenerative, cardiovascular, renal, and infectious diseases, as well as other diseases, including cancer and aging diseases, has been investigated in clinical trials involving many of these categories of patients.Citation24,Citation65,Citation67,Citation74 In particular, the introduction of L1 for the treatment of non-iron-loaded patients by targeting focal toxic iron deposits, eg, in Friedreich’s ataxia, and also of toxic labile iron, eg, in diabetic and nondiabetic glomerular disease, is a reflection of the antioxidant and safety potential of this drug.Citation24,Citation82,Citation83 In these two non-iron-loaded conditions, L1 was used at doses of 50–75 mg/kg/day for 6–9 months with no serious toxic side effects.Citation82,Citation83 Similarly, the safety of L1 in many other categories of non-iron-loaded diseases has also been confirmed in clinical trials involving patients with normal iron stores, including the anemia of chronic disease, renal dialysis, infections, Parkinson’s, and other neurodegenerative diseases.Citation24,Citation65,Citation67,Citation74
The most serious toxic side effects of L1 are reversible agranulocytosis (<1%) and neutropenia (<5%).Citation24,Citation84 Weekly or fortnightly mandatory blood count is recommended for patients using L1 for prophylaxis against this toxicity. A few fatal cases of agranulocytosis in TM have been reported for patients treated with L1 who did not follow this safety procedure.Citation24,Citation39,Citation84 Less serious toxic side effects include gastric intolerance, joint pains, and Zn deficiency. Zn supplements are used for prophylaxis for patients on long-term treatment with L1 and DFO, including TM and IL-NTDT patients.Citation24,Citation84
It should be noted that the rate of fatalities associated with the use of iron-chelating drugs is different. An estimated 11.7% mortality (1,935 of 16,514 patients) was reported by the European Medicines Agency for DFX, whereas for DFO and L1, the rate of mortality is less than 0.1%.Citation77,Citation78 Toxicity-vigilance and prophylactic measures are important steps for ensuring the safety of TM, IL-NTDT, and other iron-loaded patients treated with chelation therapy, especially when using high doses in patients with low or normal iron stores.
Effective iron-chelation therapy in thalassemia intermedia and in other iron-loaded non-transfusion-dependent thalassemias
Iron overload and related toxicity as a result of increased gastrointestinal absorption in TI and other IL-NTDT conditions was identified more than 40 years ago.Citation12,Citation13 Diagnostic methods, such as serum ferritin, serum iron, increased urinary excretion in response to DFO, and postmortem examination, revealed the extent of the iron load and toxicity in these categories of patients.Citation12–Citation18,Citation85
Iron-chelation therapy has been widely and successfully used from the early 1980s for the treatment of iron overload in TI, other IL-NTDT, and other similar conditions.Citation86 Initial reports indicated the efficacy of DFO in increasing iron excretion in these categories of patients. For example, increased urinary iron excretion in response to continuous 12-hour subcutaneous DFO (20–100 mg/kg/day, maximum 2 g/day) and decrease in serum ferritin (initial 68–940 μg/L and final 48–420 μg/L) after 6 months of treatment was observed in ten TI patients (age 1–17 years, five males, two splenectomized) with different Hb levels (7–10.5 g/dL) and variable iron load.Citation86 Four patients participating in the study had previous transfusions to a total in each case of 0.5, 1.5, 2.0, and 10.0 red blood-cell units, respectively. Dose adjustment was carried out by assessing 24-hour urinary iron excretion in response to the different doses of DFO, which were administered over 3 days. Different levels of iron load were detected in this cohort of young TI patients, including a 5-year-old patient. A tailored chelation therapy was recommended using this DFO-response method for reducing chelator toxicity. It was also suggested that iron-chelation treatment should begin from an early age to prevent iron accumulation.Citation86
Many nontransfused iron-loaded patients have also been treated with DF. In one case, subcutaneous DFO (2 g/day for 9–11 months) was shown to be effective in hereditary hemochromatosis patients where phlebotomy was not possible.Citation87 Monitoring of liver iron by repeated noninvasive liver iron-concentration measurements using superconducting quantum interference-device biosusceptometry has shown that DFO was as effective in liver-iron elimination (12 mg/day) as normal phlebotomy treatment (14.3 mg/day) of weekly 500 mL blood removals.Citation87
Successful iron-chelation treatments in TI and other IL-NTDT patients have also been reported using L1. For example, L1 at low doses of 25–50 mg/kg/day was studied for 17 weeks and 86 weeks in seven HbE β-thalassemia and two TM iron-loaded nontransfused patients.Citation88 Substantial reductions in serum ferritin (from mean 2,168 to 418 μg/L) and hepatic iron (from mean 20.3 to 11.7 mg/g dry weight) were observed in these patients.Citation88 Similar results were obtained in another study in 16 NTDT HbH disease (3/4 α-globin gene deletion) patients (29–76 years) using L1 at 50 mg/kg/day initially for 4–8 months. The dose then increased in the majority of cases to 75 mg/kg/day.Citation89 In 18 months, mean serum ferritin decreased from 1,492 to 519 μg/L, and in nine of the patients was below the normal cutoff point of 397 μg/L before the 18-month monitoring period. Significant improvement in cardiac function and decrease in liver-iron content measured by MRI T2 was also detected in 12 of the patients.Citation89 No reduction in iron load was observed in a control group of 16 patients with HbH disease who received no chelation treatment in the same study.Citation89
The use of higher doses of L1 (75–100 mg/kg/day) in IL-NTDT can result in the rapid normalization of iron stores. For example, administration of L1 at 75 mg/kg/day for 9 months in an iron-loaded, nontransfused TI patient with cardiac and other problems caused a substantial reduction to normal levels in serum ferritin (from 2,174 to 251 μg/L) and hepatic iron (from 14.6 to 1.9 mg/g dry weight).Citation90 An increase in cardiac and liver MRI T2 relaxation time was also observed following the 9-month treatment, indicating the effective removal of iron from these organs by L1.Citation90
In a 5-year long-term randomized clinical trial of 88 occasionally transfused TI patients (median age 41 years, 48 females), the effectiveness of oral L1 (50–75 mg/kg/day) on 47 patients with median serum ferritin 1,221 μg/L was compared to a group of 41 patients with median serum ferritin 1,122 μg/L treated with subcutaneous DFO (30–50 mg/kg/day, 5 days per week).Citation91 Both chelators were found to be effective in reducing iron load, with median serum ferritin levels reaching 579 μg/L in the L1 group and 484 μg/L in the DFO group. It was concluded in this >200 patient-year-long study that L1 was associated with an efficacy and safety profile similar to that of DFO.Citation91 Toxic side effects of L1 were gastric intolerance, joint pains, neutropenia in six, and agranulocytosis in four patients. Compliance was 85% for patients on L1 and 76% for DFO. The cost of chelation between L1 and DF was not significantly different, and was estimated to be about ten times lower than that of DFX.Citation56,Citation57,Citation91 A follow-up 5-year trial using L1 and DFO continued with members of the same group of TI patients, with similar results.Citation91
Rapid clearance of excess iron in iron-loaded nontransfused patients has also been observed using the L1–DFO combination, which appears to be easily achieved and also to be faster in comparison to TM patients.Citation65–Citation69 For example, a combination of intravenous DFO (30 mg/kg/day for 5 months) and oral L1 (75 mg/kg/day for 2 months) was used in a juvenile hemochromatosis nontransfused iron-loaded patient (serum ferritin 3,773 μg/L) with severe cardiomegaly and congestive heart failure with an ejection fraction of 25%.Citation92 The combination L1–DFO chelation treatment caused an improvement in the ejection fraction to 49% and blood parameters, and also removal of antiarrhythmic drugs and normalization of serum ferritin.Citation92 Increase in the ejection fraction has also been observed in different groups of TM patients using L1 and the L1–DFO combination. Citation24,Citation65–Citation69 Overall, effective and safe chelation therapy using DFO, L1, and their combination could be applied to TI and other IL-NTDT patients.
A number of clinical trials have been carried out to test the effect of DFX in TI and other IL-NTDT patients.Citation22 The efficacy and toxicity of DFX appear to be dose-dependent. For example, in a randomized 1-year clinical trial involving 110 TI patients, some NTDT and others receiving occasional transfusions, DFX (5–10 mg/kg/day) caused no significant change in serum ferritin at 24 weeks and only significant change at 52 weeks at 10 mg/kg/day compared to baseline.Citation93 Similar changes were reported for liver-iron concentration. Adverse events were reported in 78% of the patients, and only 24% were thought to be drug-related, namely nausea, rash, and diarrhea.Citation93 In a continuation study for a further year, the results of the use of DFX (5–20 mg/kg/day) on efficacy and toxicity in a selected group of six not previously chelated of the placebo group and 18 of the 110 chelated patients (12 TI, six α-thalassemia, and six HbE β-thalassemia) were reported.Citation93 Median serum ferritin before the study was 825 (range: 393–2,169) μg/L, and by the end of the study had decreased to 427 (range: 102–1,010) μg/L. Similar changes were reported for liver-iron concentration.Citation93 Approximately 29% of the adverse events reported were similar to the previous study and thought to be drug-related.Citation93 In a different 1-year study involving a cohort of 50 TI patients, DFX (10–40 mg/kg/day) caused a decrease of serum ferritin from baseline mean 1,700 to 835 μg/L in 8 months, but thereafter remained stable or slightly increased to 880 μg/L on the 12th month.Citation94
In contrast to these studies, lower efficacy and higher toxicity was reported for DFX in TI and other IL-NTDT patients by investigators not related or funded by the manufacturers of DFX.Citation95 For example, in a retrospective study of 72 TI and TM patients of mean age 20 years, the use of DFX (20–40 mg/kg/day) for a mean 16.7 months resulted in a 25% dropout due to toxicity and low efficacy.Citation95 In particular, adverse effects of renal function were reported, including a >33% increase in serum creatinine in 43 patients. Persistent increase in serum creatinine in seven of the 43 patients caused discontinuation of DFX.Citation95 No significant change in serum fer-ritin was observed at the end of the study (initial 3,059±309 versus final 2,965±309 μg/L). Similarly, no change in cardiac iron load but a decrease in liver iron was observed in this cohort of patients. The patients affected by the DFX toxicity reverted back to L1, DFO, or the L1–DFO combination.Citation95 High incidence of renal toxicity using DFX has also been reported in many other groups of patients, including young patients, by independent investigators not sponsored or without connections with the manufacturers of DFX.Citation96–Citation98
The variation in the iron overload in patients of IL-NTDT and the restriction imposed on the safety for the use of the chelating drugs in different iron-overload levels, as well as the target iron-loaded organ(s), requires the design of specific chelation protocols for each patient case. Similar chelation protocols apply in the treatment of TM patients with equivalent iron overload to IL-NTDT patients. It appears that the selection of the most effective and safest chelation protocol together with the monitoring of the iron load using serum ferritin and MRI T2 or T2*, as well as the use of prophylactic measures for chelating drug toxicity, could ensure the effective reduction and maintenance of iron load to normal levels and lead to an overall decrease in morbidity and mortality in IL-NTDT patients.
Future therapeutic approaches of iron-chelation therapy in iron-loaded nontransfused thalassemia patients caused by increased iron absorption
Patients with TI and other IL-NTDT have a diverse genotypic and clinical picture of variable severity, including different levels of iron overload and associated toxicity in the liver, heart, and other organs.Citation10,Citation11,Citation17,Citation19,Citation27,Citation32,Citation73 Within this context, several approaches in relation to chelation therapy could be envisaged. The design and application of optimal chelation therapies will be required in each case based on a risk–benefit assessment of the drug(s) and other factors similar to TM patients.Citation23,Citation24 In particular, the cost of chelating drugs is very important, since most TI and other IL-NTDT patients live in developing countries and cannot afford expensive drug treatments.Citation56,Citation57,Citation99,Citation100
DFO and L1 appear to be effective in the removal of excess iron load in TI and other IL-NTDT patients, even at doses lower than those used in TM and other transfused patients. Selected combinations of DFO and L1, such as the International Committee on Chelation protocol, could be used as a first-line treatment not only for the rapid removal of excess cardiac iron in cases of iron-overload cardiomyopathy in TI and other IL-NTDT patients but also in the rapid clearance of liver-iron overload.Citation23,Citation24,Citation65,Citation66,Citation86
A critical question in relation to the treatment of iron overload in TI and other IL-NTDT is the timing of the initiation of the chelation therapy. Recent studies in TM patients suggest that the removal of iron by chelating drugs in TI and other IL-NTDT should be initiated as soon as the diagnosis is confirmed and with a major aim the complete removal of all excess iron and prevention of its accumulation in organs.Citation23,Citation24,Citation86 In studies of TM patients with normal-range iron-store levels, lower overall doses of chelating drugs and in particular L1 were used for achieving this aim.Citation65,Citation67 Furthermore, the advantage of this approach is that in patients with normal-range iron stores, the damage to organs is prevented and clinical management can continue without related organ complications, such as liver cirrhosis and cardiomyopathy.Citation23,Citation24
L1 should be considered as a first-line treatment for TI and other IL-NTDT patients with low or normal iron stores, since it would appear from studies in these categories that included cohorts of TM patients to be safer and more effective in comparison to DFO and DFX.Citation74–Citation77 The latter two chelating drugs are recommended by the manufacturers to be used in patients with serum ferritin levels higher than 500 μg/L.Citation74–Citation77 Similarly, the prophylactic use of low doses of L1 (eg, 10 mg/kg/day) may be sufficient in preventing iron overload in young and older patients with TI and other IL-NTDT.
Many safety, efficacy, and financial questions are raised regarding the use of DFX in TI and IL-NTDT patients. Additional toxicity issues can be envisaged in patients with low iron stores, where DFX may cause an increase in the absorption of dietary iron and other toxic metals, eg, aluminum.Citation23,Citation48 DFX is also very expensive and cannot be afforded by the vast majority of patients, especially those living in developing countries.Citation3,Citation23,Citation56,Citation57,Citation99,Citation100 The introduction of combinations of DFX with DFO and L1 increases the prospect of more effective and less toxic therapies for TM, TI, IL-NTDT, and other related conditions in comparison to DFX monotherapies.Citation100–Citation105 Pharmacological and toxicological data are important parameters for the construction of such combination therapies.Citation101,Citation106,Citation107
Although current interest in TI and other IL-NTDT patients is mainly related to the marketing efforts of the manufacturers of DFX and their academic partners, there are many other fields of investigation that need to be explored in addition to iron-chelation therapy. Some of the strategies for the treatment of the other clinical complications may also influence the rate of iron overload and chelation therapy. Within this context, the time of initiation and the rate of red blood-cell transfusions, as well as splenectomy, will have a direct effect on iron loading and change in the chelation strategies.Citation85,Citation105 Similarly, the role of iron overload and chelation therapy on the activation of HbF production by hydroxyurea, which also has iron-chelation properties, has not yet been fully investigated.Citation11,Citation28,Citation34–Citation37,Citation52,Citation108 The effects of modulators of erythropoietin, hepcidin, ferroportin, and other regulatory molecules on iron metabolism and erythropoietic activity are also expected to influence future chelation-therapy strategies in TI and other IL-NTDT.Citation38,Citation109,Citation110
A major therapeutic strategy in TI, other IL-NTDT, and especially hereditary hemochromatosis is the prevention or decrease of gastrointestinal iron absorption. This can be accomplished by new, synthetic, or naturally occurring iron chelators that can bind iron in the gastrointestinal tract and inhibit or decrease the rate of its transfer and storage in the body. In particular, chelators and chelator–iron complexes that are not orally absorbed and chelators that form insoluble iron complexes could be considered for such treatments.Citation33,Citation111 Inhibitors of DMT1 activity and activators of hepcidin production could also be used for minimizing iron absorption. The combination of chela-tors and of chelators with regulators of DMT1, hepcidin, and ferroportin could also be considered for potential use in reducing increased iron absorption and associated iron overload in IL-NTDT.Citation33,Citation109–Citation111
The inhibition of iron absorption in IL-NTDT could be accompanied by relocation and reutilization of stored iron for erythropoiesis and other iron-metabolism activities. Within this context, L1 has been shown to facilitate this process by the redistribution of iron stored in hepatocytes and macrophages to hemopoietic tissues via iron exchange with transferrin.Citation112–Citation119
Overall, several treatments could become available for the future management of TI, other IL-NTDT, and hereditary hemochromatosis. These could involve the use of chelation therapies for increasing iron excretion, chelation therapies for inhibiting iron absorption, chelation therapies for iron redistribution, selected diets with lower and less absorbable forms of iron, the use of regulators of DMT1, hepcidin, and ferroportin and other proteins of iron metabolism involved in the transport of iron, activators of HbF production, and combinations of such treatments.
Acknowledgments
This study was supported from internal funds of the Postgraduate Research Institute of Science, Technology, Environment and Medicine, a nonprofit, charitable organization. The article was a result of an invitation by the journal editors.
Disclosure
The authors report no conflicts of interest in this work.
References
- PantopoulosKPorwalSKTartakoffADevireddyLMechanisms of mammalian iron homeostasisBiochemistry201251295705572422703180
- AndrewsNCForging a field: the golden age of iron biologyBlood2008112221923018606887
- No authors listedCommunity control of hereditary anaemias: memorandum from a WHO meetingBull World Health Organ198361163806601544
- WeatherallDJGleggJBThe Thalassaemia Syndromes3rd edOxfordBlackwell Scientific Publications1981
- FederJNGnirkeAThomasWA novel MHC class I-like gene is mutated in patients with hereditary haemochromatosisNat Genet1996134339408
- BartonJCEdwardsCQHemochromatosis: Genetics, Pathophysiology, Diagnosis and TreatmentCambridgeCambridge University Press2000
- HalliwellBGutteridgeJMFree radicals and antioxidant protection: mechanisms and significance in toxicology and diseaseHum Toxicol1988717133278973
- KontoghiorghesGJProspects for introducing deferiprone as potent pharmaceutical antioxidantFront Biosci (Elite Ed)2009116117819482634
- SingerSTVariable clinical phenotypes of α-thalassemia syndromesScientificWorldJournal2008961562519618088
- GalanelloRCaoARelationship between genotype and phenotype: thalassemia intermediaAnn NY Acad Sci19988503253339668554
- Borgna-PignattiCModern treatment of thalassaemia intermediaBr J Haematol2007138329130417565568
- PippardMJCallenderSTWarnerGTWeatherallDJIron absorption and loading in β-thalassaemia intermediaLancet19792814781982190918
- PootrakulPKitcharoenKYansukonPThe effect of erythroid hyperplasia on iron balanceBlood1988714112411293355891
- KontoghiorghesGJEracleousEEconomidesCKolnagouAAdvances in iron overload therapies. Prospects for effective use of deferiprone (L1), deferoxamine, the new experimental chelators ICL670, GT56-252, L1NAll and their combinationsCurr Med Chem200512232663268116305464
- KontoghiorghesGJSpyrouAKolnagouAIron chelation therapy in hereditary hemochromatosis and thalassemia intermedia: regulatory and non regulatory mechanisms of increased iron absorptionHemoglobin201034325126420524815
- AessoposAKatiMFarmakisDHeart disease in thalassemia inter-media: a review of the underlying pathophysiologyHaematologica200792565866517488690
- SonakulDPachareePWasiPFucharoenSCardiac pathology in 47 patients with β-thalassaemia/haemoglobin ESoutheast Asian J Trop Med Public Health19841545545636242076
- AessoposAFarmakisDTrompoukisCCardiac involvement in sickle β-thalassemiaAnn Hematol200988655756419107483
- AessoposAFarmakisDKaragiorgaMCardiac involvement in thalassemia intermedia: a multicenter studyBlood200197113411341611369631
- Borgna-PignattiCGaraniMCForniGLHepatocellular carcinoma in thalassaemia: an update of the Italian registryBr J Haematol2014167112112624992281
- FragatouSTsourveloudisIManesisGIncidence of hepatocel-lular carcinoma in a thalassemia unitHemoglobin201034322122620524812
- TaherATPorterJBViprakasitVApproaching low liver iron burden in chelated patients with non-transfusion-dependent thalassemia: the safety profile of deferasiroxEur J Haematol201492652152624460655
- KontoghiorghesGJA new era in iron chelation therapy: the design of optimal, individually adjusted iron chelation therapies for the complete removal of iron overload in thalassemia and other chronically transfused patientsHemoglobin200933533233819814679
- KolnagouAKontoghiorgheCNKontoghiorghesGJTransition of thalassaemia and Friedreich ataxia from fatal to chronic diseasesWorld J Methodol20144419721825541601
- KontoghiorgheCNAndreouNConstantinouKKontoghiorghesGJWorld health dilemmas: orphan and rare diseases, orphan drugs and orphan patientsWorld J Methodol20144316318825332915
- KontoghiorghesGJKolnagouAMolecular factors and mechanisms affecting iron and other metal excretion or absorption in health and disease: the role of natural and synthetic chelatorsCurr Med Chem200512232695270916305466
- FucharoenSKetvichitPPootrakulPSiritanaratkulNPiankijagumAWasiPClinical manifestation of β-thalassemia/hemoglobin E diseaseJ Pediatr Hematol Oncol200022655255711132229
- VichinskyEHemoglobin E syndromesHematology Am Soc Hematol Educ Program2007798318024613
- PantopoulosKFunction of the hemochromatosis protein HFE: lessons from animal modelsWorld J Gastroenterol200814456893690119058322
- ByrnesVBarrettSRyanEIncreased duodenal DMT-1 expression and unchanged HFE mRNA levels in HFE-associated hereditary hemochromatosis and iron deficiencyBlood Cells Mol Dis200229325126012547214
- MavrogeniSGotsisELadisVMagnetic resonance evaluation of liver and myocardial iron deposition in thalassemia intermedia and β-thalassemia majorInt J Cardiovasc Imaging200824884985418581254
- AuWYLamWWChuWWOrgan-specific hemosiderosis and functional correlation in Chinese patients with thalassemia intermedia and hemoglobin H diseaseAnn Hematol2009881094795019165482
- KontoghiorghesGJChelators affecting iron absorption in miceArzneimittelforschung19904012133213352095129
- ItaliaKYJijinaFJMerchantRResponse to hydroxyurea in β-thalassemia major and intermedia: experience in western IndiaClin Chim Acta20094071–2101519545554
- SingerSTKuypersFAOlivieriNFFetal haemoglobin augmentation in E/β0 thalassaemia: clinical and haematological outcomeBr J Haematol2005131337838816225658
- PatrinosGPGrosveltFGPharmacogenomics and therapeutics of haemoglobinopathiesHemoglobin2008321–222923618275000
- OlivieriNFFreedmanMHPerrineSPTrial of recombinant human erythropoietin: three patients with thalassemia intermediaBlood19928012325832601467532
- AdekileADAzabAFAl-SharidaSIClinical and molecular characteristics of non-transfusion-dependent thalassemia in KuwaitHemoglobin201539532032626076396
- KontoghiorghesGJNeocleousKKolnagouABenefits and risks of deferiprone in iron overload in thalassaemia and other conditions: comparison of epidemiological and therapeutic aspects with deferoxamineDrug Saf200326855358412825969
- KontoghiorghesGJStructure/red blood cell permeability activity of iron(III) chelator complexesInorganica Chim Acta19881512101106
- KontoghiorghesGJMayAUptake and intracellular distribution of iron from transferrin and chelators in erythroid cellsBiol Met199033–41831872073459
- ForsbeckKNilssonKKontoghiorghesGJVariation in iron accumulation, transferrin membrane binding and DNA synthesis in the K-562 and U-937 cell lines induced by chelators and their iron complexesEur J Haematol19873943183253691756
- YamamotoRSWilliamsGMFrangelHHWeisburgerJH8-Hydroxyquinoline:chronic toxicity and inhibitory effect on the carcinogenicity of N-2-fluorenylacetamideToxicol Appl Pharmacol19711946876985132036
- KontoghiorghesGJThe Design of Orally Active Iron Chelators for the Treatment of Thalassaemia [doctoral thesis]Colchester, UKUniversity of Essex1982
- HarveyRSReffittDMDoigLAFerric trimaltol corrects iron deficiency anaemia in patients intolerant of ironAliment Pharmacol Ther19981298458489768526
- GascheCAhmadTTulassayZFerric maltol is effective in correcting iron deficiency anemia in patients with inflammatory bowel disease: results from a phase-3 clinical trial programInflamm Bowel Dis201521357958825545376
- KontoghiorghesGJDo we need more iron-chelating drugs?Lancet2003362938249549612927446
- KontoghiorghesGJTransparency and access to full information for the fatal or serious toxicity risks, low efficacy and high price of deferasirox, could increase the prospect of improved iron chelation therapy worldwideHemoglobin200832660861519065341
- DresowBFischerRNielsenPGabbeEEPigaAEffect of oral iron chelator L1 on iron absorption in manAnn N Y Acad Sci19988504664689668586
- BerkovitchMLivneALushkovGThe efficacy of oral defer-iprone in acute iron poisoningAm J Emerg Med2000181364010674529
- DjaldettiMFishmanPNottiIBesslerHThe effect of tetracycline administration on iron absorption in miceBiomedicine19813551501527317563
- KonstantinouEPashalidisIKolnagouAKontoghiorghesGJInteractions of hydroxycarbamide (hydroxyurea) with iron and copper: implications on toxicity and therapeutic strategiesHemoglobin201135323724621599436
- ZurloMGDe StefanoPBorgna-PignattiCSurvival and causes of death in thalassaemia majorLancet19892865327302567801
- ModellBKhanMDarlisonMSurvival in β-thalassaemia major in the UK: data from the UK Thalassaemia RegisterLancet200035592202051205210885361
- TelferPTWarburtonFChristouSImproved survival in thalassemia major patients on switching from desferrioxamine to combined chelation therapy with desferrioxamine and deferiproneHaematologica200994121777177819815834
- KontoghiorghesGJEthical issues and risk/benefit assessment of iron chelation therapy: advances with deferiprone/deferoxamine combinations and concerns about the safety, efficacy and costs of deferasiroxHemoglobin2008321–211518274978
- CohenARIron chelation therapy: you gotta have heartBlood2010115122333233420339104
- DézsiLVécseiLClinical implications of irregular ADMET properties with levodopa and other antiparkinson’s drugsExpert Opin Drug Metab Toxicol201410340942424437461
- KontoghiorghesGJBartlettANSheppardLBarrJNorteyPOral iron chelation therapy with deferiprone. Monitoring of biochemical, drug and iron excretion changesArzneimittelforschung199545165697893273
- AessoposAKatiMFarmakisDPolonifiEDeftereosSTsironiMIntensive chelation therapy in β-thalassemia and possible adverse cardiac effects of desferrioxamineInt J Hematol200786321221517988985
- MavrogeniSIGotsisEDMarkussisVT2 relaxation time study of iron overload in β-thalassemiaMAGMA1998617129794284
- AndersonLJHoldenSDavisBCardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overloadEur Heart J200122232171217911913479
- KolnagouAYazmanDEconomidesCEracleousEKontoghiorghesGJUses and limitations of serum ferritin, magnetic resonance imaging T2 and T2* in the diagnosis of iron overload and in the ferrikinetics of normalization of the iron stores in thalassemia using the International Committee on Chelation deferiprone/deferoxamine combination protocolHemoglobin200933531232219814677
- PennellDJT2* magnetic resonance and myocardial iron in thalassemiaAnn N Y Acad Sci2005105437337816339685
- KolnagouAKleanthousMKontoghiorghesGJReduction of body iron stores to normal range levels in thalassaemia by using a deferiprone/deferoxamine combination and their maintenance thereafter by deferiprone monotherapyEur J Haematol201085543043820662901
- FarmakiKTzoumariIPappaCChouliarasGBerdoukasVNormalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia majorBr J Haematol2010148346647519912219
- KolnagouAKontoghiorghesGJMaintenance of normal range body iron store levels for up to 4.5 years in thalassemia major patients using deferiprone monotherapyHemoglobin201034320420920524810
- KolnagouAKleanthousMKontoghiorghesGJEfficacy, compliance and toxicity factors are affecting the rate of normalization of body iron stores in thalassemia patients using the deferiprone and deferoxamine combination therapyHemoglobin201135318619821599431
- TannerMAGalanelloRDessiCCombined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunctionJ Cardiovasc Magn Reson2008101218298856
- WoodJCKangBPThompsonAThe effect of deferasirox on cardiac iron in thalassemia major: impact of total body iron storesBlood2010116453754320421452
- VlachakiEAgapidouASpanosGFive years of deferasirox therapy for cardiac iron in β-thalassemia majorHemoglobin201539529930426177199
- MeerpohlJJAntesGRückerGDeferasirox for managing iron overload in people with thalassaemiaCochrane Database Syst Rev20122CD00747622336831
- WasiPPootrakulPFucharoenSWinichagoonPWilairatPPromboonAThalassemia in southeast Asia: determination of different degrees of severity of anemia in thalassemiaAnn N Y Acad Sci19854451191263893274
- KontoghiorghesGJKolnagouAPengCTShahSVAessoposASafety issues of iron chelation therapy in patients with normal range iron stores including thalassaemia, neurodegenerative, renal and infectious diseasesExpert Opin Drug Saf20109220120620059374
- US Food and Drug AdministrationClinical review: Exjade (deferasirox, ICL-670)2005 Available from: http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4177B1_02_b.pdfAccessed December 1, 2015
- Exjade (deferasirox) tablets for oral suspension [prescribing information]East Hanover, NJNovartis Pharmaceutical Corporation2011 Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021882s010lbl.pdfAccessed December 1, 2015
- KontoghiorghesGJA record number of fatalities in many categories of patients treated with deferasirox: loopholes in regulatory and marketing procedures undermine patient safety and misguide public funds?Expert Opin Drug Saf201312560560923688354
- KontoghiorghesGJTurning a blind eye to deferasirox’s toxicity?Lancet201338198731183118423561999
- BoelaertJRFenvesAZCoburnJWDeferoxamine therapy and mucormycosis in dialysis patients: report of an international registryAm J Kidney Dis19911866606671962650
- OrtonRBde VeberLLSulhHMOcular and auditory toxicity of long-term, high-dose subcutaneous deferoxamine therapyCan J Ophthalmol19852041531564052864
- CasesAKellyJSabaterFOcular and auditory toxicity in hemodialyzed patients receiving desferrioxamineNephron199056119232234245
- BoddaertNSangKHRötigASelective iron chelation in Friedreich ataxia: biologic and clinical implicationsBlood2007110140140817379741
- RajapurkarMMHegdeUBhattacharyaAAlamMGShahSVEffect of deferiprone, an oral iron chelator, in diabetic and non-diabetic glomerular diseaseToxicol Mech Methods201323151022978744
- CohenARGalanelloRPigaADe SanctisVTrictaFSafety and effectiveness of long-term therapy with the oral iron chelator deferiproneBlood200310251583158712763939
- PootrakulPVongsmasaVLaongpanichPWasiPSerum ferritin levels in thalassemias and the effect of splenectomyActa Haematol19816642442506800190
- CossuPToccafondiCVardeuFIron overload and desferriox-amine chelation therapy in β-thalassemia intermediaEur J Pediatr198113732672717318837
- NielsenPFischerRBuggischPJanka-SchaubGEffective treatment of hereditary haemochromatosis with desferrioxamine in selected casesBr J Haematol2003123595295314632789
- PootrakulPSirankaprachaPSankoteJClinical trial of deferiprone iron chelation therapy in β-thalassaemia/haemoglobin E patients in ThailandBr J Haematol2003122230531012846901
- ChanJCChimCSOoiCGUse of the oral chelator deferiprone in the treatment of iron overload in patients with Hb H diseaseBr J Haematol2006133219820516611312
- OlivieriNFKorenGMatsuiDReduction of tissue iron stores and normalization of serum ferritin during treatment with the oral iron chelator L1 in thalassemia intermediaBlood19927910274127481586721
- CalvarusoGVitranoADi MaggioRDeferiprone versus deferoxamine in thalassemia intermedia: results from a 5-year long-term Italian multicenter randomized clinical trialAm J Hematol201590763463825809173
- FabioGMinonzioFDelbiniPBianchiACappelliniMDReversal of cardiac complications by deferiprone and deferoxamine combination therapy in a patient affected by a severe type of juvenile hemo-chromatosis (JH)Blood2007109136236416960153
- TaherATPorterJViprakasitVDeferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled studyBlood2012120597097722589472
- KarimiMArandiNHaghpanahSEfficacy of deferasirox (Exjade®) in modulation of iron overload in patients with β-thalassemia intermediaHemoglobin201539532732926114738
- Al-KhaboriMBhandariSAl-HuneiniMAl-FarsiKPanjwaniVDaarSSide effects of deferasirox iron chelation in patients with beta thalassemia major or intermediaOman Med J201328212112423599881
- DeeCMCheukDKHaSYChiangAKChanGCIncidence of deferasirox-associated renal tubular dysfunction in children and young adults with β-thalassaemiaBr J Haematol2014167343443624989901
- ChuangGTTsaiIJTsauYKLuMYTransfusion-dependent thalas-saemic patients with renal Fanconi syndrome due to deferasirox useNephrology (Carlton)2015201293193526016559
- NaderiMSadeghi-BojdSValeshabadAKA prospective study of tubular dysfunction in pediatric patients with β-thalassemia major receiving deferasiroxPediatr Hematol Oncol201330874875424134694
- TeawtrakulNChansungKSirijerachaiCWanitpongpunCThepsuthammaratKThe impact and disease burden of thalassemia in Thailand: a population-based study in 2010J Med Assoc Thai201295Suppl 7S211S21623130457
- ViprakasitVNuchprayoonIChuansumritADeferiprone(GPO-L-ONE®) monotherapy reduces iron overload in transfusion-dependent thalassemias: 1-year results from a multicenter prospective, single arm, open label, dose escalating phase III pediatric study (GPO-L-ONE; A001) from ThailandAm J Hematol201388425126023460233
- KontoghiorghesGJFuture chelation monotherapy and combination therapy strategies in thalassemia and other conditions: comparison of deferiprone, deferoxamine, ICL670, GT56-252, L1NAll and starch deferoxamine polymersHemoglobin200630232994716798657
- TotadriSBansalDBhatiaPAttriSVTrehanAMarwahaRKThe deferiprone and deferasirox combination is efficacious in iron overloaded patients with β-thalassemia major: a prospective, single center, open-label studyPediatr Blood Cancer20156291592159625820920
- CassinerioEOrofinoNRoghiACombination of deferasirox and deferoxamine in clinical practice: an alternative scheme of chelation in thalassemia major patientsBlood Cells Mol Dis201453316416724846580
- ElalfyMSAdlyAMWaliYTonySSamirAElhenawyYIEfficacy and safety of a novel combination of two oral chelators deferasirox/ deferiprone over deferoxamine/deferiprone in severely iron overloaded young beta thalassemia major patientsEur J Haematol201595541142025600572
- KolnagouAMichaelidesYKontoghiorgheCNKontoghiorghesGJThe importance of spleen, spleen iron, and splenectomy for determining total body iron load, ferrikinetics, and iron toxicity in thalassemia major patientsToxicol Mech Methods2013231344123039902
- KontoghiorghesGJGoddardJGBartlettANSheppardLPharma-cokinetic studies in humans with the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-oneClin Pharmacol Ther19904832552612401124
- WaldmeierFBruinGJGlaenzelUPharmacokinetics, metabolism, and disposition of deferasirox in β-thalassemic patients with transfusion-dependent iron overload who are at pharmacokinetic steady stateDrug Metab Dispos201038580881620097723
- CaoHPharmacological induction of fetal hemoglobin synthesis using histone deacetylase inhibitorsHematology20049322323315204104
- RuchalaPNemethEThe pathophysiology and pharmacology of hepcidinTrends Pharmacol Sci201435315516124552640
- ArezesJNemethEHepcidin and iron disorders: new biology and clinical approachesInt J Lab Hematol201537Suppl 1929825976966
- KontoghiorghesGJBarrJNorteyPSheppardLSelection of a new generation of orally active α-ketohydroxypyridine iron chelators intended for use in the treatment of iron overloadAm J Hematol19934243403498493983
- KontoghiorgheCNKolnagouAKontoghiorghesGJPotential clinical applications of chelating drugs in diseases targeting transferrin-bound iron and other metalsExpert Opin Investig Drugs2013225591618
- VreugdenhilGSwaakAJde Jeu-JaspersCvan EijkHGCorrelation of iron exchange between the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one (L1) and transferrin and possible antianaemic effects of L1 in rheumatoid arthritisAnn Rheum Dis199049119569572256752
- EvansRWSharmaMOgwangWPatelKJBartlettANKon-toghiorghesGJThe effect of α-ketohydroxypyridine chelators on transferrin saturation in vitro and in vivoDrugs Today199228Suppl A923
- SmeetsMEVreugdenhilGHoldrinetRSImprovement of erythropoiesis during treatment with deferiprone in a patient with myelofibrosis and transfusional hemosiderosisAm J Hematol19965132432448619408
- ChangYHShawCFWuKHHsiehKHSuYNLuPJTreatment with deferiprone for iron overload alleviates bone marrow failure in a Fanconi anemia patientHemoglobin200933534635119814681
- VreugdenhilGSwaakAJKontoghiorghesGJvan EijkHGEfficacy and safety of oral iron chelator L1 in anaemic rheumatoid arthritis patientsLancet198928676139813992574342
- MostertLJVan DorstJAKosterJFVan EijkHGKontoghiorghesGJFree radical and cytotoxic effects of chelators and their iron complexes in the hepatocyteFree Radic Res Commun1987363793883508452
- BrockJHLicéagaJArthurHMKontoghiorghesGJEffect of novel 1-alkyl-3-hydroxy-2-methylpyrid-4-one chelators on uptake and release of iron from macrophagesAm J Hematol199034121252327400