Abstract
Neuromyelitis optica or Devic disease is an inflammatory disorder of the central nervous system. It is caused by antibodies that attack aquaporin 4 water channels in the cell membrane of astrocytic foot processes at the blood brain barrier. It can involve the optic nerve, the spinal cord and beyond. Here we review its pathophysiology, clinical features, and therapy.
Introduction
Neuromyelitis optica (NMO) or Devic disease is an inflammatory disorder of the central nervous system characterized by episodes of optic neuritis and transverse myelitis. The condition was defined by Dr Eugene Devic, a physician practising in Lyon, France, in 1894.Citation1–Citation3 It was subsequently thought that there was no clinical involvement beyond the optic nerve and spinal cord and that the disorder might be, where monophasic, a variant of acute disseminated encephalomyelitis (ADEM) and, where relapsing, a variant of classic multiple sclerosis (MS). However symptoms in other neurological axes have been repeatedly reported in NMO cases. NMO is also currently considered as an entity distinct from classic multiple sclerosis because it has clinical manifestations, antibodies, radiological and pathological features that differ from MS.Citation4 The prognosis of NMO has been poor without appropriate treatment as many patients were left with severe disability and many died within weeks or months.Citation5 Early recognition and treatment of NMO, therefore, are essential. In this article we review the pathogenesis and clinical manifestations of NMO with an emphasis on the optic nerve and spinal cord involvement, and beyond. Clinicopathological correlation and treatment are also discussed.
Optic nerve and spinal cord involvement
Pathophysiology
In NMO, the involvement of the spinal cord and optic nerves or chiasm has been commonly reported but there is increasing evidence that isolated or multiple cerebral lesions can occur.Citation6 NMO is characterized by neuronal destruction with glial (astrocyte and oligodendrocyte) and connective tissue reaction in the spinal cord and optic nerve or chiasm.Citation6,Citation7 In the acute phase, leukocytes, particularly neutrophils and eosinophils, infiltrate perivascular areas.Citation7,Citation8 Demyelination, axonal loss, and macrophage infiltration are commonly seen in the optic nerve and the grey and white matter of the spinal cord.Citation6–Citation12 The changes vary in degree from lacunar degeneration to frank necrosis which has been observed particularly in the spinal cord.Citation7,Citation13 Small foci of necrosis have been demonstrated in the optic nerve.Citation7 In severely affected cases resulting in blindness or paraplegia, swelling and softening of the affected parenchyma has been identified at an early stage in the evolution of an attack.Citation6,Citation10,Citation13 The inflammation usually occurs over several cord segments.Citation6,Citation9,Citation10 The spinal cord may be affected partially or entirely in cross-section and the inflammation may extend to the pial surface.Citation6,Citation9,Citation10 In some chronic cord lesions, a cyst or a long cavity is formed.Citation6,Citation10,Citation14 Astrocytosis and atrophy develop in later stages,Citation7,Citation10,Citation14,Citation15 up to 10 months after the onset in one study.Citation16 The walls of medium-sized vessels in the cord lesions appear thick and hyalinized, giving a ‘rubber band’ appearance.Citation6 Studies have revealed hyalinization of small arteries and veins as they enter affected parenchyma with the lumen greatly obliterated.Citation10,Citation13 The sclerosis of the vessels is located in close proximity to the necrotic cord area and also observed in degenerated nerve roots.Citation13 However the vascular changes have not been observed in every case.Citation16 It is notable that patients in postmortem studies were severely affected and their pathological findings seem to be at the extreme end of the spectrum of the disorder. More recent reports indicate that NMO may manifest in a lesser clinical severity.Citation17 To aid the understanding of the clinical syndrome, we include two examples of clinical and pathological correlation from the literature ().
Figure 1 Sections from the 11th thoracic and 3rd lumbar segments, using Loyez myelin stain, revealed irregular regions of degeneration in the white matter.
Reprinted from Greenfield JG, Turner JWA. Acute and subacute necrotic myelitis. Brain. 1939;62:227–252 with permission from Oxford University Press.
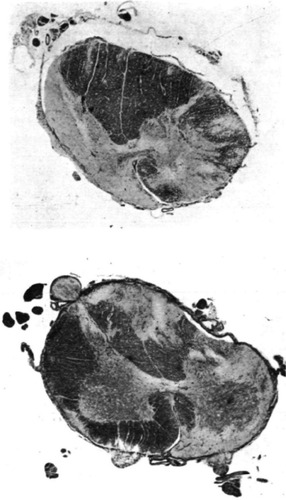
Ortiz de Zarate et alCitation6 described a 49-year-old man who presented with bilateral blurred vision and became blind within 5 days. Cerebrospinal fluid (CSF), urine, and blood tests were all unremarkable except for a high erythrocyte sedimentation rate. Two and a half months later, right lower limb weakness developed, followed by paraplegia after a further week. Sensory impairment to the level of the costal margin began a month later and progressed to the second rib within the next few days. CSF remained normal. He died from sepsis in the 5th month of his illness. A postmortem study revealed T2 to L1 spinal cord damage with the entire thickness of the cord affected in the central portion of the lesion, and necrosis was evident from T6 to T12. Myelomalacia was prominent centrally in the spinal cord. All stages of neuronal degeneration, astrocyte hyperplasia, plenty of scavenger cells (white blood cells that engulfed debris), and interstitial edema were widespread. The walls of medium-sized vessels within the lesions appeared thick and hyalinized, giving a “rubber tube” appearance. Similar to the cord lesions, there was central malacia and several scavenger cells were observed in the optic chiasm. Groups of lymphocytes were demonstrated in the hypothalamus and crus cerebri.
The NMO antibody and aquaporin 4
Evidence of humoral immunity such as perivascular deposition of immunoglobulin and complement in a vasculocentric pattern around thickened hyalinized blood vessels, and the infiltration of neutrophils or eosinophils, is prominent in NMO lesions.Citation8 In 2004 researchers at the Mayo Clinic discovered an NMO immunoglobulin (NMO-IgG, not IgM), later referred to as anti-aquaporin 4 (AQP4) antibody, that is specific to NMO.Citation18,Citation19 CSF protein electrophoresis in NMO is often negative for oligoclonal bands in patients with NMO,Citation19 however, there is at least one study demonstrating evidence for possible intrathecal production of AQP4 specific autoimmunity.Citation20 AQP4 Ab is not detected in MS patients. The antibody selectively binds to the extracellular domain of (AQP4) water channels in the cell membrane of astrocytic foot processes at the blood–brain barrier.Citation19,Citation21 AQP4 forms a macromolecular complex with excitatory amino acid transporter 2 (EAAT2) on astrocyte plasma membranes.Citation22 It regulates CNS water and ion homeostasis.Citation23,Citation24 The distribution of AQP4 in the brain is wide. It occurs along the entire surface of plasma membrane of the astrocytic foot processes that face vessels and the pia mater; on the basolateral membrane of ependymal cells; and in the glia lamellae of the supraoptic nucleus and other osmosensitive regions in the hypothalamus.Citation24 A recent study has revealed that AQP4 is prominent at the glia limitans externa, the cerebral cortex, the grey-white matter junction, the subependymal region, the abluminal surface of penetrating cortical blood vessels in a rim pattern, the astrocytic foot processes abutting vessels in a rosette pattern, the floor of the fourth ventricle including the area postrema, but is minimal in normal cerebral white matter.Citation25 AQP4 is diffusely expressed in the entire spinal cord with the greatest concentration in the central grey matter and pia mater,Citation26 and EAAT2 is found abundantly in the grey matter.Citation22,Citation25 AQP4 is also highly present in the optic nerve and retinal Müller cells, a type of astrocyte; but absent in myelin, neurons, and oligodendrocytes.Citation19,Citation24,Citation25 Characteristic NMO lesions, with or without necrosis, lack AQP4 and show vasculocentric depositions of IgG, IgM, and complement.Citation22,Citation25,Citation27 These findings coincide with the inflammation, edema, necrosis, cavitation, cord atrophy, perivascular cuffing, and thickened vessel walls.Citation26 The regions of AQP4 depletion colocalize with the immune complex deposition in NMO lesions.Citation25 The loss of AQP4 parallels the loss of EAAT2 in NMO.Citation22,Citation28 In contrast to the stage-dependent loss of AQP4 in MS lesions, the loss of AQP4 in NMO lesions depends on neither stage of disease nor CNS region.Citation25 The binding of NMO-IgG to AQP4 initiates two events: AQP4 endolysosomal degradation; and complement activation.Citation19 In the presence of active complement in vitro, the binding of NMO-IgG to the AQP4 channel in astrocytes increases plasma membrane permeability.Citation22 In the absence of active complement in vitro, the astrocytic membranes are still intact but NMO-IgG down-regulates AQP4, Na+-dependent glutamate transport, and EAAT2, leading to glutamate homeostasis impairment.Citation22 Hinson et al have hypothesized that the disrupted EAAT2 would increase extracellular glutamate levels in AQP4-rich regions, which is toxic to oligodendroglia and neurons leading to subsequent demyelination.Citation19
Seropositivity for NMO-IgG is useful to differentiate NMO from MSCitation21 and is 76% sensitive and 94% specific to NMO.Citation29 It predicts relapses and NMO development in patients with a first episode of severe longitudinally extensive transverse myelitis (LETM).Citation30 Around 70% of NMO patients have antibody detectable in the serum. A Japanese group has recently developed a new NMO-IgG assay which has 91% sensitivity and 100% specificity to NMO.Citation17 The serum NMO-IgG titre correlates with the severity of the disease with higher titers in patients with complete blindness, LETM, or extensive cerebral lesions and low titers following corticosteroid and immunosuppressive treatments.Citation17 The NMO-IgG titer in the serum is consistently higher than in the CSF.Citation17 A study revealed that CSF NMO-IgG was detectable in three seronegative cases and that this measurement might improve the sensitivity of the test.Citation31
Demyelination and axonopathy develop in both MS and NMO but astrocytic damage or astrocytopathy predominates only in NMO.Citation32 Misu et al revealed that the depletion of AQP4 was associated with astrocytic impairment as decreased glial fibrillary acidic protein or GFAP, an astrocyte specific protein, accompanies the loss of AQP4, particularly where there is immune complex deposition in the early stage of NMO lesions.Citation26 However myelin basic protein (MBP) staining is relatively intact, suggesting that demyelination is not the primary pathology. These findings differ from what has been observed in ischemic infarction and MS lesions. The surrounding areas of NMO lesions exhibit reactive gliosis as AQP4 and GFAP expressions rise. However the relationship between AQP4 expression, GFAP expression, and perivascular deposition of activated complement and immunoglobulin was found to be heterogeneous.Citation33 A recent NMO study revealed AQP4 and GFAP loss in demyelinating areas in some patients, but total preservation of AQP4 in areas with demyelination and GFAP loss in other patients even in cases seropositive for NMO-IgG.Citation33 The findings indicate that AQP4 loss is independent of the presence of the NMO-IgG antibody.Citation33 In addition, perivascular immune complex deposition was demonstrated in active and chronic NMO lesions but not tightly related to perivascular AQP4 depletion; and not found in any demyelinating MS lesions.Citation33 Matsuoka et al, hence, have hypothesized that there might be two pathological types of NMO: AQP4 autoimmunity-related and AQP4 autoimmunity-unrelated, and the latter may account for seronegative NMO.Citation33
Two studies revealed that the GFAP level in the CSF (CSF-GFAP) increased during clinical relapse and remarkably decreased following intravenous methylprednisolone treatment.Citation32,Citation34 The level was greater in myelitis than optic neuritis and cerebral lesions. The level in NMO was significantly higher than that in MS; acute disseminated encephalomyelitis (which is identified by monophasic encephalomyelitis with multifocal white matter lesions by brain and spinal MRI); neuro-Behçet disease; meningitis; cord infarction; headache; sinusitis; and conversion disorder.Citation32 The findings are suggestive of astrocytopathy in NMO but not in the other conditions stated above. CSF-GFAP in NMO correlates well with the Expanded Disability Status Scale (EDSS) and has 90% sensitivity and 76.9% specificity when compared with the other conditions.Citation32 CSF-GFAP level has a potential role as a biomarker for NMO activity. It is noted that the level is also high in cerebral ischemia and mildly elevated in Alzheimer’s disease.Citation32 Additionally, serum GFAP was found to be significantly higher in patients with NMO-related optic neuritis (ON) as compared to MS-type ON in one recent study.Citation35
Clinical manifestations: world studies
Since the discovery of NMO-IgG, various additional clinical manifestations of NMO have been identified. Revised NMO criteria have been proposed by Wingerchuk et al in 2006.Citation29 The criteria include optic neuritis, acute myelitis, and at least two out of three supportive criteria: contiguous spinal cord MRI lesion extending over three or more vertebral segments; brain MRI not meeting MS diagnostic criteria; and seropositivity for NMO-IgG. NMO-IgG seropositivity has been incorporated in recent diagnostic criteria for NMO.Citation36 It can therefore include limited forms, including isolated or recurrent optic neuritis,Citation37 isolated or recurrent transverse myelitis, and a wide range of clinical presentations (NMO spectrum disorders).
Prevalence of NMO is greater in females. Mean age of onset is the late 30s in NMO as opposed to the early 30s in MS. However both childhood and elderly onset (up to 80 years of age), has been reported.Citation8,Citation18,Citation38 NMO is usually sporadic but familial occurrences have been reported, involving about 3% of all NMO patients in a series.Citation39 The clinical course of NMO patients is mostly relapsing-remitting rather than monophasic.Citation4,Citation38,Citation40 Conversion to a secondary progressive course is uncommon.Citation40–Citation42 A series showed that patients with a relapsing course had a poorer prognosis than the less common monophasic type and one third of these patients died from respiratory failure as a consequence of cervical cord lesions.Citation43 Patients can present with either ON or transverse myelitis or both.Citation38,Citation40 The mean interval between the first two attacks was significantly shorter if the initial lesion was bifocal (11.1 months), compared to either a spinal cord (33.1 months) or an isolated optic nerve lesion (34 months) in a French cohort.Citation40 The mean number of relapses decreased after the first two years in this study.Citation40 The median time from the onset of NMO to EDSS score 4, 6, and 7 was 7, 10, and 21 years respectively.Citation40 Serum NMO-IgG was detectable in 54% of 111 cases tested.Citation40
In a Caucasian study from Denmark 163 cases were ascertained who had diagnoses of ON, MS, NMO, and TM without MR changes of MS. Twenty-six percent were diagnosed as NMO on clinical criteria (62% of whom were AQP4 positive).Citation44 In a Thai series, seropositive NMO-IgG was demonstrated in almost 40% of patients (53/135 cases) with idiopathic inflammatory demyelinating CNS disease which included NMO, other NMO spectrum disorders, conventional MS, optico-spinal MS (OSMS), and clinical isolated syndromes.Citation45 NMO-IgG was detected in 9% of Western MS patientsCitation18 and 15% of northern Japanese conventional MS (CMS) patients.Citation46 A study in Cuba and the French West Indies (an ethnically mixed population) revealed seropositivity to NMO-IgG in 33.3% of relapsing NMO cases and 4.8% of relapsing remitting MS.Citation47 It was shown in this study that seropositive patients had more attacks and more disability as evaluated by EDSS than seronegative cases, particularly in the motor and sensory systems.
OSMS was considered as an Asian form of MS in the past but recent studies have shown otherwise. It has been suggested that there is no important difference between OSMS and NMO and that they are the same disease.Citation48 A study in the northern part of Japan revealed that NMO-IgG was detected in 63% (12/19) of OSMS cases who experienced optic neuritis, myelitis, and fulfilled Wingerchuk criteria, and 15% (2/13) of CMS cases.Citation47 The majority of these patients showed long spinal cord involvement (>3 vertebral segments) and no perception of light in one or both eyes. A study in Kyushu, the southernmost part of Japan, recruited relapsing remitting MS patients according to Poser criteria and subdivided them into OSMS (58 cases) and CMS (90 cases).Citation49 Patients who had both optic nerve and spinal cord involvement, without any clinical evidence of disease in either the cerebrum or the cerebellum, were considered to be OSMS. They could manifest additionally with minor brainstem signs, such as transient double vision and nystagmus. Patients who were not included in the criteria, were considered as CMS. None was seropositive for human T-cell leukemia virus type I (HTLV-1). Half of the OSMS patients fulfilled Wingerchuk 1999 NMO criteria. The AQP4 antibody positivity rate was 36.2% in patients with OSMS, 6.7% in CMS, and 0% in healthy individuals.Citation49 AQP4 antibody-positive patients with OSMS and CMS showed higher relapse rate and EDSS than AQP4 antibody-negative CMS patients and were mostly female. The AQP4 antibody-negative OSMS patients had results for these parameters in between the two groups. Both clinical features and peripheral blood cytokine production patterns of low-titer anti-AQP4 antibody patients were similar to those of anti-AQP4 antibody-negative OSMS patients with LETM. Thus it is clear that the precise case definition for OSMS will result in a differing proportion of cases fulfilling diagnostic criteria for classical NMO on the one hand and classical MS on the other.
In classical NMO the CSF usually shows pleocytosis (50–1000 × 106 WBC/L) with neutrophil predominance and infrequent oligoclonal bands.Citation41 Syndromic NMO is found in other autoimmune disorders such as SLE and Sjögren disease.Citation41
Optic neuritis (ON)
Optic neuritis presents with subacute loss of vision with or without orbital pain (especially on eye movement), decreased colour vision, and signs of optic neuropathy.Citation50 It is a clinical diagnosis but MRI of the optic nerves is confirmatory in most cases ().Citation50 Involvement of different segments of the anterior visual pathway influences the clinical presentation. For instance, pain usually occurs with involvement of the intraorbital segment of the optic nerve; is quite severe in optic nerve sheath inflammation; but is not a feature of disease limited to the intracranial segment of the optic nerve. In optic chiasm involvement, the presentation may mimic bilateral simultaneous optic neuritis. Clinical ophthalmic assessments include visual acuity; Ishihara colour test; visual field; pupillary light reaction; anterior segments; retina; and optic disc. ON has been classified as typical and atypical types. The “typical” type implies ON as seen in MS but it should be noted that this is typical only in Caucasian populations and it would preferable to use a more specific designation such as DON (demyelinating ON) or MS-ON, depending on whether criteria for MS are fulfilled.Citation50 Other alternative ON phenotypes are considered as atypical including NMO; chronic relapsing inflammatory optic neuropathy (CRION); ADEM; neuroretinitis; post-infection and post-vaccination ON; infectious diseases such as neuroborreliosis, tuberculosis, syphilis, and cytomegalovirus; and other systemic diseases such as sarcoidosis, autoimmune disease such as systemic lupus erythematosus, Sjögren syndrome, and Behçet disease.Citation50 Features of typical and atypical ON in adults are shown in and respectively and more reviews on MS-ON can be consulted.Citation50–Citation55 A question that should be addressed in a first presentation of optic neuritis is whether the case is typical or not, as the management differs.Citation53 When atypical ON is suspected, further investigations are recommended such as MRI of the brain and orbits with gadolinium, lumbar puncture, chest x-ray, blood tests including full blood count, liver function, renal function, calcium and electrolyte, erythrocyte sedimentation rate, autoimmune screening, ACE, syphilis and viral serological studies. An investigation of the pattern of visual field defect in NMO revealed that both central and non-central scotoma (altitudinal, quadrant, three quadrant, hemianopia, and bitemporal hemianopia) were observed in seropositive NMO patients and the incidence of non-central scotoma was higher than in MS patients.Citation56 Among non-central scotomata in NMO cases, an altitudinal defect was the most common.Citation56
Figure 2 A postcontrast axial T1-weighted fat-suppressed MRI of the orbits in a NMO patient showed enhanced left optic nerve. The finding was consistent with acute left optic neuritis.
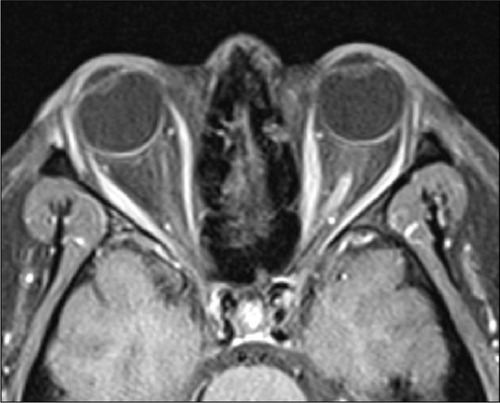
Table 1 Summary of typical symptoms and signs of optic neuritisCitation54
Table 2 Summary of features of atypical optic neuritisCitation54
Patients with NMO-type ON have a poorer visual outcome and a thinner peripapillary retinal nerve fibre layer, measured with optical coherence tomography, than MS-ON.Citation57 Furthermore the NMO-IgG titer in ON patients with permanent complete blindness at least in one eye has been shown to be higher than that in ON patients without blindness.Citation17 A study of ON in patients with NMO in an African population showed that ON was the first manifestation in 23 cases (76.6%), being retrobulbar ON in 20 cases, and papillitis in 3.Citation58 Seventy percent of patients had bilateral involvement. Thirty percent of patients with a first episode of ON experienced a residual loss of visual acuity ≤ 20/200.Citation58 In the cohort studied by Collongues et al, this visual acuity loss affected 22% of patients.Citation40 Patients in this study who first presented with bifocal disease experienced a worse visual prognosis than those who first presented with either an i solated cord or optic nerve lesion.Citation40 Moreover, a high number of lesions on brain MRI was related to a shorter duration from the onset of NMO to severe residual visual loss.Citation40 A study performed in central London revealed that African and African- Caribbean patients who presented with acute isolated ON had a threefold increase in the risk of developing NMO-type ON than Caucasian patients.Citation59 Optic neuritis in Chinese patients seems to be aggressive and normal MRI scans of the brain were commonly observed in one study.Citation60
Isolated ON with clinical, radiological, and serological evidence of neither MS nor NMO can be subdivided into three groups: CRION; recurrent isolated ON (RION); and a solitary episode of isolated ON (SION). CRION has been defined in an article published in 2003 based in London.Citation61 It is a form of ON which is usually bilateral but sequential rather than simultaneous associated with pain and with no clinical myelitis at extended follow up. It is characterized by repeated ON relapses immediately or soon after withdrawing immunosuppression, which is a feature of neither MS nor NMO nor RION. In CRION, MRI of the brain is normal; the optic nerves reveal hyperintensity on T2 weighted images and contrast enhancement; and CSF oligoclonal bands are rare.Citation61 RION refers to relapsing remitting isolated ON in one or both optic nerves in patients who have no clinical or laboratory-supported evidence of MS.Citation62 SION is a unilateral or bilateral ON in patients with evidence of neither MS nor NMO, who do not require immunosuppression to prevent relapse, and do not have recurrence of ON or another neurological episode after extended follow up.Citation51 A RION series from the Mayo clinic demonstrated that 20% of 34 patients were seropositive for NMO-IgG and 50% (6/12) of the seropositive cases developed a myelitis attack and fulfilled criteria for a diagnosis of NMO within 8.9 years (median) following the first ON episode.Citation63 MRI brain of all cases was unremarkable or showed nonspecific abnormalities. Non-Caucasian patients were more commonly seropositive than seronegative. Another study revealed that 20.8% (5/24) of patients with RION converted to NMO due to the occurrence of myelitis within 2–12 years follow-up (mean 5.8 years) and 25% (6/24) of RION cases had seropositive NMO-IgG.Citation64 Four of 21 French patients (19%) with bilateral ON and/or RION were seropositive for NMO-IgG.Citation65 In a Japanese cohort, 26.9% (7/26) of RION patients had positive serum NMO-IgG.Citation49 A study in Cuba and the French West Indies (mixed population) revealed seropositivity to NMO-IgG in 16.6% of 12 RION cases.Citation47 In a prospective series involving 114 ON patients from London, serum NMO-IgG was detected in 5% of SION, 6% of RION, 5% of CRION, 56% of NMO-ON, and no MS-ON cases.Citation62 In an investigation performed in China, the rate of seropositivity was 50% in patients who presented with RIONCitation66 and 32% (11/34) in patients with severe ON (acuity 20/200 or worse).Citation67 Higher titers had significantly more ON relapses than lower titers and 18.2% (2/11) of seropositive ON cases developed transverse myelitis within 32 months.Citation67 Using a recently developed fluorescence immunoprecipitation assay employing recombinant human AQP4 in 224 subjects from multiple centers in Europe,Citation68 seropositive NMO-IgG was demonstrated in 5.8% of SION, 58.8% of NMO-ON, and 0% in MS-ON and normal subjects. Seropositive NMO-IgG in SION was rare with a high rate of NMO conversion according to Wingerchuk’s revised criteria; whereas none of the seronegative SION cases (0/60) converted to NMO within 26 month follow-up (median) and 13/60 NMO-IgG negative SION patients were diagnosed with MS later in the disease course.Citation68 Seropositive NMO-IgG predicted a poor visual outcome.Citation68 The Danish study revealed that serum NMO-IgG was detectable in 3/12 (25%) of RION, 1/28 (0.04%) of SION, and 1/4 (25%) of bilateral ON.Citation44 In summary, seropositive NMO-IgG in isolated ON confidently excludes an MS type of ON but seronegativity does not rule out subsequent NMO conversion.Citation62 Seropositive NMO-IgG RION is a limited form of NMO and predicts a potentially poor visual outcome.Citation63 Moreover, these patients are at high risk of developing transverse myelitis and severe disability in the future.Citation63 Hence long-term immunosuppression should be considered in RION patients seropositive for NMO-IgG.Citation63
Transverse myelitis
Acute transverse myelitis (ATM) is a focal inflammation of the spinal cord having numerous potential causes and resulting in motor, sensory, and autonomic dysfunction.Citation69 It is paramount to exclude compressive lesions and secondary ATM which includes autoimmune disorders such as NMO, systemic lupus erythematosus (SLE), Sjögren’s syndrome, and antiphospholipid syndrome; inflammation such as MS, ADEM, Behçet disease, and sarcoidosis; infection; neoplasm; metabolic disorders such as B12 deficiency and copper deficiency; vascular conditions such as infarction and dural fistula; and radiation myelopathy.Citation30,Citation69,Citation70 Idiopathic ATM refers to the situation where no cause has been found.Citation69 Patients with NMO usually present subacutely over weeks to months with moderate to severe spinal cord involvement.Citation71 When the condition involves three or more vertebral segments, it is termed LETM which is a part of the Wingerchuk 2006 criteria ().Citation29 Such lesions may extend from the level of C2 to the conus medularis.Citation72 NMO patients complain of sensory disturbance such as numbness, tingling, dissociated sensory loss, pain, weakness, and bowel and bladder dysfunction.Citation6,Citation13,Citation16 These symptoms can involve one leg initially before progressing to the other leg and armsCitation6 over a matter of weeksCitation13 or the two legs simultaneously.Citation9 Patients present with signs of white matter dysfunction such as spasticity, hyperreflexia and a positive Babinski sign.Citation72–Citation74 Mild muscle wasting along with pyramidal tract signs, a mixed picture of gray and white matter dysfunction, has been reported.Citation13 A study of myelitis in SLE revealed that patients with clinical “white matter” myelitis were more likely to fulfill NMO criteria than those with “gray matter” myelitis.Citation75 Paroxysmal symptoms can occur, such as intermittent sensory disturbance becoming constant,Citation13 tonic spasms, and Lhermitte’s symptom.Citation43 Spinal MRI usually reveals T2 signal hyperintensity, edema, and gadolinium enhancement of acutely inflamed regions. Unlike MS, NMO lesions tend to be located at the central region rather than the peripheral white matter Citation26,Citation76 and holocord involvement is also observed.Citation47 In chronic cases, MRI scans demonstrate atrophic spinal cord and occasionally cavities.Citation15,Citation77 Recurrent transverse myelitis, commonly seen in NMO,Citation30 can occur in the same location in the spinal cord.Citation74 A progressive course is unusual but still possible. For example, in one report a NMO-IgG seropositive female experienced a 4-month history of progressive quadriplegia and respiratory failure following nausea, vomiting, and hiccupping.Citation78 Her weakness worsened in spite of intravenous steroids but improved with plasma exchange. The case represents an extreme end of the clinical spectrum of myelitis in NMO.
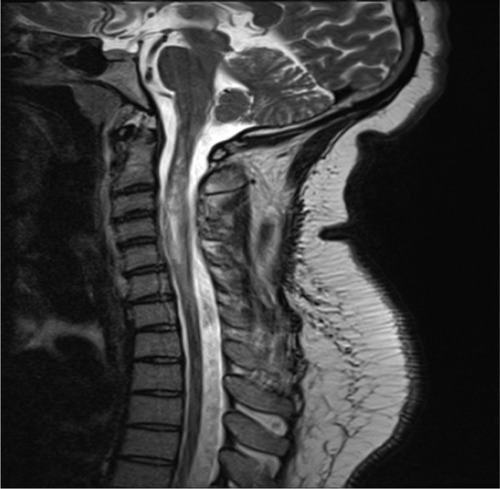
Figure 3 A sagital T2-weighted MRI of the spinal cord in a NMO patient showed hyperintense signal and swelling of several cervical and thoracic cord levels. The finding was consistent with acute myelitis.
Serum NMO-IgG was detected in 52% (14/27) of patients with recurrent isolated longitudinal extended transverse myelitis (R-LETM) in North American Caucasian series.Citation18 The rate of seropositivity in R-LETM varied in other studies, rates being 33% in a French study,Citation65 60% in a Japanese study,Citation76 23.5% in another Japanese series,Citation49 and 14.2% in a Cuban and French West Indies series (mixed population).Citation47 Thirty-eight percent (11/29) of patients in a Mayo clinic series who presented with single episode of LETM were seropositive for NMO-IgG; 56% of these seropositive cases developed a second episode, either TM or ON, within a year.Citation30 Serum antinuclear antibody and extractable nuclear antigen were demonstrated in three of these relapsed cases. No further relapse occurred in seronegative LETM cases.Citation30 R-LETM may be considered another limited form of NMO.Citation30 Early immunosuppression has been recommended in patients who present with single LETM and seropositive NMO-IgG in order to prevent the second event.Citation30
In contrast to LETM, patients who presented with acute partial transverse myelitis (APTM) involving ≤2 vertebral segments and who had normal cerebral MRI scans showed a much lower frequency of NMO-IgG seropositivity (5% or 1/22 in one study).Citation79 However the NMO-IgG titre of APTM appeared low in another study.Citation17 NMO cannot be ruled out completely in cases with APTM as it could represent another end of the clinical spectrum.
Beyond the optic nerve and spinal cord
Brain MRI scans are usually normal or show nonspecific white matter lesions at the onset of NMO.Citation43 There is increasing evidence for the occurrence of lesions outside the optic nerve and spinal cord (). A pathological study of a NMO case with seropositive NMO-IgG, who presented with generalized numbness, tongue numbness, vomiting, hiccups, and double vision, has been reported.Citation11 MRI brain demonstrated several lesions in the dorsal medulla, basal ganglia, and an enhancing lesion in the right temporal lobe. She recovered spontaneously. Within two years she had exhibited two episodes of LETM. A MRI brain during the last attack showed new brain lesions with a recurrent gadolinium-enhancing right temporal lobe lesion. Similar to the spinal cord pathology reported above, a temporal lobe biopsy revealed lymphocyte, macrophage and eosinophil infiltration, myelin and axonal loss, and perivascular complement deposition. The patient responded well to Rituximab. Other cerebral lesions that have been reported so far include periventricular and white matter lesions.Citation47 A large cavity in the white matter of the right frontal lobe associated with meningitis,Citation14 and extensive cerebral white matter lesions along with lesions in the corpus callosum.Citation12 Extensive brain lesions are frequent in children with NMO.Citation80
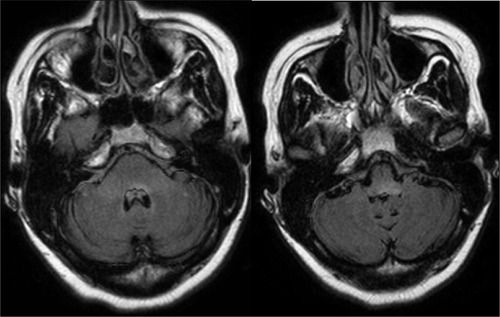
Figure 4 Axial fluid-attenuated inversion recovery (FLAIR) MRI scans of the brain showed hyperintense signal in the pons and medulla in a NMO patient who presented with ataxia, nausea, and vomiting.
Posterior reversible encephalopathy has been reported in five seropositive NMO patients.Citation81 These patients, who presented with confusion, double vision, and cortical blindness, had bilateral T2-weighted hyperintense signals in the frontal, parietal and occipital lobes, and cerebellum. These might be due to NMO or medication related.Citation82 An unenhanced tumefactive lesion in the left temporo-parietal region and ovoid lesions in the pericallosal areas with normal CSF profiles have been observed in a NMO-IgG seropositive case who experienced motor aphasia, finger agnosia, right-left disorientation, and right hand paresis.Citation83 Extensive brain lesions like tumors or ADEM and MS-like ovoid lesions on MRI have been identified in other reports on NMO.Citation76,Citation84
Brainstem lesions have included a typical NMO case with right deviation of the tongue and right lower facial weakness;Citation16 lesions in the floor of fourth ventricle involving the subependymal region and area postrema associated with intractable vomiting;Citation25,Citation84,Citation85 periaqueductal lesions associated with intractable hiccup and nausea;Citation86 medulla oblongata lesions associated with nausea, vomiting, and acute respiratory failure;Citation43 abnormal eye movement such as opsoclonus, downbeat nystagmus, upbeat nystagmus, central vestibular nystagmus;Citation87 one and a half syndrome;Citation88 and bilateral internuclear ophthalmoplegia.Citation89 Hypothalamic and thalamic lesions have also been reported in patients presenting with hypersomnia and confabulation, weight loss, and behavioral change.Citation84,Citation90,Citation91
Treatment
One investigation demonstrated that the serum NMO-IgG level was associated with clinical disease activity, in that it rose during relapse and decreased during remission or following treatment with steroid and immunosuppressive drugs such as azathioprine, rituximab, and cyclophosphamide.Citation92 The low antibody titer was sustained for about a year in two patients who received a combination of azathioprine and prednisolone following intravenous methylprednisolone for acute relapse.Citation92
Acute relapse
Intravenous methylprednisolone
Intravenous methylprednisolone 1 g daily for 3–6 days has been recommended for acute myelitis or optic neuritis, following by a course of oral prednisolone.Citation93,Citation94 A prospective case series revealed that hyperacute treatment with corticosteroids may prevent visual loss in patients with optic neuritis who present with prodromal retro-orbital pain but no visual deterioration.Citation95 The patients included NMO cases, all had a prior history of optic neuritis, and the acute recurrence was confirmed by MRI.
Plasma exchange
Plasma exchange should be carried out quickly in a severe progressive steroid refractory event, as a rescue therapy.Citation93,Citation94 The response rate was 42% in one study.Citation96 The usual regimen is five exchanges of 1.5 plasma volumes over 10 days.Citation97 Early treatment seems to be associated with moderate to marked improvement.Citation98 It was also found to be useful in other smaller studies.Citation99–Citation101 The improvement in responsive cases appears to occur rapidly after the treatment and to be sustained.Citation98
Prophylactic immunosuppressive treatment
Azathioprine
Azathioprine is effective in preventing relapses in NMO spectrum disorders when used alone or in combination with prednisolone and has been classified as a class IV treatment.Citation102 Annualized relapse rate reduction was 76% in a Mayo Clinic studyCitation102 and 70% in a Brazilian study.Citation103 EDSS and visual scores of NMO patients have been shown to improve.Citation102–Citation104 The suggestion is to initiate the medication after the first attack in all seropositive patients and seronegative cases with a high chance of NMO relapse.Citation102 Thiopurine methyl transferase (TPMT) should be checked in all cases. If the TPMT level is within a normal range, the recommended dose is 2.5–3 mg/kg/day in patients.Citation102 A lower dose should be initiated in case of low TPMT (heterozygous) and an alternative treatment should be sought if the level is very low (homozygous deficiency).Citation102 Full blood count, liver function, and renal function tests are conducted regularly. The target is a rise in mean corpuscular volume for at least 5 points from the baselineCitation102 or a slight decline in lymphocyte count. Adverse effects include immunosuppression, myelosuppression, allergy, gastrointestinal disturbance, hepatic toxicity, and myalgia.Citation94
Mycophenolate mofetil ()
Mycophenolate mofetil is a noncompetitive inhibitor of the enzyme inosine 5′-monophosphate dehydrogenase. It inhibits purine synthesis and controls lymphocyte proliferation and T-cell-dependent antibody.Citation105 A few studies (class IV) showed a good efficacy on reducing relapse rate and improving disability.Citation106,Citation107 Side effects include immunosuppression, myelosuppression, gastrointestinal disturbance, infection, early pregnancy loss and congenital malformation.Citation94,Citation108
Table 3 Treatment summary
Rituximab ()
Rituximab is a chimeric monoclonal antibody directed against antiCD20 which is expressed on pre-B cells and mature B cells.Citation109 Its use in NMO treatment was first described by Cree at al in 2005 and was found to be effective in reducing the relapse rate and improving disability.Citation109 Further studies showed more or less the same results.Citation109–Citation111 It is recommended to monitor CD19, CD27, or CD 20 B cells regularly and to maintain the level at zero.Citation108,Citation110–Citation112 A study demonstrated that CD19 reappeared within 251–350 days following the last rituximab infusion, which correlated well with the rise of NMO-IgG and clinical relapse.Citation92 A repeat treatment should be given when the number of these B cells increases, regardless of clinical relapse.Citation112 The NMO-IgG titer declined markedly and rapidly following re-infusion, consistent with clinical improvement;Citation92,Citation110 but did not disappear even when CD19 was undetectable in a study.Citation92 It is notable that a seropositive NMO patient experienced agammaglobulinemia due to the depression of B-lymphocytes associated with carbamazepine and was relapse-free when CD19 was low (0.11%).Citation113 However resistance was demonstrated in a case with CD19 cell depletion.Citation114 Plasma cells produce a variety of antibodies and do not express CD20. Perhaps the resistance might be because rituximab does not suppress plasma cells.Citation114 Side effects include immunosuppression, myelosuppresion,Citation94 progressive multifocal leucoencephalopathyCitation115 and posterior reversible encephalopathy syndrome.Citation116 Transient hypotension and influenza-like symptoms have been reported and can be managed with intravenous methylprednisolone.Citation110 Infection has been recorded, including urinary and respiratory tract infection, and septicemia.Citation110,Citation117
Cyclophosphamide ()
Cyclophosphamide is an antineoplastic alkylating agent. Its active metabolite binds with the guanine bases of DNA to interfere with mitosis.Citation94 Some case reports revealed that it was useful especially when a patient did not show any response to other medications and had other autoimmune diseases,Citation72,Citation118–Citation120 whereas two reports showed a non-satisfactory outcome.Citation121,Citation122 The relapse rate declined significantly in one study, consistent with a decrease in serum NMO-IgG level.Citation92 Side effects include immunosuppression, myelosuppression, alopecia, gastrointestinal disturbance, hemorrhagic cystitis, bladder cancer, leukemia, and infertility.Citation94
Conclusion
NMO is an inflammatory disorder of the central nervous system. Following the discovery of the NMO-IgG (anti AQP4) antibody, it has become evident that the spectrum of the disease is wider than previously thought and not limited to the optic nerve and the spinal cord. It can behave aggressively and fatalities occur from respiratory failure. Blindness is also a significant risk. Early recognition and treatment, when clinical symptoms are mild, could prevent further severe disability and relapses. In addition, not all patients with clinical NMO have NMO-IgG seropositivity. Perhaps this is due to low titers of NMO-IgG or alternatively there might be other unidentified antibodies or causes.
Disclosure
The authors report no conflicts of interest in this work.
References
- DevicESubacute myelitis complicated by optic neuritis [Myelite subaigue compliquee de nevrite optique]Bull Med1894810331034 French
- DevicEAcute thoracolumbar myelitis with optic neurits, autopsy [Myelite aigue dorse-lombaire avec nevrite optique, autopsie.]Congress Francais Medicine (Premiere Session)Lyon18951434439 French
- GaultFNeuromyelitis optica [De la neuromyelite optique aigue]ThesisLyon1894 French
- O’RiordanJIGallagherHLThompsonAJClinical, CSF, and MRI findings in Devic’s neuromyelitis opticaJ Neurol Neurosurg Psychiatry19966043823878774400
- CloysDENetskyMGNeuromyelitis opticaVinkenPJBruynGWHandbook of Clinical NeurologyNewYorkElsevier North Holland Inc19709426436
- Ortiz de ZarateJCTamaroffLSicaRERodriguezJANeuromyelitis optica versus subacute necrotic myelitis. II. Anatomical study of two casesJ Neurol Neurosurg Psychiatry19683166416455709852
- StansburyFCNeuromyelitis optica (Devic’s disease): presentation of five cases with pathologic study, and review of the literatureArch Ophthal1949423292335
- LucchinettiCFMandlerRNMcGavernDA role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis opticaBrain2002125Pt 21450146112076996
- LeonardiAArataLFarinelliMCerebrospinal fluid and neuropathological study in Devic’s syndrome. Evidence of intrathecal immune activationJ Neurol Sci1987821–32812903440870
- MandlerRNDavisLEJefferyDRKornfeldMDevic’s neuromyelitis optica: a clinicopathological study of 8 patientsAnn Neurol19933421621688338340
- JacobsDRoemerSWeinshenkerBThe pathology of brain involvement in neuromyelitis optica spectrum disorderMult Scler200612S155
- AlmekhlafiMAClarkAWLucchinettiCFZhangYPowerCBellRBNeuromyelitis optica with extensive active brain involvementArch Neurol201168450851221482930
- GreenfieldJGTurnerJWAAcute and subacute necrotic myelitisBrain193962227252
- NakamuraMEndoMMurakamiKFujiharaKItoyamaYAn autopsied case of neuromyelitis optica with a large cavitary cerebral lesionMult Scler200511673573816320738
- O’RiordanJIWalkerMPlantGTGrahamEMNon-communicating syringomyelia and neuromyelitis opticaJ Neurol1999246431431610367703
- HoffmanHLAcute necrotic myelopathyBrain195578337739313269598
- TakahashiTFujiharaKNakashimaIAnti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titreBrain2007130Pt 51235124317449477
- LennonVAWingerchukDMKryzerTJA serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosisLancet200436494512106211215589308
- HinsonSRPittockSJLucchinettiCFPathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis opticaNeurology200769242221223117928579
- BennettJLLamCKalluriSRIntrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis opticaAnn Neurol200966561762919938104
- LennonVAKryerTJPittockSJVerkmanASHinsonSRIgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channelJ Exp Med2005202447347716087714
- HinsonSRRoemerSFLucchinettiCFAquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by downregulating EAAT2J Exp Med2008205112473248118838545
- AbbottNJRönnbäckLHanssonEAstrocyte–endothelial interactions at the blood–brain barrierNat Rev Neurosci20067415316371949
- Amiry-MoghaddamMOttersenOPThe molecular basis of water transport in the brainNat Rev Neurosci20034991100114682361
- RoemerSFParisiJELennonVAPattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosisBrain20071301194120517282996
- MisuTFujiharaKKakitaALoss of aquaporin 4 in lesions in neuromyelitis optica: distinction from multiple sclerosisBrain20071301224123417405762
- MisuTFujiharaKNakamuraMLoss of aquaporin-4 in active perivascular lesions in neuromyelitis optica: a case reportTohoku J Exp Med2006209326927516778375
- ZengXNSunXLGaoLAquaporin-4 deficiency downregulates glutamate uptake and GLT-1 expression in astrocytesMol Cell Neurosci200734343917074507
- WingerchukDMLennonVAPittockSJLucchinettiCFWeinshenkerBGRevised diagnostic criteria for neuromyelitis opticaNeurology200666101485148916717206
- WeinshenkerBGWingerchukDMVukusicSNeuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitisAnn Neurol200659356656916453327
- KlawiterECAlvarezE3rdXuJNMO-IgG detected in CSF in seronegative neuromyelitis opticaNeurology200972121101110319307546
- TakanoRMisuTTakahashiTSatoSFujiharaKItoyamaYAstrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker studyNeurology201075320821620644148
- MatsuokaTSuzukiSOSuenagaTIwakiTKiraJIReappraisal of aquaporin-4 astocytopathy in Asian neuromyelitis optica and multiple sclerosis patientsBrain Pathol201121551653221241398
- TakanoRMisuTTakahashiTIzumiyamaMFujiharaKItoyamaYA prominent elevation of glial fibrillary acidic protein in the cerebrospinal fluid during relapse in neuromyelitis opticaTohoku J Exp Med20082151555918509235
- StoroniMPetzoldAPlantGTThe use of serum glial fibrillary acidic protein measurements in the diagnosis of neuromyelitis optica spectrum optic neuritisPLoS One201168e2348921876753
- SaizAZulianiLBlancoYRevised diagnostic criteria for neuromyelitis optica (NMO): Application in a series of suspected patientsJ Neurol200725491233123717401734
- ZulianiLBlancoYTavolatoBNeuromyelitis optica autoantibody (NMO-IgG) in patients with suspected NMO or limited forms of NMOMult Scler200612S156
- CreeBAGoodinDSHauserSLNeuromyelitis opticaSemin Neurol200222210512212524556
- MatielloMKimHJKimWFamilial neuromyelitis opticaNeurology201075431031520660861
- CollonguesNMarignierRZephirHNeuromyelitis optica in France: a multicenter study of 125 patientsNeurology201074973674220194912
- WingerchukDMDiagnosis and treatment of neuromyelitis opticaNeurologist200713121117215722
- WingerchukDPittockSLennonVLucchinettiCFWeinshenkerBThe rate of conversion to a secondary progressive course is lower in neuromyelitis optica than multiple sclerosisMult Scler200612S155
- WingerchukDMHogancampWFO’BrienPCWeinshenkerBGThe clinical course of neuromyelitis optica (Devic’s syndrome)Neurology19995351107111410496275
- AsgariNLillevangSTSkejoeHPFalahMStenagerEKyvikKOA population-based study of neuromyelitis optica in CaucasiansNeurology201176181589159521536639
- SirithoSNakashimaITakahashiTFujiharaKPrayoonwiwatNAQP4 anitbody-positive Thai cases: clinical features and diagnostic problemsNeurology201177982783421813785
- NakashimaIFujiharaKMiyazawaIClinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgGJ Neurol Neurosurg Psychiatry20067791073107516505005
- Cabrera-GómezJABonnanMGonzalez-QuevedoANeuromyelitis optica positive antibodies confer a worse course in relapsing-neuromyelitis optica in Cuba and French West IndiesMult Scler200915782883319498017
- WeinshenkerBGWingerchukDMNakashimaIFujiharaKLennonVAOSMS is NMO, but not MS: proven clinically and pathologicallyLancet Neurol20065211011116426985
- MatsushitaTIsobeNMatsuokaTAquaporin-4 autoimmune syndrome and anti-aquaporin-4 antibody-negative opticospinal multiple sclerosis in JapaneseMult Scler200915783484719465451
- ShamsPNPlantGTOptic neuritis: a reviewInt MS J2009163828919878630
- HickmanSJDaltonCMMillerDHPlantGTManagement of acute optic neuritisLancet200236093491953196212493277
- PauDAl ZubidiNYalamanchiliSPlantGTLeeAGOptic neuritisEye (Lond)201125783384221527960
- PlantGTOptic neuritis and multiple sclerosisCurr Opin Neurol2008211162118180647
- HickmanSJKoMChaudhryFOptic neuritis: an update typical and atypical optic neuritisNeuro-Ophthalmology200832237248
- FraserCPlantGTOptic neuritis: pathophysiology, clinical features, and managementMinagaANeuroinflammationCambridge, UKElsevier2011253276
- NakajimaHHosokawaTSuginoMVisual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosisBMC Neurology2010104520565857
- JindahraPHedgesTRMendoza-SantiestebanCEPlantGTOptical coherence tomography of the retina: applications in neurologyCurr Opin Neurol2010231162320009925
- MerleHOlindoSBonnanMNatural history of the visual impairment of relapsing neuromyelitis opticaOphthalmology2007114481081517141316
- StoroniMPittockSJWeinshenkerBGPlantGTOptic neuritis in an ethnically diverse population: Higher risk of atypical cases in patients of African or African-Caribbean heritageJ Neurosci20123121–22125
- ZhangXWangWWangQClinical features of optic neuritis in ChinaNeuro-Ophthalmology200731133136
- KiddDBurtonBPlantGTGrahamEMChronic relapsing inflammatory optic neuropathy (CRION)Brain2003126Pt 227628412538397
- PetzoldAPittockSLennonVMaggioreCWeinshenkerBGPlantGTNeuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritisJ Neurol Neurosurg Psychiatry201081110911120019228
- MatielloMLennonVAJacobANMO-IgG predicts the outcome of recurrent optic neuritisNeurology200870232197220018434643
- de SezeJArndtCJeanjeanLRelapsing inflammatory optic neuritis: is it neuromyelitis optica?Neurology200870222075207618505981
- MarignierRDe SezeJVukusicSNMO-IgG and Devic’s neuromyelitis optica:a French experienceMult Scler200814444044518208892
- LaiCTianGLiuWWeiWTakahashiTZhangXClinical characteristics, therapeutic outcomes of isolated atypical optic neuritis in ChinaJ Neurol Sci20113051–2384021470641
- LaiCTianGTakahashiTLiuWYangLZhangXNeuromyelitis optica antibodies in patients with severe optic neuritis in ChinaJ Neuroophthalmol2011311161921150455
- JariusSFrederiksonJWatersPFrequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritisJ Neurol Sci20102981–215816220850793
- Transverse Myelitis Consortium Working GroupProposed diagnostic criteria and nosology of acute transverse myelitisNeurology200259449950512236201
- KitleyJLLeiteMGeorgeJPalaceJThe differential diagnosis of longitudinally extensive transverse myelitisMult Scler201218327128521669935
- ScottTFNosology of idiopathic transverse myelitis syndromesActa Neurol Scand2007115637137617511844
- GadzeZPPatient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case reportBMC Neurology200995619852774
- TomizawaYFukaeJNodaKLinear medullary pericanal lesion with longitudinal myelitis can be diagnostic of neuromyelitis opticaInter Med2009483175176
- MatsumotoHShimizuTOkabeSRecurrent spinal cord attacks in a patient with a limited form of neuromyelitis opticaIntern Med201150550951321372469
- BirnbaumJPetriMThompsonRIzbudakIKerrDDistinct subtypes of myelitis in systemic lupus erythematosusArthritis Rheum200960113378338719877037
- MatsuokaTMatsushitaTKawanoYHeterogeneity of aquaporin-4 autoimmunity and spinal cord lesions in multiple sclerosis in JapaneseBrain2007130Pt 51206122317439988
- RavagliaSBogdanovEIPichiecchioABergamaschiRMogliaAKikhaylovIMPathogenic role of myelitis for syringomyeliaClin Neurol Neurosurg2007109654154617467892
- KimJParkYHKimSMA case of chronic progressive myelopathyMult Scler201016101255125720798133
- ScottTFKassabSLPittockSJNeuromyelitis optica IgG status in acute partial transverse myelitisArch Neurol200663101398140017030654
- LotzeTENorthropJLHuttonGJRossBSchiffmanJSHunterJVSpectrum of pediatric neuromyelitis opticaPediatrics20081225e1039e104718838462
- MagañaSMMatielloMPittockSJPosterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disordersNeurology200972871271719237699
- KarceskiSNeuromyelitis opticaNeurology2009728e40e4119237693
- IkedaKItoHHidakaTRepeated non-enhancing tumefactive lesions in a patient with a neuromyelitis optica spectrum disorderIntern Med20115091061106421532234
- PittockSJLennonVAKreckeKWingerchukDMLucchinettiCFWeinshenkerBGBrain abnormalities in neuromyelitis opticaArch Neurol200663339039616533966
- ApiwattanakulMPopescuBFMatielloMIntractable vomiting as the initial presentation of neuromyelitis opticaAnn Neurol201068575776121031587
- MisuTFujiharaKNakashimaISatoSItoyamaYIntractable hiccup and nausea with periaqueductal lesions in neuromyelitis opticaNeurology20056591479148216275842
- HageRJrMerleHJeanninSCabrePOcular oscillations in the neuromyelitis optica spectrumJ Neuroophthalmol201131325525921623230
- KitthaweesinKVongkulsiriSOne-and-a-half syndrome in neuromyelitis opticaNeuro-Ophthalmol2008326305308
- ShinodaKMatsushitaTFurutaKWall-eyed bilateral internuclear ophthalmoplegia (WEBINO) syndrome in a patient with neuromyelitis optica spectrum disorder and anti-aquaporin-4 antibodyMult Scler201117788588721300735
- SamartKPhanthumchindaKNeuromyelitis optica with hypothalamic involvement: a case reportJ Med Assoc Thai201093450550920462097
- ViegasSWeirAEsiriMSymptomatic, radiological and pathological involvement of the hypothalamus in neuromyelitis opticaJ Neurol Neurosurg Psychiatry200980667968219448094
- JariusSAboul-EneinFWatersPAntibody to aquaporin-4 in the long-term course of neuromyelitis opticaBrain2008131Pt 113072308018945724
- WingerchukDMWeinshenkerBGNeuromyelitis opticaCurr Treat Options Neurol2008101555618325300
- CollonguesNde SezeJCurrent and future treatment approaches for neuromyelitis opticaTher Adv Neurol Disord20114211112121694808
- PlantGTSibtainNAThomasDHyperacute corticosteroid treatment of optic neuritis at the onset of pain may prevent visual loss: a case seriesMultiple Sclerosis International201110.1155/2011/815068
- MagañaSMKeeganBMWeinshenkerBGBeneficial plasma exchange response in central nervous sytem inflammatory demyelinationArch Neurol201168787087821403003
- CarrollWMFujiharaKNeuromyelitis opticaCurr Treat Options Neurol201012324425520842585
- KeeganMPinedaAAMcClellandRLDarbyCHRodroguezMWeinshenkerBGPlasma exchange for severe attacks of CNS demyelination: predictors of responseNeurology200258114314611781423
- GwathmeyKBalogunRABurnsTNeurologic indications for therapeutic plasma exchange: an updateJ Clin Apher201126526126821915895
- WeinshenkerBGO’BrienPCPettersonTMRandomized trial of plasma exchange in acute central nervous system inflammatory demyelinating diseaseAnn Neurol199946687888610589540
- WatanabeSNakashimaIMisuTTherapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis opticaMult Scler200713112813217294622
- CostanziCMatielloMLucchinettisCFAzathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis opticaNeurology201177765966621813788
- BichuettiDBLobato de OliveiraEMOliveiraDMAmorin de SouzaNGabbaiAANeuromyelitis optica treatment: analysis of 36 patientsArch Neurol20106791131113620837859
- MandlerRNAhmedWDencoffJEDevic’s neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprineNeurology1998514121912209781568
- GinzlerEMAranowCMerrillJTOrloffKHenryDToxicity and tolerability of mycophenolate mofetil (MMF) vs intravenous cyclophosphamide (IVC) in a multicenter trial as induction therapy for lupus nephritis (LN)Arthritis Rheum200348S647
- JacobAMatielloMWeinshenkerBGTreatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patientsArch Neurol20096691128113319752302
- FalciniFTrapaniSRicciLRestiMSimoniniGde MartinoMSustained improvement of a girl affected with Devic’s disease over 2 years of mycophenolate mofetil treatmentRheumatology (Oxford)200645791391516638802
- US Food and Drug AdministrationInformation on mycophenolate mofetil (marketed as CellCept) and mycophenolic acid (marketed as Myfortic) (FDA alert). Last updated March 26, 2010. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm111294.htm
- CreeBALambSMorganKChenAWaubantEGenainCAn open label study of the effects of rituximab in neuromyelitis opticaNeurology20056471270127215824362
- KimSKimWLiXFJungIJKimHJRepeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 yearsArch Neurol201168111412142021747007
- BediGSBrownADDelgadoSRUsmaniNLamBLSheremataWAImpact of rituximab on relapse rate and disability in neuromyelitis opticaMult Scler201117101225123021622594
- PellkoferHLKrumbholzMBertheleALong-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximabNeurology201176151310131521482945
- TanakaYKimuraKKawachiIInuzukaTNo relapse of neuromyelitis optica during drug-induced B-lymphopenia with hypogammaglobulinemiaNeurology201075191745174721060099
- QianPCrossAHNaismithRTLack of response to monoclonal antibody therapy in neuromyelitis opticaArch Neurol20116891207120921911704
- US Food and Drug AdministrationDrugs [homepage on the Internet]Silver Spring MDRituximab (marketed as Rituxan) information (FDA alert) Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm109106.htm12182006
- Sanchez-CarteyronAAlarciaRAraJRMartineJPosterior reversible encephalopathy syndrome after rituximab infusion in neuromyelitis opticaNeurology201074181471147320439850
- JacobAWeinshenkerBGViolichITreatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patientsArch Neurol200865111443144818779415
- MokCCToCHMakAPoonWLImmunoablative cyclophosphamide for refractory lupus-related neuromyelitis opticaJ Rheumatol200835117217418176991
- PolgárARozsaCMullerVMatolcsiJPoorGKissEVDevic’s syndrome and SLE: challenges in diagnosis and therapeutic possibilities based on two overlapping casesAutoimmun Rev201110317117420920613
- ChiaWCWangJNLaiMCNeuromyelitis optica: a case reportPediatr Neonatol201051634735221146800
- BirnbaumJKerrDOptic neuritis and recurrent myelitis in a woman with systemic lupus erythematosusNat Clin Pract Rheumatol20084738138618493269
- ArabshahiBPollockANSherryDDAlbertDAKreigerPAPeslerFDevic disease in a child with primary Sjögren syndromeJ Child Neurol200621428528616900921
- CapobiancoMMalucchiSdi SapioAVariable responses to rituximab treatment in neuromyelitis optica (Devic’s disease)Neurol Sci200728420921117690854
- NasirSKerrDABirnbaumJNineteen episodes of recurrent myelitis in a woman with neuromyelitis optica and systemic lupus erythematosusArch Neurol20096691160116319752308
- MahmoodNASilverKOnelKKoMJavedAEfficacy and safety of rituximab in pediatric neuromyelitis opticaJ Child Neurol201126224424721183724