
Abstract
Background
Inhaled nanoparticles can cross pulmonary air–blood barrier into circulation and cause vascular endothelial injury and progression of cardiovascular disease. However, the molecular mechanism underlying the vascular toxicity of copper oxide nanoparticles (CuONPs) remains unclear. We have recently demonstrated that the release of copper ions and the accumulation of superoxide anions contributed to CuONPs-induced cell death in human umbilical vein endothelial cells (HUVECs). Herein, we further demonstrate the mechanism underlying copper ions-induced cell death in HUVECs.
Methods and Results
CuONPs were suspended in culture medium and vigorously vortexed for several seconds before exposure. After treatment with CuONPs, HUVECs were collected, and cell function assays were conducted to elucidate cellular processes including cell viability, oxidative stress, DNA damage and cell signaling pathways. We demonstrated that CuONPs uptake induced DNA damage in HUVECs as evidenced by γH2AX foci formation and increased phosphorylation levels of ATR, ATM, p53 and H2AX. Meanwhile, we showed that CuONPs exposure induced oxidative stress, indicated by the increase of cellular levels of superoxide anions, the upregulation of protein levels of heme oxygenase-1 (HO-1) and glutamate-cysteine ligase modifier subunit (GCLM), the elevation of the levels of malondialdehyde (MDA), but the reduction of glutathione to glutathione disulfide ratio. We also found that antioxidant N-acetyl-L-cysteine (NAC) could ameliorate CuONPs-induced oxidative stress and cell death. Interestingly, we demonstrated that p38 mitogen-activated protein kinase (MAPK) signaling pathway was activated in CuONPs-treated HUVECs, while p38α MAPK knockdown by siRNA significantly rescued HUVECs from CuONPs-induced DNA damage and cell death. Importantly, we showed that copper ions chelator tetrathiomolybdate (TTM) could alleviate CuONPs-induced oxidative stress, DNA damage, p38 MAPK pathway activation and cell death in HUVECs.
Conclusion
We demonstrated that CuONPs induced oxidative DNA damage and cell death via copper ions-mediated p38 MAPK activation in HUVECs, suggesting that the release of copper ions was the upstream activator for CuONPs-induced vascular endothelial toxicity, and the copper ions chelator TTM can alleviate CuONPs-associated cardiovascular disease.
Introduction
Copper oxide nanoparticles (CuONPs) is important metal oxide nanomaterial, which exhibits good conductive, catalytic and antibacterial properties. It has been widely used as semiconductors,Citation1 oxidation catalysts,Citation2 energy storage materials,Citation3 and antibacterial agents.Citation4 Despite its widespread industrial and biomedical application, concerns regarding potential toxic impacts of CuONPs have increased rapidly. CuONPs have been reported as the most toxic metal oxide nanoparticles.Citation5,Citation6 Previous in vitro studies have reported that CuONPs can induce cytotoxicity in numerous cell types, including human A549 lung epithelial cells,Citation7–Citation9 MCF7 breast cancer cells,Citation10 HL6 0promyelocytic leukemic cells,Citation11 HepG2 liver hepatocellular carcinoma cells,Citation12 frog A6 kidney epithelial cellsCitation13 and rat primary brain astrocytes.Citation14 Still, the molecular mechanism underlying CuONPs cytotoxicity remains incompletely understood.
Cardiovascular disease is the number one cause of death globally.Citation15 Vascular endothelial cells form the inner surface of blood vessels, whose dysfunction leads to the development of cardiovascular disease.Citation16 Increasing evidence has indicated that engineered nanoparticles exposure may penetrate pulmonary air–blood barrier into circulation. Recently, Miller et al, further clarified that inhaled nanoparticles selectively accumulated at sites of vascular inflammation in both animal models of disease and in human volunteers,Citation17 suggesting that inhaled nanoparticles can directly interact with vascular endothelial cell, resulting in endothelial injury and acceleration of cardiovascular diseases.
It has been suggested that the toxicity of metal oxide nanoparticles results from the release of metal ions or/and the nanoparticles themselves. Our recent study demonstrated that autophagy-dependent release of zinc ions was involved in human A549 lung epithelial cell death and acute lung injury in mice induced by zinc oxide nanoparticles exposure.Citation18,Citation19 Hanagata et al reported that the released copper ions, rather than CuONPs themselves, mediated CuONPs toxicity in A549 lung epithelial cells.Citation9 Recently, we also demonstrated that lysosomal deposition of CuONPs facilitated copper ions release and triggered CuONPs-induced human umbilical vein endothelial cells (HUVECs) death.Citation20 However, the exact roles of copper ions in CuONPs toxicity need further be investigated.
Reactive oxygen species (ROS) are chemically reactive molecules, which mainly originate from damaged mitochondria. ROS are mainly composed of superoxide anions (O2−), hydrogen peroxide (H2O2) and hydroxyl free radical (·OH). Oxidative stress, indicated by increased intracellular ROS, is associated with nanoparticles cytotoxicity.Citation21 Oxidative stress can result in oxidized nucleic acids, amino acids, carbohydrates and lipids.Citation22 Previous studies have suggested that nanoparticles-induced oxidation of nucleic acids could cause DNA damage, cell cycle arrest and subsequent cell death.Citation23,Citation24 Our recent study also demonstrated that CuONPs exposure caused mitochondrial dysfunction and the accumulation of mitochondrial superoxide anions in HUVECs.Citation25
In the current study, we clarified the potential links between the release of copper ions, oxidative stress response and p38 MAPK activation in CuONPs-induced cytotoxicity in vascular endothelial cells. We demonstrated that the copper ions released from CuONPs induced oxidative stress and p38 MAPK signaling activation, which subsequently caused DNA damage and cell death in HUVECs. Our findings contribute to clarifying the mechanisms underlying CuONPs toxicity in the circulation system and suggest that the chelation of copper ions may be an effective means of alleviating CuONPs toxicity.
Materials and Methods
Materials and Cell Culture
CuONPs (<50 nm particle size, #544868) were purchased from Sigma Aldrich (St. Louis, MO, USA) and characterized as previously described.Citation20 Human umbilical vein endothelial cells (HUVECs), the most widespread in vitro model for studying endothelial physiology and pathology, was obtained from the American Type Culture Collection (Rockville, MD, USA) and was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 100 U penicillin-streptomycin at 37°C in a 5% CO2 humidified incubator.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) was used to characterize CuONPs morphology. Briefly, a few drops of CuONPs solution in deionized water were added to carbon-coated copper electron microscope grids and observed under a TEM (Hitachi-7500, Japan) after drying at room temperature. Cell ultrastructure was also observed under TEM. After treatment and subsequent trypsinization, cells were centrifuged at 1200 rpm/min for 5 min and fixed using 4% glutaraldehyde for 2 h at 4°C. Then, cells were postfixed using 1% osmium tetroxide for 1 h at 4°C, dehydrated in a graded series of alcohol and acetone and embedded in Epon 816 (Electron Microscopy Sciences, Hatfield, PA, USA). Ultrathin sections were prepared using a Leica ultramicrotome (Leica Microsystems, Buffalo Grove, IL, USA) and stained with uranyl acetate-lead citrate. TEM images were obtained using a JEM-1400Plus transmission electron microscope (JEOL Ltd. Tokyo, Japan).
MTS Cell Viability Assay
Cell viability was assayed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) kit (#G3581, Promega, Madison, WI, USA) as described previously.Citation19 Briefly, HUVECs were seeded in 96-well plates overnight. After treatment with CuONPs or other reagents for indicated times, the MTS solution was diluted 10-fold in DMEM and subsequently added to each well of the cultured plate. The plate was incubated at 37°C for 1 h and the absorbance of each well at 490 nm was measured using a VERS Amax Microplate Reader (Molecular Devices Corp, Sunnyvale, CA, USA). All assays were conducted in triplicate.
Fluorescence Activated Cell Sorting (FACS) Assay
FACS assays were performed as previously described.Citation20,Citation25,Citation26 Calcein AM (50 nM, Thermo Fisher Scientific) was used to determine cell viability. DHE (5 nM, Beyotime, Jiangsu, China) and MitoSOX (5μM, Thermo Fisher Scientific) were used to measure cellular superoxide anions and mitochondrial superoxide anions, respectively. All FACS assays were performed using CytoFLEX Platform (Beckman Coulter, Miami, FL, USA), and the results were analyzed using FlowJo Version VX software (TreeStar, Ashland, OR, USA). All assays were conducted in triplicate.
Comet Tail Assay
Comet tail assay was conducted as described previously.Citation27 Briefly, HUVECs seeded in 12-well plates were treated using CuONPs (20 μg/mL) for 12 h. Then, the cells were collected and added a few droplets of low-melting agarose (Amresco, Solon, USA). The cell mixture was then spread onto a slide precoated with normal-melting agarose (Amresco, Solon, OH, USA), and incubated in lysis buffer at 4°C for 1 h. Then, the slides were placed in electrophoresis buffer for 30 min and neutralized in water. After dehydration at room temperature, the slides were stained using ethidium bromide (Amresco, Solon, OH, USA) and observed under a fluorescence microscope (Eclipse TiU, Nikon Corp., Tokyo, Japan).
Immunofluorescence Staining
Cells were fixed using cold methanol for 15 min and subsequently permeabilized with 0.2% Triton X-100 in phosphate buffer saline (PBS) for 10 min at room temperature. After blocking with 2% bovine serum albumin in PBS for 1 h at room temperature, cells were incubated with a primary antibody against phospho-histone H2AX (S139) (γH2AX, 1:100, Cell Signaling Technology, #9508) diluted in a blocking solution at room temperature for 1 h. After three washes with PBS, the cells were incubated with Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary antibody was used (1:500, Molecular Probes, #A-21207) and 4ʹ,6-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR, USA) diluted in PBS for 1 h at room temperature. After washing with PBS, the coverslips were sealed using nail polish. Fluorescence images were obtained using a Nikon A1R+/A1 Confocal Microscope (Nikon, Tokyo, Japan).
Western Blotting Assay
Cells were directly lysed using 2 × protein lysing buffer (2% SDS, 5% β-mercaptoethanol, 0.5% sucrose and 0.2% bromophenol blue) at 4°C for 20 min, and then the lysates were heated in a metal bath at 98°C for 5 min. Cell lysates were separated on 4–12% SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, Billerica, MA, USA). The membranes were blocked using 5% non-fat milk and immunoblotted using indicated antibodies. The protein signals were detected using a New-SUPER ECL Substrate Kit (Keygen Biotech, Nanjing, China). The primary antibodies used in this study targeted phospho-ATR (S428) (1:1000, abcam, ab178407), phospho-ATM (S1981) (1:1000, Cell Signaling Technology, #5883), phospho-p53 (S15) (1:1000, abcam, ab223868), phospho-histone H2AX (S139) (1:1000, Cell Signaling Technology, #9508), GCLM (1:1000, abcam, ab126704), HO-1 (1:1000, abcam, ab68477), phospho-p38 (T180/Y182) (1:500, Santa Cruz Biotechnology, sc-17852-R), phospho-Hsp27 (S82) (1:1000, abcam, ab155987), phospho-ATF2 (T71) (1:1000, abcam, ab32019) and Actin (1:3000, Bioss, #bs-0061R). The secondary antibodies used in this study were horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:10,000, TransGen Biotech, Beijing, China, #HS201-01) and HRP-conjugated goat anti-rabbit IgG (1:10,000, TransGen Biotech, Beijing, China, #HS101-01).
GSH/GSSG Assay
Reduced (GSH) and oxidized (GSSG) glutathione in CuONPs-treated HUVECs were measured spectrophotometrically using a GSH and GSSG Assay Kit (Beyotime, Shanghai, China). Briefly, HUVECs were added in a three-fold volume of protein removal reagent M and fully vortexed. Then, the cell samples were freeze-thawed in liquid nitrogen and a 37°C water bath. After centrifuging at 10,000 g for 10 min at 4°C, the supernatant was used for the determination of the GSH/GSSG ratio in accordance with the manufacturer’s protocol. All assays were conducted in triplicate.
MDA Assays
The level of MDA in the CuONPs-treated HUVECs was assayed using a Lipid Peroxidation MDA Assay Kit (Beyotime, Shanghai, China). Briefly, HUVECs seeded into 12-well plates were directly lysed using cell lysis buffer (Beyotime, Shanghai, China). The lysate was centrifuged for 10 min at 12,000 g and the supernatant was collected. The protein concentrations were measured using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Then, an equal volume of supernatant was incubated with thiobarbituric acid buffer at 100°C for 15 min. Subsequently, the absorbance was measured at 532 nm using a VERS Amax Microplate Reader (Molecular Devices Corp, Sunnyvale, CA, USA). The values were expressed as MDA (nmol/mg protein). All assays were conducted in triplicate.
RNA Interference
A small interfering RNA (siRNA) targeting human p38α (GCCCAUAAGGCCAGAAACUtt) and a negative control siRNA (siNC, UUCUCCGAACGUGUCACGUtt) were obtained from GenePharma (Shanghai, China). Cells were transfected with the siRNAs using Lipofectamine RNAiMAX (#13778150, Invitrogen) for 48 h in accordance with the manufacturer’s instructions. The knockdown efficiencies of the siRNAs were determined by Western blotting analysis.
Statistical Analysis
Comparisons between two groups were made through unpaired Student’s t-test. Comparisons of parameters among multiple groups were made using one-way ANOVA followed by Tukey’s test. The data are shown as the mean ± standard deviation (S.D.). Each experiment was repeated at least three times. All statistical tests were conducted using Prism 6 software (GraphPad Software, San Diego, CA, USA). **p<0.05 indicates differences.
Results
Physical Properties and Cellular Uptake of CuONPs
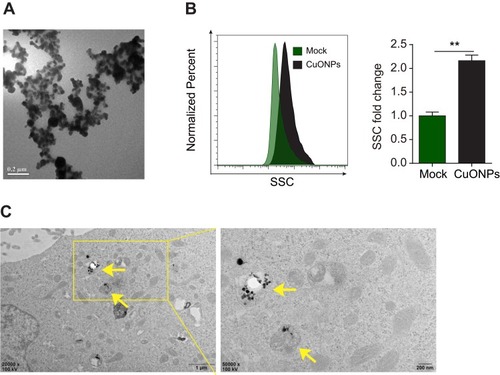
The energy dispersive spectroscopy and dynamic light scattering properties of CuONPs were examined in our previous study.Citation20 The morphology of CuONPs in deionized water was characterized by transmission electron microscope (TEM). TEM images showed that CuONPs was subglobose shape with an average size of <50 nm (). Cellular uptake of CuONPs was confirmed by FACS assay as described previously.Citation28 FACS results showed that CuONPs induced an increase of SSC intensity of HUVECs, indicating the endocytosis of CuONPs into HUVECs (). Furthermore, TEM images showed that CuONPs mainly aggregated in the lysosomes of HUVECs ().
Figure 1 Physical properties and cellular uptake of CuONPs. (A) A representative transmission electron microscope (TEM) image of CuONPs dissolved in distilled water. (B) Representative FACS data for SSC intensity of HUVECs treated with 20 μg/mL CuONPs for 12 h. Mock, untreated HUVECs. Unpaired Student’s t-tests were performed for statistical analysis. **p < 0.05 versus untreated HUVECs. (C) Representative TEM images of HUVECs treated with 20 μg/mL CuONPs for 12 h. Yellow arrows indicate lysosomal deposition of CuONPs.
CuONPs Triggered DNA Damage and Cell Death in HUVECs
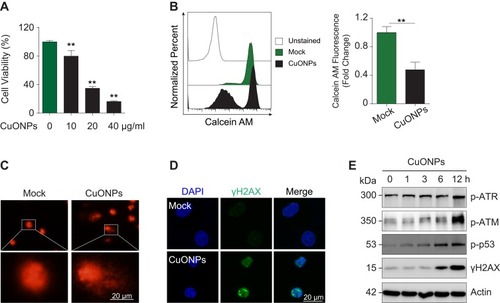
Cytotoxic effects of CuONPs on vascular endothelial cells were investigated with MTS assay. The results showed that CuONPs reduced HUVECs viability in a dose-dependent manner (). FACS assay after Calcein AM labeling also confirmed that CuONPs exposure caused more than 50% of cell death in HUVECs (). Comet assay results revealed that CuONPs treatment caused DNA damage in HUVECs (). DNA damage in CuONPs-treated HUVECs were further evaluated by γH2AX foci formation (a sensitive molecular marker of DNA damage). The results from immunofluorescence staining demonstrated that CuONPs treatment resulted in the formation of γH2AX foci in the nuclei of HUVECs (). Furthermore, we showed that the phosphorylation levels of ATR, ATM, p53 and H2AX increased obviously, suggesting that CuONPs caused DNA damage in HUVECs in an ATM/ATR-p53-dependent manner ( and Figure S1A–D). Collectively, these results indicate that CuONPs exposure trigger DNA damage and cell death in HUVECs.
Figure 2 DNA damage and cell death in CuONPs-treated HUVECs. (A) MTS assay of HUVECs treated with different concentrations of CuONPs (0, 10, 20, or 40 μg/mL) for 24 h. (B) Representative FACS data for CuONPs-treated HUVECs after Calcein AM (live cell fluorescent probe) staining. (C) Comet assay to evaluate CuONPs-induced DNA damage in HUVECs. (D) Immunofluorescence assay of γH2AX foci formation in CuONPs-treated HUVECs. (E) Western blotting assay of phospho-ATR, phospho-ATM, phospho-p53 and phospho-H2AX (γH2AX) in HUVECs treated with CuONPs (20 μg/mL) for 0, 1, 3, 6 and 12 h. Actin was used as a loading control. In (A), one-way ANOVA followed by Tukey’s test was performed for statistical analysis. In (B), unpaired Student’s t-tests were performed for statistical analysis. **p < 0.05 versus untreated HUVECs.
CuONPs Induced Oxidative Stress in HUVECs
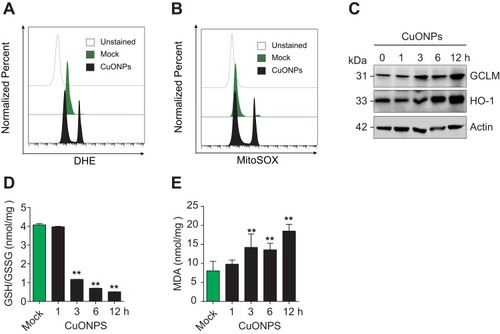
To investigate whether CuONPs trigger oxidative stress in HUVECs, the level of superoxide anions in CuONPs-treated HUVECs were detected using the fluorescent probe DHE and MitoSOX. FACS data showed that CuONPs exposure significantly increased intracellular level of superoxide anions in HUVECs ( and Figure S2A–B). Then, the protein levels of antioxidant enzymes GCLM and HO-1 in HUVECs were determined. Our results revealed that GCLM and HO-1 were upregulated in CuONPs-treated HUVECs in a dose-dependent manner ( and Figure S2C–D). Analysis of GSH and GSSG showed that CuONPs caused a decrease in GSH/GSSG ratio in HUVECs (). As lipid oxidation occurs when cells undergo oxidative stress, the cellular level of lipid oxidation in CuONPs-treated HUVECs were examined. The results showed that CuONPs also caused MDA accumulation in HUVECs (). In brief, our results suggest that CuONPs induced oxidative stress in HUVECs.
Figure 3 CuONPs treatment caused oxidative stress in HUVECs. (A) Representative FACS data for cellular superoxide anions in CuONPs-treated HUVECs after staining with the fluorescent probe DHE. (B) Representative FACS data for mitochondrial superoxide anions in CuONPs-treated HUVECs after staining with the fluorescent probe MitoSOX. (C) Western blotting analysis of protein levels of GCLM and HO-1. Actin was used as a loading control. (D) GSH/GSSG ratio assays of HUVECs treated with CuONPs (20 μg/mL) for 0, 1, 3, 6 and 12 h. (E) Lipid peroxidation (MDA) assay of HUVECs treated with CuONPs (20 μg/mL) for 0, 1, 3, 6 and 12 h. In (D) and (E), one-way ANOVA followed by Tukey’s test was performed for statistical analysis. **p < 0.05 versus untreated HUVECs.
CuONPs-Induced Oxidative Stress Mediated DNA Damage and Cell Death in HUVECs
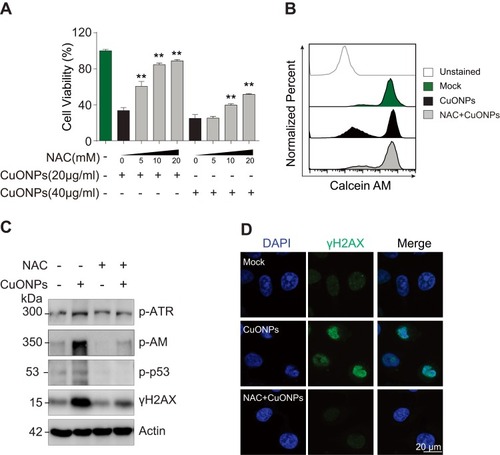
To determine the role of oxidative stress in CuONPs-treated HUVECs, we pretreated HUVECs with antioxidant N-Acetyl-L-cysteine (NAC). The results demonstrated that NAC ameliorated CuONPs-induced cell death in a dose-dependent manner (). Calcein AM assay also showed that NAC rescued HUVECs from CuONPs-induced cell death ( and S3A). Moreover, NAC significantly prevented the CuONPs-induced upregulation of phosphorylation levels of ATR, ATR, p53 and H2AX in HUVECs ( and S3B–E). NAC pretreatment also reduced γH2AX foci formation in HUVECs induced by CuONPs (). All these data indicate that oxidative stress-mediated DNA damage and cell death in CuONPs-treated HUVECs.
Figure 4 Oxidative stress-mediated DNA damage and cell death in CuONPs-treated HUVECs. (A) MTS assay of HUVEC cell viability after pretreatment with different concentrations of NAC (0, 5, 10 or 20 mM) for 1 h and then treatment with CuONPs (20 or 40 μg/mL) for 24 h. one-way ANOVA followed by Tukey’s test was performed for statistical analysis. **p < 0.05. (B) Representative FACS data for Calcein AM fluorescence of CuONPs-treated HUVECs. (C) Western blotting assay of phospho-ATR, phospho-ATM, phospho-p53 and γH2AX in HUVECs, which were pretreated with or without NAC (10 mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. Actin was used as a loading control. (D) Immunofluorescence assay of γH2AX foci formation in CuONPs-treated HUVECs.
p38 MAPK Contributed to CuONPs-Induced DNA Damage and Cell Death in HUVECs
The p38 MAPK is activated in response to cellular stress. In this study, we showed that CuONPs also induced a robust activation of p38 MAPK and subsequent phosphorylation of HSP27 and ATF-2, two downstream effectors of the p38 MAPK pathway ( and S4A). Next, we examined whether p38 MAPK plays a role in CuONPs toxicity on vascular endothelial cells. The results showed that p38α MAPK knockdown by siRNA prevented CuONPs-induced changes of the phosphorylation levels ATR, H2AX, HSP27 and ATF-2 in HUVECs ( and S4B–C). Then, we demonstrated that p38α MAPK knockdown significantly rescued HUVECs from CuONPs-induced cell death in a dose-dependent manner (). Collectively, these data demonstrate that p38 MAPK signaling is directly involved in CuONPs-induced DNA damage and cell death in HUVECs.
Figure 5 p38 MAPK was involved in DNA damage and cell death in CuONPs-treated HUVECs. (A) Western blotting assay of phospho-p38, phospho-Hsp27 and phosphor-ATF-2 in HUVECs treated with CuONPs (20 μg/mL) for 0, 1, 3, 6 and 12 h. Actin was used as a loading control. (B) Western blotting assay of p38α in HUVECs transfected with 75 nM siNC (negative control siRNA) or si-p38α for 48 h. Actin was used as a loading control. (C) Western blotting assay of phospho-ATR, γH2AX, phospho-Hsp27 and phospho-ATF-2 in HUVECs, which were transfected with siNC or si-p38α for 48 h and then treated with CuONPs (20 μg/mL) for 12 h. Actin was used as a loading control. (D) MTS assay of HUVEC after transfection with siNC or si-p38α for 48 h and then treatment with different concentrations of CuONPs (0, 10, 20 or 40 μg/mL) for 24 h. One-way ANOVA followed by Tukey’s test was performed for statistical analysis. **p < 0.05 versus untreated HUVECs.
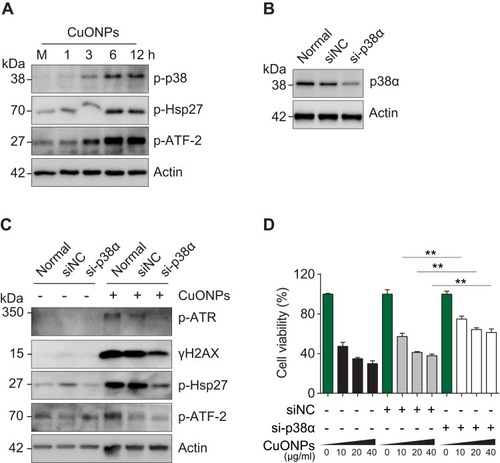
The Copper Ions Chelator Tetrathiomolybdate Alleviated P38 MAPK-Mediated DNA Damage and Cell Death in CuONPs-Treated HUVECs
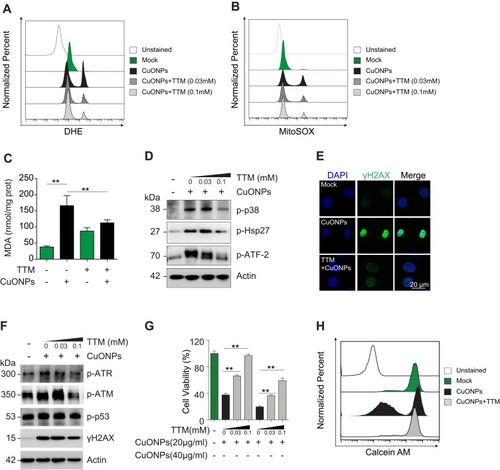
In our previous study, we demonstrated that CuONPs exposure resulted in the release of copper ions in HUVECs.Citation20 Therefore, in the current study, we determined whether the released copper ions mediate p38 MAPK-mediated DNA damage and cell death in CuONPs-treated HUVECs. We showed that copper ions chelator TTM prevent the upregulation of superoxide anions in CuONPs-treated HUVECs ( and S5A–B). The results from MDA assay showed that TTM alleviated CuONPs-induced oxidative stress in HUVECs (). In addition, TTM inhibited the activation of p38 MAPK, indicated by the reduction of p38 MAPK, HSP27 and ATF-2 phosphorylation in TTM-treated HUVECs ( and S5C). Furthermore, the chelation of copper ions also attenuated CuONPs-induced γH2AX foci formation and the upregulation of ATR, ATM, p53 and H2AX phosphorylation in HUVECs ( and S5D). Finally, we showed that the chelation of copper ions rescued HUVECs from CuONPs-induced cell death in a dose-dependent manner (). Calcein AM assay further confirmed that TTM treatment reduced HUVECs death induced by CuONPs ( and S5E). Collectively, these findings indicate that copper ions-superoxide anions-p38 MAPK-DNA damage axis mediated CuONPs-induced cell death in HUVECs.
Figure 6 Copper ions chelator TTM alleviated HUVECs DNA damage and cell death induced by CuONPs. (A) and (B) Representative FACS data regarding cellular superoxide anions and mitochondrial superoxide anions after staining with the fluorescent probe DHE and MitoSOX, respectively. HUVECs were pretreated with different concentrations of the copper ions chelator TTM (0, 0.03 and 0.1 mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. (C) Lipid peroxidation (MDA) assay of HUVECs pretreated with or without TTM (0.1 mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. (D) Western blotting assay of phosphor-p38, phosphor-Hsp27 and phosphor-ATF-2 in HUVECs, pretreated with or without TTM (0, 0.03, and 0.1mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. Actin was used as a loading control. (E) Immunofluorescence assay of the level of γH2AX in CuONPs-treated HUVECs, pretreated with or without TTM (0.1 mM). (F) Western blotting assay of phospho-ATR, phospho-ATM, phospho-p53 and γH2AX in HUVECs, which were pretreated with or without TTM (0, 0.03, and 0.1 mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. Actin was used as a loading control. (G) MTS assay of HUVECs viability after pretreatment with different concentrations of the copper ions chelator TTM (0, 0.03, and 0.1 mM) for 1 h and then treatment with CuONPs (20 or 40 μg/mL) for 24 h. (H) Representative FACS data for Calcein AM fluorescence in HUVECs, which were pretreated with or without TTM (0.1 mM) for 1 h and then treated with CuONPs (20 μg/mL) for 12 h. In (C) and (G), one-way ANOVA followed by Tukey’s test was performed for statistical analysis. **p < 0.05.
Discussion
CuONPs have been widely applied in multiply areas.Citation29,Citation30 Air–blood barrier translocation of inhaled CuONPs may directly modulate endothelial dysfunction and the pathogenesis of atherosclerosis,Citation17 thus it is important to elucidate the molecular mechanism underlying CuONPs-induced vascular endothelial dysfunction.
In our previous studies, we showed that CuONPs exposure induced excessive accumulation of superoxide anions in vascular endothelial cells, which are mainly attributed to CuONPs lysosomal deposition-induced lysosomal dysfunction and disruption of protective mitophagy.Citation20,Citation25 In the current study, we further confirmed that CuONPs induced oxidative stress in HUVECs, indicated by the increase of cellular levels of superoxide anions, the upregulation of protein levels of HO-1 and GCLM, the elevation of the levels of MDA, but the reduction of GSH/GSSG ratio (). Furthermore, we revealed that CuONPs-induced oxidative stress directly triggered DNA damage and cell death in HUVECs ( and ). This finding is consistent with previous studies reporting that oxidative stress is a mechanism of CuONPs toxicity.Citation31–Citation33 Oxidative stress may lead to irreversible oxidation of proteins, lipids and nucleic acids, consequently trigger DNA damage and cell death in HUVECs.
Copper is one of the important metal elements in maintaining homeostasis.Citation34 Copper deficiency can result in a wide variety of clinical manifestations including impaired growth, neurological dysfunction and other possible abnormalities.Citation35 Conversely, copper overload can also cause many diseases such as Wilson’s diseaseCitation36 and Alzheimer’s disease.Citation37 Previous studies have demonstrated that the toxicity of metal nanomaterials may originate from the nanoparticles themselves, the released metal ions or a combination of both.Citation18,Citation38 In our previous study, we showed that the copper ions released from CuONPs was involved in CuONPs-induced cell death.Citation20 In the current study, we confirmed that TTM, a copper chelator for the treatment of Wilson’s disease,Citation39 alleviated CuONPs-induced lipid oxidation, HUVECs DNA damage and cell death in HUVECs (). Our data suggest that the release of copper ions were the upstream factor of CuONPs-induced DNA damage and cell death in HUVEC, indicating that TTM may be an effective treatment for CuONPs-cardiovascular diseases.
The exact roles of copper ions in CuONPs toxicity are also explored in the present study. We demonstrated that the release of copper ions was involved in oxidative stress response in CuONPs-treated HUVECs (). It has been reported that the levels of serum copper ions are increased under inflammatory conditions to trigger oxidative stress responses that activate inflammatory responses.Citation40 Conversely, copper ions selective chelators can alleviate copper-induced oxidative stress.Citation41 The p38 MAPK signaling pathway has been implicated in cellular responses including inflammation, metabolism, cell death, development, cell differentiation, senescence, and tumorigenesis.Citation42,Citation43 The p38 MAPK signaling pathway has been implicated in cellular responses to exposure to CuONPs in vivo and in vitro model.Citation25,Citation44,Citation45 In this study, we further determined that p38 MAPK pathway activation was implicated in copper ions-induced oxidative stress in HUVECs (). All these data indicate that the CuONPs exposure causes a significant increase of cellular copper ions, which induce oxidative stress and activate MAPK pathways. Nevertheless, the upstream regulators and downstream effectors of p38 MAPK in CuONPs-treated HUVECs need further investigations.
It is well documented that p38 MAPK is activated in response to DNA double strand breaks (DSBs).Citation46 DSBs, in turn, promoted nuclear translocation of p38 MAPK to facilitate the phosphorylation of p38 MAPK downstream effectors to induce G2/M cell cycle checkpoint and DNA repair.Citation47 There is increasing evidence that p38 MAPK signaling pathway is also involved in nanoparticle-induced cytotoxicity. Gold nanoparticles exposure induced mechanical stress and activation of p38 MAPK pathway in mesenchymal stem cells.Citation48 In the current study, our results revealed that CuONPs increased the phosphorylation levels of p38 MAPK and its downstream targets Hsp27 and ATF-2, while p38 MAPK knockdown could prevent CuONPs-induced cytotoxicity in HUVECs (). This supports the previous findings showing that silver nanoparticles exposure activates p38 MAPK through the nuclear factor-E2-related factor-2 and nuclear factor-kappaB signaling pathways, subsequently inducing DNA damage, cell cycle arrest and apoptosis. These findings also indicate that p38 MAPK may play dual roles in nanoparticle-induced cytotoxicity and genotoxicity. Nanoparticle-induced DNA damage stress may activate p38 MAPK to induce DNA repair or trigger cell death directly.
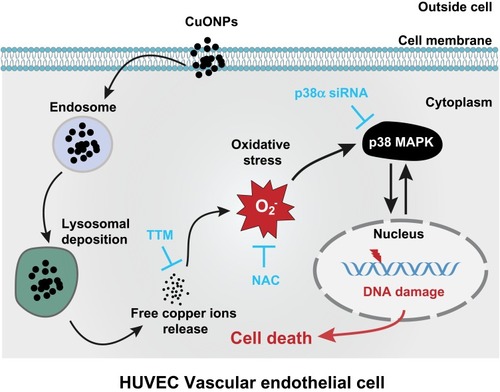
In summary, our findings demonstrate that the release of copper ions contributes to CuONPs-induced vascular endothelial cell death. The released copper ions induced oxidative stress and the activation of the p38 MAPK signaling pathway in HUVECs, while the chelation of copper ions significantly reduced the oxidative stress response and inhibited the activation of p38 MAPK signaling. Furthermore, CuONPs-induced oxidative stress may trigger DNA damage and apoptotic cell death in HUVECs through the p38 MAPK signaling pathway, because p38α knockdown significantly rescued CuONPs-induced oxidative DNA damage and HUVECs death (). These findings provide an insight into the underlying links between the release of copper ions, oxidative stress and activation of downstream p38 MAPK signaling in CuONPs-induced cytotoxicity in the cardiovascular system.
Figure 7 Schematic representation of the role of released copper ions in p38 MAPK-mediated DNA damage and cell death in CuONPs-treated HUVECs. Cellular uptake and lysosomal deposition of CuONPs lead to the release of copper ions from CuONPs, which triggers oxidative stress response and p38 MAPK signaling activation. The activation of p38 MAPK signaling pathway contributes to CuONPs-induced DNA damage and cell death in HUVECs. Meanwhile, CuONPs-induced DNA damage may feedback-regulate p38 MAPK signaling pathway, resulting in aggravation of DNA damage and cell death in HUVECs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81500343 and 81903358), the Chongqing Research Program of Basic Research and Frontier Technology (cstc2018jcyjAX0355, cstc2017jcyjAX0162) and the Science and Technology Project Affiliated to the Education Department of Chongqing, China (KJQN201800434 and KJQN201900421).
Disclosure
The authors report no conflicts of interest in this work.
References
- Jiang XC, Herricks T, Xia YN. CuO nanowires can be synthesized by heating copper substrates in air. Nano Lett. 2002;2(12):1333–1338. doi:10.1021/nl0257519
- Diao FY, Tian FH, Liang WS, Feng HL, Wang YQ. Mechanistical investigation on the self-enhanced photocatalytic activity of CuO/Cu2O hybrid nanostructures by density functional theory calculations. Phys Chem Chem Phys. 2016;18(40):27967–27975. doi:10.1039/C6CP03977D27711427
- Choi KJ, Jang HW. One-dimensional oxide nanostructures as gas-sensing materials: review and issues. Sensors. 2010;10(4):4083–4099. doi:10.3390/s10040408322319343
- Oberdorster G. Pulmonary effects of inhaled ultrafine particles. Int Arch Occ Env Hea. 2001;74(1):1–8. doi:10.1007/s004200000185
- Xu M, Fujita D, Kajiwara S, et al. Contribution of physicochemical characteristics of nano-oxides to cytotoxicity. Biomaterials. 2010;31(31):8022–8031. doi:10.1016/j.biomaterials.2010.06.02220688385
- Karlsson HL, Cronholm P, Gustafsson J, Moller L. Copper oxide nanoparticles are highly toxic: a comparison between metal oxide nanoparticles and carbon nanotubes. Chem Res Toxicol. 2008;21(9):1726–1732. doi:10.1021/tx800064j18710264
- Sun T, Yan Y, Zhao Y, Guo F, Jiang C, Srinivasula SM. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One. 2012;7(8):e43442. doi:10.1371/journal.pone.004344222916263
- Wongrakpanich A, Mudunkotuwa IA, Geary SM, et al. Size-dependent cytotoxicity of copper oxide nanoparticles in lung epithelial cells. Environ Sci Nano. 2016;3(2):365–374. doi:10.1039/C5EN00271K27347420
- Hanagata N, Zhuang F, Connolly S, Li J, Ogawa N, Xu M. Molecular responses of human lung epithelial cells to the toxicity of copper oxide nanoparticles inferred from whole genome expression analysis. ACS Nano. 2011;5(12):9326–9338. doi:10.1021/nn202966t22077320
- Laha D, Pramanik A, Maity J, et al. Interplay between autophagy and apoptosis mediated by copper oxide nanoparticles in human breast cancer cells MCF7. Biochim Biophys Acta. 2014;1840(1):1–9. doi:10.1016/j.bbagen.2013.08.01123962629
- Rodhe Y, Skoglund S, Odnevall Wallinder I, Potacova Z, Moller L. Copper-based nanoparticles induce high toxicity in leukemic HL60 cells. Toxicol In Vitro. 2015;29(7):1711–1719. doi:10.1016/j.tiv.2015.05.02026028147
- Piret JP, Jacques D, Audinot JN, et al. Copper(II) oxide nanoparticles penetrate into HepG2 cells, exert cytotoxicity via oxidative stress and induce pro-inflammatory response. Nanoscale. 2012;4(22):7168–7184.23070296
- Thit A, Selck H, Bjerregaard HF. Toxic mechanisms of copper oxide nanoparticles in epithelial kidney cells. Toxicol in Vitro. 2015;29(5):1053–1059. doi:10.1016/j.tiv.2015.03.02025862124
- Bulcke F, Thiel K, Dringen R. Uptake and toxicity of copper oxide nanoparticles in cultured primary brain astrocytes. Nanotoxicology. 2014;8(7):775–785. doi:10.3109/17435390.2013.82959123889294
- Franklin BA, Brook R, Pope III CA. Air pollution and cardiovascular disease. Curr Probl Cardiol. 2015;40(5):207–238. doi:10.1016/j.cpcardiol.2015.01.00325882781
- Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438(7070):937. doi:10.1038/nature0447916355211
- Miller MR, Raftis JB, Langrish JP, et al. Inhaled nanoparticles accumulate at sites of vascular disease. ACS Nano. 2017;11(5):4542–4552. doi:10.1021/acsnano.6b0855128443337
- Jiang X, Tang Q, Zhang J, et al. Autophagy-dependent release of zinc ions is critical for acute lung injury triggered by zinc oxide nanoparticles. Nanotoxicology. 2018;12(9):1068–1091. doi:10.1080/17435390.2018.151309430317896
- Zhang J, Qin X, Wang B, et al. Zinc oxide nanoparticles harness autophagy to induce cell death in lung epithelial cells. Cell Death Dis. 2017;8(7):e2954. doi:10.1038/cddis.2017.33728749469
- Zhang J, Zou Z, Wang B, et al. Lysosomal deposition of copper oxide nanoparticles triggers HUVEC cells death. Biomaterials. 2018;161:228–239. doi:10.1016/j.biomaterials.2018.01.04829421558
- von Moos N, Slaveykova VI. Oxidative stress induced by inorganic nanoparticles in bacteria and aquatic microalgae–state of the art and knowledge gaps. Nanotoxicology. 2014;8(6):605–630. doi:10.3109/17435390.2013.80981023738945
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–R462. doi:10.1016/j.cub.2014.03.03424845678
- Wan R, Mo Y, Feng L, Chien S, Tollerud DJ, Zhang Q. DNA damage caused by metal nanoparticles: involvement of oxidative stress and activation of ATM. Chem Res Toxicol. 2012;25(7):1402–1411. doi:10.1021/tx200513t22559321
- Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis. 2012;17(8):852–870. doi:10.1007/s10495-012-0705-622395444
- Zhang Y, Ding Z, Zhao G, et al. Transcriptional responses and mechanisms of copper nanoparticle toxicology on zebrafish embryos. J Hazard Mater. 2018;344:1057–1068. doi:10.1016/j.jhazmat.2017.11.03930216965
- Qin X, Zhang J, Wang B, Xu G, Zou Z. LAMP-2 mediates oxidative stress-dependent cell death in Zn2+-treated lung epithelium cells. Biochem Biophys Res Commun. 2017;488(1):177–181. doi:10.1016/j.bbrc.2017.05.03028483530
- Chen C, Jiang X, Lai Y, Liu Y, Zhang Z. Resveratrol protects against arsenic trioxide-induced oxidative damage through maintenance of glutathione homeostasis and inhibition of apoptotic progression. Environ Mol Mutagen. 2015;56(3):333–346. doi:10.1002/em.2191925339131
- Park J, Ha MK, Yang N, Yoon TH. Flow cytometry-based quantification of cellular au nanoparticles. Anal Chem. 2017;89(4):2449–2456. doi:10.1021/acs.analchem.6b0441828192941
- Grigore ME, Biscu ER, Holban AM, Gestal MC, Grumezescu AM. Methods of synthesis, properties and biomedical applications of CuO nanoparticles. Pharmaceuticals. 2016;9(4):75. doi:10.3390/ph9040075
- Suleiman M, Mousa M, Hussein A, Hammouti B, Hadda TB, Warad I. Copper (II)-oxide nanostructures: synthesis, characterizations and their applications-review. J Mater Environ Sci. 2013;4(5):792–797.
- Jing X, Park JH, Peters TM, Thorne PS. Toxicity of copper oxide nanoparticles in lung epithelial cells exposed at the air–liquid interface compared with in vivo assessment. Toxicol in Vitro. 2015;29(3):502–511. doi:10.1016/j.tiv.2014.12.02325575782
- Niska K, Santos-Martinez MJ, Radomski MW, Inkielewicz-Stepniak I. CuO nanoparticles induce apoptosis by impairing the antioxidant defense and detoxification systems in the mouse hippocampal HT22 cell line: protective effect of crocetin. Toxicol in Vitro. 2015;29(4):663–671. doi:10.1016/j.tiv.2015.02.00425701151
- Ko J-W, Shin N-R, Park J-W, et al. Copper oxide nanoparticles induce collagen deposition via TGF-β1/Smad3 signaling in human airway epithelial cells. Nanotoxicology. 2018;12(3):239–250. doi:10.1080/17435390.2018.143277829383958
- Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21(21):R877–R883. doi:10.1016/j.cub.2011.09.04022075424
- Means RT. Nutritional Anemia: Scientific Principles, Clinical Practice, and Public Health. Cambridge University Press; 2019.
- Wooton-Kee CR, Jain AK, Wagner M, et al. Elevated copper impairs hepatic nuclear receptor function in Wilson’s disease. J Clin Invest. 2015;125(9):3449–3460. doi:10.1172/JCI7899126241054
- Quinn JF, Crane S, Harris C, Wadsworth TL. Copper in alzheimer’s disease: too much or too little? Expert Rev Neurother. 2009;9(5):631–637.19402774
- Wang D, Lin Z, Wang T, et al. Where does the toxicity of metal oxide nanoparticles come from: the nanoparticles, the ions, or a combination of both? J Hazard Mater. 2016;308:328–334. doi:10.1016/j.jhazmat.2016.01.06626852208
- Medici V, Sturniolo GC. Tetrathiomolybdate, a copper chelator for the treatment of Wilson disease, pulmonary fibrosis and other indications. IDrugs. 2008;11(8):592–606.18683094
- Pereira TC, Campos MM, Bogo MR. Copper toxicology, oxidative stress and inflammation using zebrafish as experimental model. J Appl Toxicol. 2016;36(7):876–885. doi:10.1002/jat.330326888422
- Rakshit A, Khatua K, Shanbhag V, Comba P, Datta A. Cu(2+) selective chelators relieve copper-induced oxidative stress in vivo. Chem Sci. 2018;9(41):7916–7930. doi:10.1039/C8SC04041A30450181
- Hotamisligil GS, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8(10):a006072. doi:10.1101/cshperspect.a00607227698029
- Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15(8):369–379. doi:10.1016/j.molmed.2009.06.00519665431
- Park JW, Lee IC, Shin NR, et al. Copper oxide nanoparticles aggravate airway inflammation and mucus production in asthmatic mice via MAPK signaling. Nanotoxicology. 2016;10(4):445–452. doi:10.3109/17435390.2015.107885126472121
- Lee IC, Ko JW, Park SH, et al. Copper nanoparticles induce early fibrotic changes in the liver via TGF-beta/Smad signaling and cause immunosuppressive effects in rats. Nanotoxicology. 2018;12(6):637–651. doi:10.1080/17435390.2018.147231329848140
- Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007;26(8):2005–2014. doi:10.1038/sj.emboj.760166817396146
- Wood CD, Thornton TM, Sabio G, Davis RA, Rincon M. Nuclear localization of p38 MAPK in response to DNA damage. Int J Biol Sci. 2009;5(5):428–437. doi:10.7150/ijbs.5.42819564926
- Yi C, Liu D, Fong CC, Zhang J, Yang M. Gold nanoparticles promote osteogenic differentiation of mesenchymal stem cells through p38 MAPK pathway. ACS Nano. 2010;4(11):6439–6448. doi:10.1021/nn101373r21028783