
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that results in loss of the upper and lower motor neurons from motor cortex, brainstem, and spinal cord. While the majority of cases are sporadic, approximately 10% show familial inheritance. ALS is usually inherited in an autosomal dominant manner, although autosomal recessive and X-linked inheritance do occur. To date, 24 of the genes at 26 loci have been identified; these include loci linked to ALS and to frontotemporal dementia-ALS, where family pedigrees contain individuals with frontotemporal dementia with/without ALS. The most commonly established genetic causes of familial ALS (FALS) to date are the presence of a hexanucleotide repeat expansion in the C9ORF72 gene (39.3% FALS) and mutation of SOD1, TARDBP, and FUS, with frequencies of 12%–23.5%, 5%, and 4.1%, respectively. However, with the increasing use of next-generation sequencing of small family pedigrees, this has led to an increasing number of genes being associated with ALS. This review provides a comprehensive review on the genetics of ALS and an update of the pathogenic mechanisms associated with these genes. Commonly implicated pathways have been established, including RNA processing, the protein degradation pathways of autophagy and ubiquitin–proteasome system, as well as protein trafficking and cytoskeletal function. Elucidating the role genetics plays in both FALS and sporadic ALS is essential for understanding the subsequent cellular dysregulation that leads to motor neuron loss, in order to develop future effective therapeutic strategies.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder with an incidence of two to three per 100,000 and a lifetime risk of one per 400 individuals.Citation1 Usually with an adult onset, initial clinical symptoms, such as loss of dexterity in the fingers or a mild limp in limb onset ALS, or slurring of speech in bulbar onset ALS, are caused by loss of the upper motor neurons in the motor cortex and brainstem and lower motor neurons in the spinal cord. Over time, the progressive nature of the disease is associated with further muscle wasting, weight loss, fasciculations, and eventually death due to respiratory failure, 32 months following symptom onset.Citation1 During disease progression, cognitive impairment may develop in up to 40% of patients, and ∼5% will go on to develop frontotemporal dementia (FTD).Citation1
While the majority of cases are sporadic ALS (SALS) with no familial history of disease, ∼10% of cases are familial ALS (FALS) and are clinically indistinguishable from SALS cases. Generally, in adult onset ALS, the disease is inherited as an autosomal dominant (AD) trait, although rare cases of juvenile ALS are more commonly associated with autosomal recessive (AR) inheritance. However, there are also instances of AR inheritance of AD genes in specific populations (SOD1 in Scandinavia and FUS in Cape Verde). In addition, there is evidence of reduced penetrance of disease-associated mutations (including p.I114T SOD1 and G4C2 C9ORF72) and oligogenic inheritance,Citation2 illustrating that ALS is a highly complex genetic disorder.
The first genetic cause of ALS was identified in 1993 through linkage analysis to the Chr21 marker DS21S223. Subsequent analysis of the nearby gene, SOD1, identified multiple pathogenic mutations in these FALS families. Over the following 16 years, linkage analysis and candidate gene sequencing of ALS families have identified further genes associated with AD or AR ALS and one instance of X-linked inheritance (). With the advent of next-generation sequencing, whole exome sequencing (WES) has allowed an exponential increase in the identification of disease-associated genes (). Following the first use of WES to identify that the VCP gene was associated with the disease, WES has been used on many FALS samples and assisted in identifying seven further genes in the last 3 years.
Figure 1 Gene frequencies in ALS.
Abbreviations: ALS, amyotrophic lateral sclerosis; FALS, familial ALS.
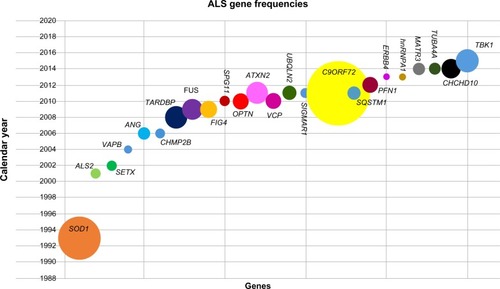
Table 1 Overview of key information available for ALS and FTDALS loci
To date, >22 ALS and four ALS + FTD (FTDALS) loci have been established, with the causative genes identified in the majority of cases. This review summarizes the current insights that have been gained from the four most common causes of FALS: SOD1, TARDBP, FUS, and C9ORF72. These genes have highlighted the roles of oxidative stress and RNA processing as contributory pathogenic mechanisms in ALS. In addition, rarer genetic variants have implicated additional biological pathways such as the ubiquitin–proteasome system (UPS), protein trafficking, and impaired cytoskeletal function. We have used the numbering of loci as described in the Online Mendelian Inheritance in Man Phenotypic Series for ALS (PS105400) and FTDALS (PS105550) in this review.Citation3
ALS1: Cu–Zn superoxide dismutase (SOD1)
Mutation of Cu–Zn superoxide dismutase 1 (SOD1) was the first described genetic cause for familial FALS.Citation4 The majority of SOD1 mutations are AD, with a highly penetrant pattern of inheritance, and are primarily associated with limb onset ALS. An exception to this rule is the D90A mutation, predominantly found in Scandinavian populations, where it is inherited in an AR manner. Frequency of SOD1 mutations varies depending on populations, from 23.5% in Scandinavia to 12% in Germany; mutations have also been identified in apparently sporadic cases.Citation5 The ALSoD database (http://alsod.iop.kcl.ac.uk)Citation6 reports that there are 183 mutations in SOD1 associated with disease (accessed November 2015), the majority of which are point mutations. Given that SOD1 encodes a 153 amino acid protein, this number is remarkable, with the mutations distributed throughout the gene and impacting upon a variety of domains within the protein. This is in contrast to some of the other ALS-associated mutations, which are more often located within a particular motif of the protein product, particularly as it is unclear whether all the reported SOD1 “mutations” are indeed pathogenic.Citation7,Citation8
The multiple mutations throughout the protein have also resulted in challenges determining how they are responsible for the disease phenotype. SOD1 is a ubiquitously expressed antioxidant protein, which catalyzes free radical superoxide to hydrogen peroxide and oxygen. As the majority of mutant proteins retain this enzymatic function, the pathogenicity is proposed to act through a toxic gain of function, although the precise nature of this toxicity remains to be determined. Many mutually compatible pathogenic mechanisms have been proposed including oxidative stress, excitotoxicity, protein aggregation, neuroinflammation, apoptosis, mitochondrial dysfunction, axonal transport dysregulation, and endoplasmic reticulum (ER) stress.Citation9 Mutant SOD1 proteins (mtSOD1) show variable states of metallation and disulfide bond formation, which leads to the ability of the demetalled and unfolded apoform to enter the intermembrane space of the mitochondrion, thereby causing mitochondrial dysfunction.Citation10 In addition, demetallation leads to increased instability and mtSOD1 shows a higher aggregation propensity than wild-type SOD1. More recently, mtSOD1, along with misfolded wild-type SOD1, has been shown to move from cell to cell and initiate a prion-like seeded aggregation of SOD1.Citation11,Citation12 While initial work demonstrated the propagation of misfolded protein in cell culture models, spinal homogenates from paralyzed mutant G93A SOD1 mice injected into 6-month-old G85R-SOD1:YFP mice (who do not usually get disease till 20 months) produced a progressive motor neuron disease within 3 months.Citation13
Normally, misfolded proteins are removed from the cell via the UPS. However, in SOD1-ALS, and also in SALS, the UPS has been shown to be impaired.Citation14,Citation15 In addition, mahogunin ring finger 1, an E3 ubiquitin ligase, which catalyzes monoubiquitination of proteins and marks them for degradation via a UPS-independent mechanism, has been shown to be decreased in the G93A mouse model. Interestingly, however, overexpression of this protein reduced SOD1 toxicity by suppressing the aggregation of SOD1. Thus, therapeutic strategies for ALS include increasing clearance of misfolded SOD1, and the heat-shock protein inducer, arimoclomol, is one such drug currently under investigation.Citation16
While initially oxidative stress was thought to be one of the primary mechanisms of mutant SOD1, the continued research on SOD1 pathogenic mechanisms has implicated UPS, protein aggregation, and degradation, as well as other aspects of protein trafficking. These pathways are also implicated by the discovery of additional FALS genes (“Protein trafficking and degradation-related genes” section).
ALS10: TAR DNA-binding protein (TARDBP)
The transactive response DNA-binding protein 43 (TDP-43) is encoded by TARDBP gene on chr1p36.22.Citation17 TARDBP is responsible for 4%–5% of FALS and nearly 1% of SALS.Citation18 Mutations in TARDBP are inherited in an AD manner and are associated with a classic ALS clinical phenotype. TARDBP encodes several isoforms of which TDP-43 is considered the most prevalent. TDP-43 is a heterogeneous nuclear ribonucleoprotein (hnRNP) with a nuclear localization signal (NLS) and nuclear export signal, which allows shuttling of the protein between the nucleus and cytoplasm. The TDP-43 protein contains three further domains, two RNA recognition motifs (RRM1 and RRM2), involved in RNA and DNA binding, and a glycine-rich domain, which is essential for interactions with other proteins and is the location of the majority of mutations.Citation19,Citation20
TDP-43 was initially identified as a transcription repressor that binds to TAR DNA in human immunodeficiency virus-1.Citation27 Subsequently, TDP-43 has been shown to play a role in RNA metabolism, including RNA transcription, alternative splicing, pre-microRNA processing, RNA transport, and messenger RNA (mRNA) stability.Citation22 TDP-43 has the ability to autoregulate its own gene expression by binding to the 3′ untranslated region (3′UTR) of its mRNA, leading to instability and decay.Citation23 TDP-43 also binds to UG-rich sequences in multiple mRNA sequences to regulate splicing.Citation24–Citation26 In addition, a novel function has recently been described, with TDP-43 able to repress the splicing of nonconserved exons known as cryptic exons.Citation27 Removal of TDP-43 allowed these cryptic exons to be incorporated into mRNA sequences, which subsequently disrupted translation and induced nonsense mediated decay. Finally, TDP-43 is also known to be a component of stress granules (SGs), although it is unclear whether this role contributes to neurodegeneration.Citation28
TDP-43 is a prominent protein in the characteristic ubiquitinated cytoplasmic inclusions found in patients with ALS and FTD.Citation29 Approximately 97% of patients with FALS and SALS are positive for TDP-43 inclusions in the motor cortex and spinal cord, thereby establishing TDP-43 as a major protein signature for disease, not just those carrying TARDBP mutations.Citation17,Citation30 The loss of TDP-43 nuclear localization in ALS is well documented, and the resulting splicing deficits in ALS cellular and animal models and in patient samples have been reported.Citation27,Citation31,Citation32 In addition to a loss of nuclear function, a cytoplasmic gain of function may also contribute to neurodegeneration. A mouse model with a mutation in the NLS of human TARDBP, thereby limiting TDP-43 to the cytoplasm, showed increased expression of transcription-related and chromatin assembly genes and histone 3′UTR processing genes.Citation33 Importantly, these transcriptional changes were not seen when an antisense oligomer was added to knockdown TDP-43 expression, thereby supporting a cytoplasmic toxic gain of function. Finally, similar to the prion-like propagation of disease described in SOD-ALS, there is also evidence that wild-type TDP-43 oligomers can spread horizontally from cell to cell via microvesicles, including the brain lysates of patients with ALS, as well as vertically along axons.Citation34 Thus, reducing the aggregation of these mutant proteins is becoming a more widely applicable therapeutic strategy.
ALS6: fused in sarcoma (FUS)
The FUS gene on chr16p11.2 was first identified as a fusion oncogene in liposarcoma. FUS belongs to the FET protein family and has been shown to be an hnRNP due to its involvement in the transcription process, transport, trafficking, alternative splicing, and microRNA processing. Similar to TDP-43, it is also present in SGs. Structurally, FUS is composed of 526 amino acids that form an N-terminal domain rich in glutamine–glycine–serine–tyrosine (QGSY), three arginine–glycine–glycine (RGG)-rich domains, an RRM, and a zinc finger motif, as well as nuclear export signal and NLS that enable nucleocytoplasmic shuttling of the protein.Citation35
Mutations in the FUS gene were initially identified in an AR Cape Verde family,Citation36 although subsequent screening established FUS to be causative also in AD ALS.Citation36,Citation37 FUS mutations account for ∼4% of FALS and 1% of SALS, with the majority of mutations located either within exons 3–6, encoding the QGSY-rich and first RGG region, or in exons 12–15, which encode the zinc finger domain, the other two RGG domains, and NLS.Citation35 While these in the C-terminal have been shown to be functional, those in exons 3–6 are more commonly found in SALS or do not always segregate with disease, suggesting incomplete penetrance or nonpathogenic variations.
Previously, depletion of RNA polymerase II from the nucleus had been shown to lead to an increase of cytoplasmic FUS, suggesting that FUS had a role in transcription.Citation38 It has subsequently been shown that FUS mediates the interaction between RNA polymerase II and the splicing factor U1 snRNP, thereby coupling transcription to splicing.Citation39 Mutations in FUS lead to mislocalization of both FUS and U1 snRNP to the cytoplasm,Citation40 and other RNA-binding proteins, including SMN1, hnRNPA1, and hnRNPA2, also colocalize in mtFUS aggregates.Citation41 The consequence of these mtFUS interactions includes dysregulated splicing and an increased binding of FUS with SMN, leading to a reduction in Gem bodies, thereby representing both a loss and a gain of function by the mtFUS.Citation42
FUS mutations may also confer pathogenicity via additional interactions. FUS has been shown to bind mRNAs and facilitate their transport down dendritesCitation43 and subsequently has been shown to bind to the polyA tail of AMPA receptor GluA1, regulating its stability, with the loss of FUS leading to a reduction of GluA1.Citation44 FUS has also been shown to translocate to the mitochondria through interaction with the mitochondrial chaperone heat shock protein 60 (HSP60) leading to mitochondrial damage.Citation45 Finally, mtFUS interacts with Pur-alpha in SGs and increases phosphorylation of the elongation initiation factor 2-alpha, consequently inhibiting protein synthesis.Citation46 However, the contribution of each of these interactions on disease pathogenesis remains to be determined.
FTDALS1: C9ORF72 (C9ORF72)
The most common cause of FALS to date is the expansion of an intronic GGGGCC repeat in C9ORF72. The region was originally identified through genome-wide association studies of SALS cases, as well as within the Finnish ALS population;Citation47,Citation48 while initial sequencing of the gene failed to identify any point mutations, targeted next-generation sequencing of the region established an intronic repeat region between the noncoding exons 1a and 1b.Citation49,Citation50 While healthy controls most commonly have <10 hexanucleotide repeats, individuals with ALS usually carry 400–2,000 repeats. The repeat expansion has been identified in 37.6% of FALS and 6.3% of SALS, as well as in up to 25.1% of familial FTD cases.Citation51 Therefore, it is not surprising that the most significant clinical phenotype associated with this genetic subtype is an increased incidence of a family history of FTD. In addition, there is evidence that there are more bulbar onset cases associated with C9ORF72-related ALS (up to 44%, compared to 25%–26% in non-C9ORF72 ALS), with some studies also reporting an earlier age of onset (by 1.8–5.0 years) and shorter disease duration (by 5.7–12.0 months).Citation52
The function of the C9ORF72 gene is currently being determined, though structural analysis has established that there is similarity to “differentially expressed in normal and neoplasic-like proteins”, which are GDP/GTP exchange factors regulating Rab-GTPases involved in vesicular trafficking.Citation53 Further work demonstrated how C9ORF72 colocalized with Rab proteins involved in autophagy and endosomal trafficking.Citation54 While the function is being established, several hypotheses have been proposed to explain how the intronic hexanucleotide repeat may cause neurodegeneration: 1) haploinsufficiency, 2) RNA toxicity, and 3) dipeptide repeat (DPR) protein toxicity.
Haploinsufficiency
Lower levels of C9ORF72 transcript were seen in patients with the repeat expansion compared to controls, and the haploinsufficiency hypothesis was supported when knockdown of the C9orf72 homolog in zebrafish caused an axonal degeneration.Citation55 In contrast, in a conditional C9orf72 knockout mouse, where C9orf72 was specifically ablated in neuronal cells, there was no evidence of a neurodegenerative phenotype.Citation56 However, a systematic investigation of the expression levels of the three C9ORF72 transcripts (variant 1 = exon 1a, 2–5; variant 2 = exon 1b, 2–11; variant 3 = exon 1a, 2–11) demonstrated significantly reduced expression of variants 1 and 2 in the cerebellum and frontal cortex of C9ORF72 expansion carriers, with a correlation between a higher level of expression of variant 1 and survival.Citation57 This suggests that antisense oligomer strategies should avoid reducing C9ORF72 expression levels.
RNA toxicity
RNA foci were identified to be located primarily in the nucleus and occasionally in the cytoplasm of motor neurons and were found to be composed of both sense and antisense RNA.Citation58 The presence of antisense RNA foci has been shown to be correlated with TDP-43 mislocalizationCitation59 but not DPRs.Citation58 The repeat sequence is thought to form G-quadruplex structures within the cell. Many RNA-binding proteins colocalize with the RNA foci, potentially sequestering them from the cell and disrupting their RNA-processing functions.Citation60,Citation61 This may underlie the significant dysregulation of RNA splicing that is seen in the presence of the expansion, where greater disruption is present in patients with a shorter survival.Citation62,Citation63 However, RNA foci have been observed in fibroblasts from asymptomatic patientsCitation60,Citation64 and in BAC transgenic mice containing an expanded allele; while RNA foci and DPRs recapitulate the neuropathology of ALS, there is no evidence of neurodegeneration.Citation65,Citation66 This is in contrast to a mouse expressing a 66-repeat G4C2 expansion specifically in the central nervous system, which exhibited neuropathological, behavioral, and motor deficits at 6 months.Citation67
DPR proteins
Finally, it was demonstrated that the GGGGCC repeat expansion was subject to repeat-associated non-ATG translation.Citation68 Both the sense and the antisense RNA are translated, forming DPR proteins composed of poly-GA, -GP, -GR, -PA, and -PR (with poly-GP generated from both antisense and sense RNA).Citation58,Citation69 These DPR proteins are found aggregated within the neuronal cytoplasmic inclusions and neuronal intranuclear inclusions in the motor cortex, cerebellum, hippocampus, and spinal cord, which also stain positive for ubiquitin and p62. Recently, antibodies raised against each of the DPR proteins have demonstrated that there is little correlation between DPR distribution/burden and clinical phenotype,Citation70,Citation71 which the authors suggest is evidence against DPR aggregation being a major pathogenic mechanism. This is in contrast to work using a Drosophila model, in which DPR expression caused neurodegeneration in the fly eye.Citation72
In summary, there appears to be growing evidence supporting some form of RNA dysregulation as a contributing mechanism to C9ORF72-ALS. However, while different cellular and animal models using different constructs are generating conflicting results on the contribution of each of the three hypotheses, the precise mechanisms still remain to be fully elucidated. Disruption of C9ORF72 protein function in endosomal trafficking may also be a contributory factor.
Other RNA-processing genes associated with ALS
Prior to the identification of TARDBP and FUS as ALS-associated genes, two RNA-processing genes had already been implicated in ALS: angiogenin (ANG) and senataxin (SETX). Subsequently, mutations in hnRNPA1 and matrin 3 (MATR3) have been identified through WES, and ataxin 2 (ATXN2) was identified as a risk factor.
ALS9: angiogenin (ANG)
Following the identification of the ANG single-nucleotide polymorphism rs11701 as overrepresented in ALS cases from Scotland and Ireland, screening of ANG identified seven missense mutations in 15 ALS cases, of which four were FALS (1.54%) and eleven were SALS (0.80%).Citation73 ANG is a member of the pancreatic ribonuclease superfamily and is neuroprotective, while in mtANG this ability is impaired.Citation74 While multiple mutations have been identified, p.K17I has not always shown disease segregation. However, a meta-analysis has demonstrated that Caucasian individuals carrying this allele have a 1.65 greater risk of ALS, which is increased to tenfold in FALS.Citation75 ANG has also been shown to induce the assembly of SGs.Citation76 Interestingly, this induction is inhibited by G-quadruplex structures, which are formed by the G4C2 C9ORF72 expanded repeat, thereby establishing a link between C9ORF72 and ANG.
ALS4: senataxin (SETX)
Mutations in SETX are associated with the juvenile onset of ALS with distal muscle weakness and absence of bulbar or sensory symptoms. Patients typically have a long and slow disease progression with a relatively normal life span.Citation77,Citation78 Rare, AD mutations in SETX occur in ALS, while recessive SETX mutations are associated with ataxia-oculomotor apraxia-2.Citation78 The mechanisms by which SETX variants lead to ALS is unknown; however, SETX encodes a DNA/RNA helicase protein proposed to play a role in DNA repair in response to oxidative stress. SETX also interacts with RNA-processing proteins regulating transcription and pre-mRNA processing suggesting that the cause of motor neuron degeneration through SETX mutations is as a result of abnormal RNA processing.Citation79
ALS13: ataxin 2 (ATXN2)
More than 36 repeats of CAG within ATXN2 causes spinocerebellar ataxia 2; however, intermediate repeats of 27–33 were found to strongly associate with ALS having established that ATXN2 modifies TDP-43 toxicity in yeast.Citation80 ATXN2 is an RNA-binding protein that is involved in RNA processing and localized to the ER, Golgi apparatus, and SGs; ATXN2 also interacts with FUS and intermediate expansions exacerbate the FUS mutant phenotype in cellular models.Citation81 A recent meta-analysis of >6,000 ALS and 7,000 controls has identified that repeat lengths of 25–28 were actually protective, whereas the significant risk was associated with CAG repeats of 31–33.Citation82 This finding is supported by an Italian study, where additionally <31 repeats were associated with spinal onset ALS and a shorter survival.Citation83
ALS20: heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)
Following the identification of hnRNPA1 and hnRNPA2B1 mutations as causative in multisystem proteinopathy families using exome sequencing, these genes were specifically analyzed in 212 FALS for which exome sequencing was available.Citation84 A single case was identified with a mutation in hnRNPA1. Interestingly, hnRNPA1 and A2/B1 are known interacting partners of TDP-43, and hnRNPA1 also interacts with ubiquilin-2.Citation85 In ALS motor neurons, there is a loss of intense hnRNPA1 nuclear staining, which also correlates with nuclear loss of TDP-43, although hnRNPA1 was not seen to colocalize with TDP-43 in the skein-like inclusions.Citation86 However, screening of 113 Italian FALS and 135 Dutch FALS and 1,084 Dutch SALS failed to find any hnRNPA1 mutations, suggesting that this is a very rare cause of FALS.
ALS21: matrin 3 (MATR3)
Exome sequencing of a large pedigree identified a mutation in the MATR3 gene; a previous family carrying a mutation in MATR3, originally diagnosed with AD distal, asymmetrical myopathy with vocal cord paralysis was reassessed and rediagnosed as having ALS.Citation87 Further screening of Italian and British ALS cases identified a further two mutations: one FALS and one SALS. MATR3 is an RNA/DNA-binding protein that interacts with TDP-43; while the p.S85C mutation enhances this interaction, two other mutations, p.F115C and p.T22A, do not. However, this difference may underlie the slow progression of the disease in the family carrying the p.S85C mutation. While no further mutations were found in 372 FALS cases from France, Taiwan, Australia, and French Canada, four mutations were found in apparent SALS cases (three in French Canadians and one in a Taiwanese).Citation88–Citation91
FET family genes
The TATA box-binding protein-associated factor 15 (TAF15) and Ewing sarcoma breakpoint region 1 (EWSR1) are RNA-binding proteins and form the FET family of proteins along with FUS. All three contain prion-like domains, a feature used to rank potential RNA-binding proteins as being involved in ALS following a functional yeast screen.Citation92 Screening of TAF15 identified five missense variants in 1,262 ALS cases, while screening of EWSR1 identified two potential mutations among 817 ALS cases.Citation93 While these variants were absent from controls, they were identified in patients with SALS, so segregation could not be demonstrated. However, both TAF15 and EWSR1 proteins show cytoplasmic mislocalization in SALS. Recently, whole-genome sequencing (WGS) identified an EWSR1 mutation in a set of monozygote twins disconcordant for ALS, suggesting that additional factors may influence disease.Citation94
Protein trafficking and degradation-related genes
From the identification of the first gene for AR ALS, ALS2, combined with the presence of characteristic ubiquitinated inclusions in ALS motor neurons, dysregulation of protein trafficking and protein degradation has been implicated in the disease process. Mutations in genes involved in endosomal transport include alsin (ALS2), vesicle-associated membrane protein (VAMP)-associated protein B (VAPB), chromatin modifying protein 2B (CHMP2B), and phosphoinositide 5-phosphatase (FIG4); those involved in the UPS include ubiquilin 2 (UBQLN2), sequestosome 1 (SQSTM1), and sigma nonopioid intracellular receptor 1 (SIGMAR1), and autophagy is primarily implicated by mutations in optineurin (OPTN), valosin-containing protein (VCP), and tank-binding kinase 1 (TBK1). There is some overlap between these three biological pathways, which have also been implicated in SOD1- and C9ORF72-related ALS.
ALS2: alsin (ALS2)
The ALS2 gene was originally identified through linkage analysis in consanguineous families from Tunisia and Saudi Arabia.Citation95,Citation96 The majority of the mutations lead to protein truncations, leading to a proposed loss of function. Alsin is thought to play a role in activating Rab5 GTPases. Rab5 is essential for endosomal trafficking, and in alsin knockout mice, neurons showed increased endosomal fusion and degradation but reduced mobility.Citation97 One of the endosome components is the AMPA receptor GluR2, levels of which are reduced in alsin knockout mice.Citation98
ALS8: vesicle-associated membrane protein (VAMP)-associated protein B (VAPB)
Linkage analysis of a large Brazilian family first established VAPB as a causative gene for ALS,Citation99 and the p.P56S mutation has been identified in multiple Brazilian pedigrees, indicative of a common founder.Citation100 Additional mutations have been reported, though not all the variations found segregated with disease.Citation101–Citation104 VAPB is a type II integral ER membrane protein, involved in intracellular trafficking and the unfolded protein responseCitation105 and in regulating ER–mitochondria interactions.Citation106 However, the p.P56S mutant protein was unable to initiate the unfolded protein response, altered calcium uptake into the mitochondria, and disrupted anterograde axonal transport of mitochondria.Citation107–Citation109
ALS17: chromatin modifying protein 2B (CHMP2B)
Mutations in CHMP2B were initially identified in two probable FALS and a further three SALS cases; the majority had a predominant lower motor neuron phenotype.Citation110,Citation111 CHMP2B is a component of the ESCRT-III endosomal trafficking system, sorting cargos into multivesicular bodies. More recently, four novel mutations were identified in apparently sporadic ALS cases, and these were located in the domain required to form the multivesicular bodies.Citation2 In cellular models, mutant CHMP2B led to the formation of large vacuoles and an increase in autophagy marker LC3-II, implicating dysregulation of autophagy as a mechanism in ALS.
ALS11: phosphoinositide 5-phosphatase (FIG4)
Mutations in FIG4 were originally identified as causative in Charcot–Marie–Tooth disease type 4J, though one family had a clinical phenotype resembling ALS. Screening of FALS and SALS cases identified nine variants, with six showing impaired function in yeast.Citation112 FIG4, also known as SAC3, regulates PI(3,5)P2 levels and thereby controls retrograde trafficking of endosomal vesicles to the Golgi. The mutant proteins showed loss of phosphatase activity, mislocalization, and inability to bind to the PI(3,5) P2 complex. Further screening in Italian and Taiwanese populations failed to find any novel variants, though only 80 SALS and 15 FALS were screened in each study.Citation113,Citation114 No pathological assessment was available on the mutation carriers; however, FIG4 was not shown to be mislocalized in SALS.Citation115
ALS15: ubiquilin 2 (UBQLN2)
UBQLN2 was identified by linkage analysis in a large multi-generational family. Screening of additional FALS cases with no male-to-male transmission found four further mutations; these were all located within the PXX repeat region of the protein.Citation116 Additional screening identified further variants adjacent or within the PXX repeat region.Citation117,Citation118 Mutations have been shown to disrupt the protein degradation pathway through defective binding to the proteasomeCitation119 and causing mislocalization of OPTN from Rab-11-positive endosomal vesicles,Citation120 as well as potentially impairing RNA metabolism, through loss of binding of UBQLN2 to hnRNP proteins, including hnRNPA1.Citation85
FTDALS3: sequestosome 1 (SQSTM1)
SQSTM1 or p62 is a ubiquitin-binding protein that plays a role in protein degradation via the proteasome and autophagy and is found within the characteristic ubiquitinated inclusions in patients with ALS. Screening of this gene found multiple mutations in both FALS and SALS cases.Citation121 Further mutations were found in patients with ALS, some in association with Paget disease of bone, which is also known to be caused by SQSTM1 mutations.Citation122,Citation123 In a zebrafish model, where endogenous SQSTM1 was knocked-down, the fish showed behavioral and axonal abnormalities, as well as disrupted autophagy, as demonstrated by increased mTOR levels.Citation124 Human SQSTM1 was able to rescue the phenotype, but the frequently found mutation, p.P392L, was unable to do so.
ALS16: sigma nonopioid intracellular receptor 1 (SIGMAR1)
Initially 3-UTR variants were identified within several AD FTDALS or FTD families and suggested that pathogenicity occurred through alteration of mRNA stability.Citation125 However, using homozygosity mapping, a missense mutation in SIGMAR1 was subsequently found to segregate in a large consanguineous family with AR juvenile ALS (ARJALS).Citation126 SIGMAR1 is an ER chaperone, a subunit of the ligand-regulated potassium channel, and enables mitochondrial calcium transport via the IP3 receptor; mutation of SIGMAR1 causes the formation of cytoplasmic aggregations, a reduction in ATP production and subsequent decrease in proteasome activity.Citation127 However, whether SIGMAR1 contributes to AD ALS is yet to be fully elucidated.
ALS12: optineurin (OPTN)
Mutations in OPTN were originally identified through homozygosity mapping of consanguineous Japanese AR ALS families, which identified a homozygous exonic deletion and a homozygous nonsense mutation.Citation128 Further screening of FALS cases identified two AD families heterozygous for a missense mutation. OPTN mediates its function through protein–protein interactions; it binds to ubiquitin and UBQLN2, is an autophagy receptor (facilitating the recruitment of cargos to autophagosomes), is required for Golgi organization (as demonstrated by the Golgi fragmentation seen in postmortem spinal motor neurons),Citation129 and also regulates NF-κB signaling.Citation130 Subsequent screening has identified additional heterozygote mutations in SALSCitation131 and AR ALS cases.Citation132,Citation133
ALS14: valosin-containing protein (VCP)
Exome sequencing of a four-generation Italian family initially suggested mutation of VCP as causative in ALS.Citation134 Subsequently, a further four variants were identified in FALS cases, thereby providing further evidence that VCP is associated with ALS. VCP is an AAA+ (extended family of ATPases associated with various cellular activities) ATPase protein involved in a variety of cellular functions including mediating the proteasomal degradation of ubiquitinated protein in multimeric complexes and the targeting of substrates to autophagosomes.Citation135 While screening failed to find any VCP mutations in some populations,Citation136–Citation138 potential mutations were found in others and also in SALS cases.Citation139,Citation140 VCP mutations are also associated with inclusion body myopathy with early onset Paget Disease and frontotemporal dementia (IBMPFD), and patient fibroblasts have shown mitochondrial uncoupling and a reduction in ATP production,Citation141 a feature also seen with SIGMAR1 mutations.
FTDALS4: tank-binding kinase (TBK1)
Mutations in TBK1 were initially identified through exome sequencing of 2,874 ALS cases; dominant variants were found in 1.097% of cases and loss of function mutations in 0.382%.Citation142 This was shortly followed by a second paper in which sequencing of 252 FALS cases identified nine loss of function and four missense mutations.Citation143 Mutations have subsequently been found in ALS, FTDALS, and FTD cases.Citation144,Citation145 TBK1 has a role in both innate immunity and NF-κB signaling, as well as in autophagy. TBK1 binds and phosphorylates ALS-related proteins OPTN and SQSTM1, whereas TBK1 mutants have been shown to no longer bind OPTN.Citation143
Impaired axonal transport and cytoskeletal dysfunction
Neurons are extremely large cells that require transport of organelles, proteins, and RNA from the cell body down the axons. Molecular motors such as kinesins and dynein guide these cargos to microtubules to mediate anterograde and retrograde transport, respectively. While mutation of the p150 dynactin subunit in mice generated a neurodegenerative phenotype, mutations in this gene were not found in human ALS.Citation146,Citation147 However, exome sequencing has identified several cytoskeletal genes with mutations reported to be causative in ALS.
ALS5: spatacsin (SPG11)
WES of two affected siblings from a nonconsanguineous family diagnosed with ARJALS identified only one gene, SPG11, in which variants were found in a compound heterozygous state.Citation148 The involvement of the hereditary spastic paraparesis gene, normally associated with HSP with thin corpus callosum, had previously been implicated by a candidate gene screening of SPG11, in which mutations in 10 families with ARJALS were identified.Citation149 While the exact function of the protein is unknown, iPSC-derived neuronal cells with SPG11 mutations demonstrated the protein colocalized with the cytoskeleton, and mutations caused axonal instability and impaired axonal transport.Citation150
ALS18: profilin 1 (PFN1)
Two multigenerational ALS families were determined to carry mutations in the PFN1 gene following WES.Citation151 Extending the screen to additional FALS cases identified a further three mutations in five FALS cases and a p.E117G variant that was identified at very low frequency in controls. Further screening of ALS cases identified additional mutations and the variant.Citation152–Citation155 A meta-analysis subsequently determined that the p.E117G was associated with ALS and proposed this variant as a risk factor.Citation156 The function of PFN1 is to convert monomeric actin to filamentous actin, and it is also found localized to SGs.Citation157 It has been demonstrated that PFN1 mutations destabilize the protein, thereby leading to a loss of function, while the mutant protein is misfolded, thereby leading to a gain of function through aberrant protein interactions.Citation158 However, the effect of the mutant protein on actin formation and SG dynamics is yet to be elucidated.
ALS22: tubulin alpha 4A (TUBA4A)
WES of 363 FALS index cases followed by rare variant burden identified five ALS cases with rare variants in TUBA4A; these included four missense mutations and one nonsense mutation, all of which were encoded in exon 4, in highly conserved amino acids, and absent from 4,300 exome variant server (EVS) controls.Citation159 While further sequencing of ALS cases only identified one further variant, functional studies demonstrated that the p.W407X nonsense mutant failed to localize to the microtubules, instead forming cytoplasmic inclusions, leading to disrupted microtubule assembly and stability, through a dominant-negative mechanism. Subsequent screening in the Chinese ALS population failed to identify any variants;Citation160 data from other populations will undoubtedly emerge as further WES experiments are completed.
Intermediate filament variants
Cytoskeletal dysfunction is further implicated in the pathogenesis of ALS through rare variants being identified in intermediate filament genes. Neurofilaments (light, medium, and heavy) are the major structural components of the neuronal cytoskeleton and are present within the characteristic ubiquitinated protein inclusions. Candidate gene screening identified rare insertion/deletion variants in the KSP repeat domains of neurofilament heavy (NEFH) gene in SALS cases,Citation161–Citation163 while a single frameshift mutation has been identified in peripherin (PRPH1).Citation164 However, the absence of mutations in known familial cases and ability to show segregation with disease has reduced the creditability of these genes as ALS loci.
Additional loci
Several additional ALS loci have been identified. Two loci are yet to have their associated genes identified: ALS3 on chr18q21 and ALS7 on chr20q13.Citation165,Citation166 A further two genes have been identified in FALS pedigrees, though their functional effects are currently predicted to disrupt neuronal development and mitochondrial function.
ALS19: Erb-b2 receptor tyrosine kinase 4 (ERBB4)
WGS of a Japanese AD ALS family identified a missense mutation in ERBB4.Citation167 Additional screening identified the same mutation in an unrelated Canadian family and a further mutation in a SALS case. ERBB4 is a receptor tyrosine kinase that is activated by neuregulin, resulting in autophosphorylation of the C-terminal. Mutations in ERBB4 reduced the level of autophosphorylation. ERBB4 was found to localize to C-boutons, arising from interneurons, which synapse with spinal motor neurons.Citation168 Interestingly, C-boutons are not found in oculomotor neurons, which are spared in ALS, while increases in neuregulin levels in C-boutons increase during disease progression of the SOD1 G93A transgenic mice.
FTDALS2: coiled-coil helix coiled-coil helix domain-containing protein 10 (CHCHD10)
CHCHD10 was initially associated with ALS through WES of a family exhibiting clinical features including ALS, FTD, cerebellar ataxia, and myopathy.Citation169 This led to ALS and ALS-FTD families being screened. Several additional mutations were found,Citation170,Citation171 though it became evident that the p.P34S mutation was nonpathogenic, as it was also found at similar frequencies in controls.Citation172 The function of CHCHD10 is unknown; it localizes to the mitochondria. Fibroblasts from family members of the original pedigree showed multiple mitochondrial DNA deletions, respiratory chain defects, and structurally abnormal mitochondria, suggesting that CHCHD10 may have a role in the respiratory chain and/or in mitochondrial genome stability.Citation169 This has been supported by additional work by Genin et al,Citation173 which found not only a loss of cristae and mitochondrial genome repair in patient fibroblasts but also a failure of apoptosis due to an inability to release cytochrome C.
Conclusion
ALS genetics is having a significant impact on our understanding of the disease and the mechanisms implicated in neurodegeneration. The majority of genes encode proteins involved in RNA processing and the protein degradation pathways, UPS, and autophagy. However, neither of these nor the other pathways implicated work in isolation but impact on other cellular processes. The proposed mechanisms are mutually compatible, and it is most likely that multiple dysregulated pathways contribute to the loss of motor neurons. This is clearly demonstrated by TDP-43, an RNA-binding protein, that is mislocalized from the nucleus, thereby causing loss of nuclear function, and is aggregated in the cytoplasm as a component of the characteristic ubiquitinated inclusions.
Along with multiple genetic causes, it is clear that these genes are also implicated in additional disorders, not only in other neurodegenerative disorders such as FTD, HSP, and ataxia, but also in myopathies, Paget disease of bone, and glaucoma (). The use of WES or WGS, in projects such as the 100,000 Genomes Project in the UK (www.genomicsengland.co.uk), or Project Mine among the International ALS community (www.projectmine.com), will potentially enable a greater understanding of why mutations in a gene in one family present with a specific clinical phenotype, while another family shows a different disease.
Table 2 Clinical phenotypes also associated with ALS genes
WES and WGS will also further our understanding of the impact of oligogenic inheritance in ALS. While family pedigrees clearly show inheritance in an AD manner for classical ALS, the move away from analyzing a single gene at a time has highlighted evidence of mutations in multiple ALS genes in some patients.Citation2 Screening of a large ALS cohort demonstrated that 14% of FALS and 2.6% of SALS cases had more than one potential pathogenic mutation in a known ALS gene, and these cases had a significantly earlier onset of disease.Citation174 This also highlights the fact that apparent SALS cases also carry genetic mutations, as has been evidenced by the identification of de novo mutations in SALS cases following WES of trios comprising of an ALS patient and their two unaffected parents.Citation175,Citation176 While in some cases, these may actually be rare AR mutations, additional WES and WGS sequencing of cases will allow the genetic contribution in SALS, estimated to be 61%,Citation177 to be elucidated.
Acknowledgments
JK and RW are funded by STRENGTH. This is an EU Joint Programme – Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND – www.jpnd.eu: Belgium, The National Funds for Scientific Research (F.R.S. FNRS); France, Agence Nationale de la Recherche (ANR); Germany, Bundesministerium für Bildung und Forschung (BMBF); Italy, Ministero della Salute; Italy, Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR); The Netherlands, The Netherlands Organisation for Health Research and Development (ZonMw); Sweden, Swedish Research Council (VR); Switzerland, Schweizerischer Nationalfonds zur Förderung der wissenschaftlichen Forschung (SNF); and United Kingdom, Medical Research Council (MRC).
JK is also funded by SOPHIA. This is an EU Joint Programme – Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND – www.jpnd.eu: France, Agence Nationale de la Recherche (ANR); Germany, Bundesministerium für Bildung und Forschung (BMBF); Ireland, Health Research Board (HRB); Italy, Ministero della Salute; The Netherlands, The Netherlands Organisation for Health Research and Development (ZonMw); Poland, Narodowe Centrum Bada i Rozwoju; Portugal, Fundação a Ciência e a Tecnologia; Spain, Ministerio de Ciencia e Innovación; Switzerland, Schweizerischer Nationalfonds zur Förderung der wissenschaftlichen Forschung (SNF); Turkey, Tübitak; and United Kingdom, Medical Research Council (MRC).
JK receives funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under the EuroMOTOR project, grant agreement no 259867 and from the European Horizon 2020 research and innovation program under grant agreement no 633413.
AAS is funded by a PhD Scholarship from the University of Dhammam, Saudi Arabia.
Disclosure
The authors report no conflicts of interest in this work.
References
- Cooper-KnockJJenkinsTShawPJClinical and molecular aspects of motor neuron diseaseColloq Series Genomic Mol Med20132160
- van BlitterswijkMvan EsMAHennekamEAEvidence for an oligogenic basis of amyotrophic lateral sclerosisHum Mol Genet2012213776378422645277
- Online Mendelian Inheritance in Man, OMIM®McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins UniversityBaltimore, MD, USA Available at: http://omim.org/Accessed March 16, 2016
- RosenDRSiddiqueTPattersonDMutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosisNature199336259628446170
- AndersenPMAmyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase geneCurr Neurol Neurosci Rep20066374616469270
- AbelOPowellJFAndersenPMAl-ChalabiAALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis geneticsHum Mutat2012331345135122753137
- FelbeckerACamuWValdmanisPNFour familial ALS pedigrees discordant for two SOD1 mutations: are all SOD1 mutations pathogenic?J Neurol Neurosurg Psychiatry20108157257720460594
- MarangiGTraynorBJGenetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challengesBrain Res20151607759325316630
- KaurSJMcKeownSRashidSMutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosisGene2016577210911826657039
- ShengYChattopadhyayMWhiteleggeJValentineJSSOD1 aggregation and ALS: role of metallation states and disulfide statusCurr Top Med Chem2012122560257223339308
- GradLIPokrishevskyESilvermanJMCashmanNRExosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfoldingPrion2014833133525551548
- MunchCBertolottiASelf-propagation and transmission of misfolded mutant SOD1: prion or prion-like phenomenon?Cell Cycle201110171121471733
- AyersJIFromholtSEO’NealVMDiamondJHBorcheltDRPrion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathwaysActa Neuropathol2016131110311426650262
- KabashiEAgarJNStrongMJDurhamHDImpaired proteasome function in sporadic amyotrophic lateral sclerosisAmyotroph Lateral Scler20121336737122632443
- KabashiEDurhamHDFailure of protein quality control in amyotrophic lateral sclerosisBiochim Biophys Acta200617621038105016876390
- KalmarBLuCHGreensmithLThe role of heat shock proteins in amyotrophic lateral sclerosis: the therapeutic potential of ArimoclomolPharmacol Ther2014141405423978556
- SreedharanJBlairIPTripathiVBTDP-43 mutations in familial and sporadic amyotrophic lateral sclerosisScience20083191668167218309045
- MillecampsSSalachasFCazeneuveCSOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlationsJ Med Genet20104755456020577002
- BaralleMBurattiEBaralleFEThe role of TDP-43 in the pathogenesis of ALS and FTLDBiochem Soc Trans2013411536154024256250
- Lagier-TourenneCPolymenidouMClevelandDWTDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegenerationHum Mol Genet201019R46R6420400460
- IgnatiusSHWuFHarrichDGarciamartinezLFGaynorRBCloning and characterization of a novel cellular protein, Tdp-43, that binds to human-immunodeficiency-virus type-1 Tar DNA-sequence motifsJ Virol199569358435967745706
- ScotterELChenHJShawCETDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targetsNeurotherapeutics20151251551825761971
- AyalaYMDe ContiLAvendano-VazquezSETDP-43 regulates its mRNA levels through a negative feedback loopEMBO J20113027728821131904
- PolymenidouMLagier-TourenneCHuttKRLong pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43Nat Neurosci20111445946821358643
- SephtonCFCenikCKucukuralAIdentification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexesJ Biol Chem20112861204121521051541
- XiaoSSanelliTDibSRNA targets of TDP-43 identified by UV-CLIP are deregulated in ALSMol Cell Neurosci20114716718021421050
- LingJPPletnikovaOTroncosoJCWongPCTDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTDScience201534965065526250685
- AulasAVande VeldeCAlterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS?Front Cell Neurosci2015942326557057
- NeumannMSampathuDMKwongLKUbiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosisScience200631413013317023659
- QinHLimLZWeiYSongJTDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNAProc Natl Acad Sci U S A2014111186191862425503365
- HighleyJRKirbyJJansweijerJALoss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neuronesNeuropathol Appl Neurobiol20144067068524750229
- De ContiLAkinyiMVMendoza-MaldonadoRRomanoMBaralleMBurattiETDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathwaysNucleic Acids Res2015438990900526261209
- Amlie-WolfARyvkinPTongRTranscriptomic changes due to cytoplasmic TDP-43 expression reveal dysregulation of histone transcripts and nuclear chromatinPLoS One201510e014183626510133
- FeilerMSStrobelBFreischmidtATDP-43 is intercellularly transmitted across axon terminalsJ Cell Biol201521189791126598621
- DengHGaoKJankovicJThe role of FUS gene variants in neurodegenerative diseasesNat Rev Neurol20141033734824840975
- KwiatkowskiTJJrBoscoDALeclercALMutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosisScience20093231205120819251627
- VanceCRogeljBHortobagyiTMutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6Science20093231208121119251628
- ZinsznerHSokJImmanuelDYinYRonDTLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttlingJ Cell Sci1997110pt 15174117509264461
- YuYReedRFUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNPProc Natl Acad Sci U S A20151128608861326124092
- YuYChiBXiaWU1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafishNucleic Acids Res2015433208321825735748
- TakanashiKYamaguchiAAggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formationBiochem Biophys Res Commun201445260060725173930
- SunSLingSCQiuJALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNPNat Commun20156617125625564
- FujiiRTakumiTTLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spinesJ Cell Sci20051185755576516317045
- UdagawaTFujiokaYTanakaMFUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilizationNat Commun20156709825968143
- DengJYangMChenYFUS interacts with HSP60 to promote mitochondrial damagePLoS Genet201511e100535726335776
- Di SalvioMPiccinniVGerbinoVPur-alpha functionally interacts with FUS carrying ALS-associated mutationsCell Death Dis20156e1943
- ShatunovAMokKNewhouseSChromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association studyLancet Neurol2010998699420801717
- LaaksovirtaHPeuralinnaTSchymickJCChromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association studyLancet Neurol2010997898520801718
- RentonAEMajounieEWaiteAA hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTDNeuron20117225726821944779
- DeJesus-HernandezMMackenzieIRBoeveBFExpanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALSNeuron20117224525621944778
- MajounieERentonAEMokKFrequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional studyLancet Neurol20121132333022406228
- Cooper-KnockJKirbyJHighleyRShawPJThe spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosisNeurotherapeutics20151232633925731823
- LevineTPDanielsRDGattaATWongLHHayesMJThe product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFsBioinformatics20132949950323329412
- FargMASundaramoorthyVSultanaJMC9ORF72, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, regulates endosomal traffickingHum Mol Genet2014233579359524549040
- CiuraSLattanteSLe BerILoss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosisAnn Neurol20137418018723720273
- KoppersMBlokhuisAMWestenengHJC9orf72 ablation in mice does not cause motor neuron degeneration or motor deficitsAnn Neurol20157842643826044557
- van BlitterswijkMGendronTFBakerMCNovel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72Acta Neuropathol201513086387626437865
- GendronTFBieniekKFZhangYJAntisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALSActa Neuropathol201312682984424129584
- Cooper-KnockJHigginbottomAStopfordMJAntisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathyActa Neuropathol2015130637525943887
- Cooper-KnockJWalshMJHigginbottomASequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansionsBrain20141372040205124866055
- LeeYBChenHJPeresJNHexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxicCell Rep201351178118624290757
- Cooper-KnockJBuryJJHeathPRC9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosisPLoS One201510e012737626016851
- PrudencioMBelzilVVBatraRDistinct brain transcriptome profiles in C9orf72-associated and sporadic ALSNat Neurosci2015181175118226192745
- Lagier-TourenneCBaughnMRigoFTargeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degenerationProc Natl Acad Sci U S A2013110E4530E453924170860
- PetersOMCabreraGTTranHHuman C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic miceNeuron20158890290926637797
- O’RourkeJGBogdanikLMuhammadAKC9orf72 BAC transgenic mice display typical pathologic features of ALS/FTDNeuron20158889290126637796
- ChewJGendronTFPrudencioMNeurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficitsScience20153481151115425977373
- MoriKWengSMArzbergerTThe C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALSScience20133391335133823393093
- MoriKArzbergerTGrasserFABidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteinsActa Neuropathol201312688189324132570
- MackenzieIRFrickPGrasserFAQuantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriersActa Neuropathol201513084586126374446
- DavidsonYRobinsonACLiuXNeurodegeneration in fronto-temporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP-43 pathology and not associated with aggregated forms of dipeptide repeat proteinsNeuropathol Appl Neurobiol Epub2015115
- MizielinskaSGronkeSNiccoliTC9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteinsScience20143451192119425103406
- GreenwayMJAndersenPMRussCANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosisNat Genet20063841141316501576
- SubramanianVCrabtreeBAcharyaKRHuman angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/path finding and survival of motor neuronsHum Mol Genet20081713014917916583
- PanLDengXDingDLengHZhuXWangZAssociation between the Angiogenin (ANG) K17I variant and amyotrophic lateral sclerosis risk in Caucasian: a meta-analysisNeurol Sci201536122163216826255299
- IvanovPO’DayEEmaraMMWagnerGLiebermanJAndersonPG-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragmentsProc Natl Acad Sci U S A2014111182011820625404306
- ChenYZBennettCLHuynhHMDNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4)Am J Hum Genet2004741128113515106121
- HiranoMQuinziiCMMitsumotoHSenataxin mutations and amyotrophic lateral sclerosisAmyotroph Lateral Scler20111222322721190393
- Skourti-StathakiKProudfootNJGromakNHuman senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent terminationMol Cell20114279480521700224
- EldenACKimHJHartMPAtaxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALSNature20104661069107520740007
- FargMASooKYWarraichSTSundaramoorthyVBlairIPAtkinJDAtaxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosisHum Mol Genet20132271772823172909
- NeuenschwanderAGThaiKKFigueroaKPPulstSMAmyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysisJAMA Neurol2014711529153425285812
- BorgheroGPugliattiMMarrosuFITALSGEN and SAR-DINALS consortiaATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestryNeurobiol Aging2015362906.e12906.e15
- KimHJKimNCWangYDMutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALSNature201349546747323455423
- GilpinKMChangLMonteiroMJALS-linked mutations in ubiquilin-2 or hnRNPA1 reduce interaction between ubiquilin-2 and hnRNPA1Hum Mol Genet2015242565257725616961
- HondaHHamasakiHWakamiyaTLoss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusionsNeuropathology201535374325338872
- JohnsonJOPioroEPBoehringerAITALSGEN ConsortiumMutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosisNat Neurosci20141766466624686783
- FifitaJAWilliamsKLMcCannEPMutation analysis of MATR3 in Australian familial amyotrophic lateral sclerosisNeurobiol Aging2015361602.e11602.e12
- LinKPTsaiPCLiaoYCMutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosisNeurobiol Aging2015362005.e12005.e4
- LeblondCSGan-OrZSpiegelmanDReplication study of MATR3 in familial and sporadic amyotrophic lateral sclerosisNeurobiol Aging201637209.e1721
- MillecampsSDe SeptenvilleATeyssouEFrench research network on FTD FTD-ALSGenetic analysis of matrin 3 gene in French amyotrophic lateral sclerosis patients and frontotemporal lobar degeneration with amyotrophic lateral sclerosis patientsNeurobiol Aging201435122882.e1315
- CouthouisJHartMPShorterJA yeast functional screen predicts new candidate ALS disease genesProc Natl Acad Sci U S A2011108208812089022065782
- CouthouisJHartMPErionREvaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosisHum Mol Genet2012212899291122454397
- Meltz SteinbergKNicholasTJKoboldtDCYuBMardisEPamphlettRWhole genome analyses reveal no pathogenetic single nucleotide or structural differences between monozygotic twins discordant for amyotrophic lateral sclerosisAmyotroph Lateral Scler Frontotemporal Degener20151638539225960086
- HadanoSHandCKOsugaHA gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2Nat Genet20012916617311586298
- YangYHentatiADengHXThe gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosisNat Genet20012916016511586297
- LaiCXieCShimHChandranJHowellBWCaiHRegulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsinMol Brain200922319630956
- LaiCXieCMcCormackSGAmyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor traffickingJ Neurosci200626117981180617093100
- NishimuraALMitne-NetoMSilvaHCA mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosisAm J Hum Genet20047582283115372378
- NishimuraALAl-ChalabiAZatzMA common founder for amyo-trophic lateral sclerosis type 8 (ALS8) in the Brazilian populationHum Genet200511849950016187141
- ChenHJAnagnostouGChaiACharacterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosisJ Biol Chem2010285402664028120940299
- KabashiEEl OussiniHBercierVInvestigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosisHum Mol Genet2013222350236023446633
- IngreCPintoSBirveANo association between VAPB mutations and familial or sporadic ALS in Sweden, Portugal and IcelandAmyotroph Lateral Scler Frontotemporal Degener20131462062723971766
- van BlitterswijkMvan EsMAKoppersMVAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patientNeurobiol Aging2012332950.e12950.e4
- LevSBen HalevyDPerettiDDahanNThe VAP protein family: from cellular functions to motor neuron diseaseTrends Cell Biol20081828229018468439
- StoicaRDe VosKJPaillussonSER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43Nat Commun20145399624893131
- De VosKJMorotzGMStoicaRVAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasisHum Mol Genet2012211299131122131369
- MorotzGMDe VosKJVagnoniAAckerleySShawCEMillerCCAmyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondriaHum Mol Genet2012211979198822258555
- KanekuraKNishimotoIAisoSMatsuokaMCharacterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8)J Biol Chem2006281302233023316891305
- ParkinsonNIncePGSmithMOMRC Proteomics in ALS StudyFReJA ConsortiumALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B)Neurology2006671074107716807408
- CoxLEFerraiuoloLGoodallEFMutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS)PLoS One20105e987220352044
- ChowCYLandersJEBergrenSKDeleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALSAm J Hum Genet200984858819118816
- TsaiCPSoongBWLinKPTuPHLinJLLeeYCFUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALSNeurobiol Aging2011323553.e1321
- VerdianiSOrigonePGeroldiAThe FIG4 gene does not play a major role in causing ALS in Italian patientsAmyotroph Lateral Scler Frontotemporal Degener20131422822923336365
- KonTMoriFTanjiKALS-associated protein FIG4 is localized in Pick and Lewy bodies, and also neuronal nuclear inclusions, in polyglutamine and intranuclear inclusion body diseasesNeuropathology201434192623888880
- DengHXChenWHongSTMutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementiaNature2011477736321121521857683
- WilliamsKLWarraichSTYangSUBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosisNeurobiol Aging201233102527.e310
- GelleraCTilocaCDel BoRSLAGEN ConsortiumUbiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and fronto-temporal dementiaJ Neurol Neurosurg Psychiatry20138418318723138764
- ChangLMonteiroMJDefective proteasome delivery of polyubiquitinated proteins by ubiquilin-2 proteins containing ALS mutationsPLoS One201510e013016226075709
- OsakaMItoDYagiTNiheiYSuzukiNEvidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosisHum Mol Genet2015241617162925398946
- FectoFYanJVemulaSPSQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosisArch Neurol2011681440144622084127
- TeyssouETakedaTLebonVMutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathologyActa Neuropathol201312551152223417734
- KwokCTMorrisAde BellerocheJSSequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDBEur J Hum Genet20142249249623942205
- LattanteSde CalbiacHLe BerIBriceACiuraSKabashiESqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLDHum Mol Genet2015241682169025410659
- LutyAAKwokJBDobson-StoneCSigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron diseaseAnn Neurol20106863964921031579
- Al-SaifAAl-MohannaFBohlegaSA mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosisAnn Neurol20117091391921842496
- FukunagaKShinodaYTagashiraHThe role of SIGMAR1 gene mutation and mitochondrial dysfunction in amyotrophic lateral sclerosisJ Pharmacol Sci2015127364125704016
- MaruyamaHMorinoHItoHMutations of optineurin in amyotrophic lateral sclerosisNature201046522322620428114
- KamadaMIzumiYAyakiTClinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutationNeuropathology201434647023889540
- BansalMSwarupGBalasubramanianDFunctional analysis of optineurin and some of its disease-associated mutantsIUBMB Life20156712012825855473
- van BlitterswijkMvan VughtPWvan EsMANovel optineurin mutations in sporadic amyotrophic lateral sclerosis patientsNeurobiol Aging2012331016.e11016.e7
- BeeldmanEvan der KooiAJde VisserMvan MaarleMCvan RuissenFBaasFDutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutationsAmyotroph Lateral Scler Frontotemporal Degener20151641041126203661
- GoldsteinONayshoolONefussyBOPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotesNeurology201686544645326740678
- JohnsonJOMandrioliJBenatarMITALSGEN ConsortiumExome sequencing reveals VCP mutations as a cause of familial ALSNeuron20106885786421145000
- MeyerHWeihlCCThe VCP/p97 system at a glance: connecting cellular function to disease pathogenesisJ Cell Sci20141273877388325146396
- MillerJWSmithBNToppSDAl-ChalabiAShawCEVanceCMutation analysis of VCP in British familial and sporadic amyotrophic lateral sclerosis patientsNeurobiol Aging201233112721.e12
- WilliamsKLSolskiJANicholsonGABlairIPMutation analysis of VCP in familial and sporadic amyotrophic lateral sclerosisNeurobiol Aging2012334837:e713
- TilocaCRattiAPensatoVSLAGEN ConsortiumMutational analysis of VCP gene in familial amyotrophic lateral sclerosisNeurobiol Aging2012333630.e12
- KoppersMvan BlitterswijkMMVlamLVCP mutations in familial and sporadic amyotrophic lateral sclerosisNeurobiol Aging2012334837.e713
- AbramzonYJohnsonJOScholzSWValosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosisNeurobiol Aging20123392231.e12231.e6
- BartolomeFWuHCBurchellVSPathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levelsNeuron201378576423498975
- CirulliETLasseigneBNPetrovskiSExome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathwaysScience20153471436144125700176
- FreischmidtAWielandTRichterBHaploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementiaNat Neurosci20151863163625803835
- GijselinckIVan MosseveldeSvan der ZeeJBELNEU ConsortiumLoss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohortNeurology2015852116212526581300
- Le BerIDe SeptenvilleAMillecampsSFrench Clinical and Genetic Research Network on FTLD/FTLD-ALSTBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohortsNeurobiol Aging2015363116.e53116.e8
- Ahmad-AnnuarAShahPHafezparastMNo association with common Caucasian genotypes in exons 8, 13 and 14 of the human cytoplasmic dynein heavy chain gene (DNCHC1) and familial motor neuron disordersAmyotroph Lateral Scler Other Motor Neuron Disord2003415015713129801
- Vilarino-GuellCWiderCSoto-OrtolazaAICharacterization of DCTN1 genetic variability in neurodegenerationNeurology2009722024202819506225
- DaoudHZhouSNoreauAExome sequencing reveals SPG11 mutations causing juvenile ALSNeurobiol Aging2012334839.e5e9
- OrlacchioABabaliniCBorrecaASPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosisBrain201013359159820110243
- Perez-BranguliFMishraHKProtsIDysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegiaHum Mol Genet2014234859487424794856
- WuCHFalliniCTicozziNMutations in the profilin 1 gene cause familial amyotrophic lateral sclerosisNature201248849950322801503
- SmithBNVanceCScotterELNovel mutations support a role for Profilin 1 in the pathogenesis of ALSNeurobiol Aging20153631602.e1727
- IngreCLandersJERizikNA novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohortsNeurobiol Aging2013341708.e17011708.e1706
- TilocaCTicozziNPensatoVSLAGEN ConsortiumScreening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in fronto-temporal dementiaNeurobiol Aging20133451517.e910
- van BlitterswijkMBakerMCBieniekKFProfilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and fronto-temporal dementiaAmyotroph Lateral Scler Frontotemporal Degener20131446346923634771
- FrattaPCharnockJCollinsTProfilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosisJ Neurol Neurosurg Psychiatry20148550650824309268
- FigleyMDBieriGKolaitisRMTaylorJPGitlerADProfilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamicsJ Neurosci2014348083809724920614
- BoopathySSilvasTVTischbeinMStructural basis for mutation-induced destabilization of profilin 1 in ALSProc Natl Acad Sci U S A20151127984798926056300
- SmithBNTicozziNFalliniCExome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALSNeuron20148432433125374358
- LiJHeJTangLTUBA4A may not be a significant genetic factor in Chinese ALS patientsAmyotroph Lateral Scler Fronto temporal Degener2015171–213
- FiglewiczDAKrizusAMartinoliMGVariants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosisHum Mol Genet19943175717617849698
- Al-ChalabiAAndersenPMNilssonPDeletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosisHum Mol Genet199981571649931323
- TomkinsJUsherPSladeJYNovel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS)Neuroreport19989396739709875737
- Gros-LouisFLariviereRGowingGA frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosisJ Biol Chem2004279459514595615322088
- HandCKKhorisJSalachasFA novel locus for familial amyotrophic lateral sclerosis, on chromosome 18qAm J Hum Genet20027025125611706389
- SappPCHoslerBAMcKenna-YasekDIdentification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosisAm J Hum Genet20037339740312858291
- TakahashiYFukudaYYoshimuraJERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19Am J Hum Genet20139390090524119685
- Gallart-PalauXTarabalOCasanovasANeuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to alpha-motoneurons and altered during motoneuron diseasesFASEB J2014283618363224803543
- BannwarthSAit-El-MkademSChaussenotAA mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvementBrain20141372329234524934289
- Dols-IcardoONebotIGorostidiADementia Genetics Spanish Consortium (DEGESCO)Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from SpainBrain2015138e40026152333
- JohnsonJOGlynnSMGibbsJRMutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosisBrain2014137e31125261972
- MarroquinNStranzSMullerKScreening for CHCHD10 mutations in a large cohort of sporadic ALS patients: no evidence for pathogenicity of the P.34S variantBrain Epub2015911
- GeninECPlutinoMBannwarthSCHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosisEMBO Mol Med201585872
- CadyJAllredPBaliTAmyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genesAnn Neurol20157710011325382069
- SteinbergKMYuBKoboldtDCMardisERPamphlettRExome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALSSci Rep20155912425773295
- ChesiAStaahlBTJovicicAExome sequencing to identify de novo mutations in sporadic ALS triosNat Neurosci20131685185523708140
- Al-ChalabiAFangFHanbyMFAn estimate of amyotrophic lateral sclerosis heritability using twin dataJ Neurol Neurosurg Psychiatry2010811324132620861059
- MillecampsSSalachasFCazeneuveCSOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlationsJ Med Genet20104755456020577002
- KirbyJGoodallEFSmithWBroad clinical phenotypes associated with TAR-DNA binding protein (TARDBP) mutations in amyotrophic lateral sclerosisNeurogenetics20101121722519760257