
Abstract
The application of nanotechnology in areas of drug delivery and therapy (ie, nanotherapeutics) is envisioned to have a great impact on public health. The ability of nanotherapeutics to provide targeted drug delivery, improve drug solubility, extend drug half-life, improve a drug’s therapeutic index, and reduce a drug’s immunogenicity has resulted in the potential to revolutionize the treatment of many diseases. In this paper, we review the liposome-, nanocrystal-, virosome-, polymer therapeutic-, nanoemulsion-, and nanoparticle-based approaches to nanotherapeutics, which represent the most successful and commercialized categories within the field of nanomedicine. We discuss the regulatory pathway and initiatives endeavoring to ensure the safe and timely clinical translation of emerging nanotherapeutics and realization of health care benefits. Emerging trends are expected to confirm that this nano-concept can exert a macro-impact on patient benefits, treatment options, and the EU economy.
Introduction
Nanomedicine is the application of nanotechnology to medicine and is envisioned to have a great impact on public health (). It uses nanosized tools for the diagnosis, prevention, and treatment of disease and encompasses several distinct application areas: drug delivery, drugs and therapies, in vivo imaging, in vitro diagnostics, biomaterials, and active implants. Over the last two decades, significant progress has been made in the field of nanomedicine, resulting in a number of products, including therapeutics and imaging agents, enabling more effective and less toxic therapeutic and diagnostic interventions.Citation1 An interdisciplinary approach resulted in the development of innovative nanomedicine preparations, which have both diagnostic and therapeutic potential and are believed to have significant potential in enabling personalized nanomedicinal treatments.Citation2,Citation3 The major applications of nanotechnology in the biomaterial field are in hard tissue implants, bone substitute materials, dental restoratives, soft tissue implants, and antibiotic materials.Citation4–Citation6
Figure 1 Rationale for the nanotherapeutic approach.
Abbreviation: R&D, research and development.
The development process in nanomedicine is clearly driven by startups and small and medium enterprises (SMEs), whereas in big corporations, nanotechnology-driven research and development is still of comparatively little importance. According to some older but comprehensive data from 2005, the EU nanomedicine industry includes 92 startups (44%), 67 SMEs (32%), and 41 large pharmaceutical or medical device companies (21%).Citation4 The 41 large pharmaceutical or medical device companies run only individual nanomedicine projects or have individual nanotechnology-based products on the market, ie, nanotechnology currently contributes little to a company’s business activities. It seems that pharmaceutical corporations with significant investments in nanomedicine will wait until a nanotechnology-enabled blockbuster shows the potential of this technology. Furthermore, most of the companies involved in nanomedicine (56%) develop products related to drug delivery and therapy. This is in line with the high proportion of nanomedicine publications in this field (76%) and the share of such products in the total nanomedicine market of about 80%.Citation4 Therefore, this review paper focuses on nanomedicine products related to drug delivery and therapy, ie, nanotherapeutics.
In the development of nanotherapeutics, nanotechnology tools are used to improve drug solubility (eg, micellesCitation7 and nanocrystalsCitation8), to guide drugs to the desired location of action with increased precision (ie, drug targetingCitation9,Citation10), to control the drug’s release (eg, nanoparticlesCitation11 and liposomesCitation12), and/or to enhance the transport across biological barriers (eg, micellesCitation13 and nanoparticlesCitation14). The main goal is to improve drug bioavailability, pharmacokinetics, efficacy, and safety to promote the treatment of diseases which currently cannot be achieved with conventional dosage forms.Citation15 Herein, we discuss the success of nanotherapeutic products, the regulatory pathway, and the initiatives endeavoring to ensure safe and timely clinical translations and the realization of health care benefits.
Nanotherapeutics with the greatest pharmaceutical and commercial potential
Application of nanotechnology in areas of drug delivery and therapy has the potential to revolutionize the treatment of many diseases.Citation16 In the past two decades, several nanotherapeutics were approved for the treatment of cancer, pain, and infectious diseases ( and ). Advanced therapy can only be achieved through the rational design of nanotherapeutics that lead to the development of nanoplatforms of particular size, shape, and surface properties that are crucial for biological interactions and consequent therapeutic effect.Citation17
Table 1 Examples of commercially available nanotherapeutic products for oral administration in the EU
Table 2 Examples for commercially available nanotherapeutic products for parenteral administration in the EU
In addition, certain drug-free nanoparticles and molecular structures can act as pharmaceutically active compounds. In addition, certain drug-free nanoparticles and molecular structures can act as pharmaceutically active compounds. At present the following candidates have proceeded to clinical trials: Vivagel™ (Starpharma, Melbourne, VIC, Australia), a dendrimer-based gel that is designed to prevent HIV/AIDS infections; NanoTherm® (MagForce, Berlin, Germany), a nanoparticle-based magnetic fluid for the local treatment of solid tumors; and the magnetic nanoparticles used in stem-cell therapies for cardiac diseases.Citation4
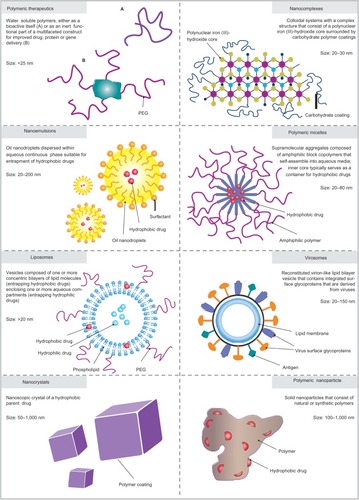
In the EU, marketed nanotherapeutic products include nanocrystals, liposomes, virosomes, nanoemulsions, polymer-protein conjugates, polymeric drugs, nanocomplexes, and nanoparticles, as shown in and and .
Figure 2 Nanotherapeutics with the greatest pharmaceutical and commercial potential.
Abbreviation: PEG, polyethylene glycol.
The majority of nanotherapeutics on the market are intended for parenteral administration. These are followed by nanotherapeutics designed for oral administration. Additionally, it is expected that a large number of ongoing preclinical and clinical investigations will result in the approval of new nanotherapeutics intended for non-parenteral routes of administration, such as ocular, pulmonary, nasal, dermal, and vaginal.Citation18 The choice of the delivery route, and consequently the barriers to be crossed, are of particular importance for drug-delivery systems.Citation19
Herein, we review nanotherapeutics and the underlying nanotechnologies that have the greatest pharmaceutical and commercial potential. Additionally, we evaluate the already developed and marketed products.
Liposomes
Liposome drug-delivery systems are nanosized spheres composed of one or more concentric lipid bilayers surrounding an aqueous core (). Thus, liposomes can serve as nanocarriers for both hydrophilic drugs, which can be entrapped in the aqueous core, and hydrophobic drugs, which can be entrapped within the lipid membrane. Increased circulation times of liposomes can be achieved through surface modification with polyethylene glycol (PEG) or other polymers.Citation20
In clinical use, liposomes are the oldest nanotherapeutic platform. During the development of liposomal products for parenteral administration, the particle size, surface charge, membrane composition, and fluidity have been recognized as crucial parameters that determine the liposomal performance in vivo. The first liposomal formulations of the anticancer drug doxorubicin were launched in the early 1990s, which included Doxil®/Caelyx® (Janssen Pharmaceutica NV, Beerse, Belgium) or Myocet® (GP Pharm SA, Barcelona, Spain/Teva Pharmaceutical Industries Ltd, Krakow, Poland).Citation21–Citation23 Doxil® is the PEGylated liposomal formulation of doxorubicin, first approved for refractory Kaposi’s sarcoma but is now also indicated in ovarian and recurrent breast cancers.Citation20 Encapsulation of cytotoxic anticancer drug doxorubicin into a liposome carrier increases its half-life and distribution in tumor tissues. In addition, doxorubicin liposomal formulations have shown significantly reduced cardiotoxicity compared with free doxorubicin.Citation24,Citation25 With DaunoXome® (Gilead Sciences Ltd, Co, Cork, Ireland), daunorubicin was incorporated into a liposomal carrier system, again to reduce the risk of acute and cumulative cardiotoxicity common to all anthracyclines.Citation26–Citation28 Liposome-based therapeutic AmBisome® (Gilead Sciences Ltd) has been approved to overcome the side effects associated with the clinical use of amphotericin B, such as infusion-related reactions and nephrotoxicity associated with chronic use.Citation29 Because of their small size and negative charge, amphotericin B-loaded liposomes avoid substantial recognition and uptake by the reticuloendothelial system. Thus, a single dose of liposomal formulation of amphotericin B provides much higher peak plasma level and much larger area under the concentration–time curve than conventional amphotericin B deoxycholate. Tissue concentrations in patients receiving liposomal formulation of amphotericin B are highest in the liver and spleen and much lower in the kidneys and lungs.Citation30
DepoCyt® (Almac Pharma Services Ltd, Craigavon, UK), a commercially available liposomal formulation of cytarabine indicated for the treatment of meningeal neoplasms, employs specific multivesicular liposome technology, referred as DepoFoam® (Pacira Pharmaceuticals, Inc.). Multivesicular liposomes consist of multiple nonconcentric aqueous chambers inside a single bilayer lipid membrane with a foam-like appearance. This highly organized multivesicular structure of liposomes enables greater drug entrapment efficiency, provides robust structural stability, and ensures sustained release of the drug. Intrathecal administration of DepoCyt® is beneficial for targeting the meninges, minimizing systemic exposure of cytarabine. Sustained release of cytarabine provides prolonged drug exposure and lower cytarabine peak levels compared with standard dosing with cytotoxic agents. Its use allows reduced dosing frequency from twice a week to once every other week and may improve the outcome, compared with frequent intrathecal injections of unencapsulated cytarabine.Citation31,Citation32 Another drug formulation exploiting DepoFoam® technology (Pacira Pharmaceuticals, Inc.) is DepoDur® (Almac Pharma Services Ltd), an extended-release injectable liposomal formulation of morphine, indicated for epidural administration for the treatment of pain following major surgery. The use of DepoDur® decreases the amount of systemically administered analgesics needed for adequate postoperative pain control. It may also provide superior pain control during the first 1–2 postoperative days compared with epidural administration of unencapsulated morphine or intravenous administration of an opioid.Citation31
Mepact® (Takeda Ireland Ltd, Wicklow, Ireland/Takeda Italia Farmaceutici S.p.A, Cerano, Italy), a liposomal formulation of mifamurtide (immunomodulator with antitumor effects mediated by activation of monocytes and macrophages), is indicated in children, adolescents, and young adults for the treatment of high-grade, non-metastatic osteosarcoma after macroscopically complete surgical resection. It is administered by intravenous infusion in conjunction with postoperative multiagent chemotherapy and has been shown to significantly increase overall survival of young patients.Citation33 After application of the liposomal infusion, the drug is cleared from the plasma within minutes and is concentrated in lung, liver, spleen, nasopharynx, and thyroid. The liposomes are phagocitosed by cells of the reticuloendothelial system. Therapeutic liposome-based products that are commercially available in the EU are listed in .
Drug nanocrystals
Approximately 40% of drug candidates are poorly soluble in water, which hinders their development and clinical applications.Citation34 One of the strategies employed to overcome this limitation is the reduction of drug crystal size to the submicron range, ie, drug nanonization.Citation8,Citation35 This strategy involves the production of drug nanocrystals by either chemical precipitation or disintegration.Citation36–Citation39 Drug nanocrystals are nanoscopic crystals of the parent compound (). According to the Noyes-Whitney equation, a decrease in particle size will result in an increase in effective surface area in the diffusion layer and a consequent increase in the drug dissolution rate.Citation40 This is one of the basic approaches of enhancing the oral bioavailability of hydrophobic drugs. Most common preparation methods include media milling, high-pressure homogenization, and nanoprecipitation.Citation34 Media milling is the oldest method employed in the production of drug nanocrystals. The disintegration of drug powders into nanoparticles is a result of strong shear forces generated by high-speed rotation of a milling chamber charged with milling pearls, dispersion media (eg, water), drug powders, and stabilizers. One of the leading nanonization platform technologies based on this method is NanoCrystal® (Elan Corporation, Dublin, Ireland). NanoCrystal® technology (Elan Corporation) has been employed in the preparation of parenteral dosages, such as nanosuspensions (eg, Zypadhera®; Lilly Pharma Fertigung und Distribution GmbH and Co, KG, Gießen, Germany), and oral dosages, such as tablets and capsules (eg, Rapamune® [Pfizer Ireland Pharmaceuticals, Dublin, Ireland], Tricor® [Recipharm, Fontaine, France], and Emend® [Merck Sharp and Dohme BV, Haarlem, the Netherlands]).Citation41 In the case of Rapamune®, nanonization of sirolimus enabled its formulation in tablets (when compared to previously used liquid dosage form) reducing the dose needed which consequently diminished influence of drug content on the tableting process (ie, the tablet contains only 1–5 mg sirolimus in the tablet, weighing about 370 mg).Citation42 Tablets are a more convenient dosage form compared with the first-marketed unpleasant-tasting lipid-based liquid solution that required cold storage and a dispensing protocol.Citation42,Citation43 Drug nanocrystal-based products that are commercially available in the EU are listed in and .
In the case of Emend®, nanocrystal fast dissolution is beneficial because aprepitant has an absorption window in the upper gastrointestinal tract.Citation42 In addition, nanocrystals eliminate the effects of food on drug absorption, as nanonization increases the surface area about 40-fold. This aspect is significant, as the commercial success of the drug might have been restricted if it had been required to be taken with food.Citation43 Similarly, in the case of Tricor®, nanocrystals significantly reduce the differences in drug absorption between the non-fed and fed states compared with non-nanocrystal tablets/capsules, because the intestinal adhesive properties of nanocrystals are not much affected by the presence of food.Citation42 In addition, tablets containing fenofibrate nanocrystals significantly reduce its required dose (145 mg) compared with tablets (160 mg) and capsules (200 mg) containing micronized and non-micronized fenofibrate, respectively.Citation44
Virosomes
Virosomes are a drug or vaccine delivery system composed of viral membranes that are reconstituted with viral lipids and proteins, allowing the fusion with target cells ().Citation45 Virosome production generally includes detergent solubilization of the influenza virus and subsequent reconstitution with two influenza envelope glycoproteins, hemagglutinin and neuraminidase. These glycoproteins have a significant role in structural stability, homogeneity, targeting, receptor- mediated endocytosis, and endosomal escape after endocytosis. Virosomes have been approved for vaccine delivery against hepatitis A virus (Epaxal®; Crucell Spain, SA, Madrid, Spain) and influenza viruses (Inflexal® V; Crucell Spain SA). In the case of Epaxal®, the adsorption of formalin-inactivated hepatitis A virus particles to immunopotentiating reconstituted influenza virosomes offers an improved seroprotection rate (100%) compared with a conventional hepatitis A virus vaccine (67.7%) in infants and children.Citation46,Citation47 Moreover, because Epaxal® does not contain aluminum as an adjuvant, the local reactions and side effects are reduced in comparison with a conventional hepatitis A virus vaccine.Citation46,Citation47 Inflexal® V offers a higher seroprotection rate (88.8%) than a conventional influenza vaccine (78.3%) and less frequent side effects due to the superior purity and biocompatible nature of Inflexal® V constituents.Citation48
Polymer therapeutics
Polymer therapeutics include the family of compounds and the drug delivery technologies that use water-soluble polymers as a common core component. The term specifically refers to polymeric drugs, polymer–drug conjugates, polymer–protein conjugates, polymeric nonviral vectors, and dendrimers. Polymer therapeutics are nanosized (eg, conjugates are typically 2–25 nm), but they are quite distinct from polymeric nanoparticles (). The polymer molecular weight, polydispersity, architecture, and conjugation chemistry have a significant impact on their safety and efficacy. Given the increase in the number of polymeric materials and in conjugate design complexity, which have been proposed for the development of polymer therapeutics, the need for an appropriate regulatory framework is at the forefront of scientific discussion.Citation49 Gaspar and DuncanCitation50 have recently reviewed the adequacy of the current tests and models used to define polymer therapeutic preclinical safety. Their review discussed the current status and future challenges for regulatory agencies. Additionally, they stressed the importance of considering each polymer therapeutic on its own merit based on doses, administration routes, dosing frequency, and proposed clinical use. Examples of such commercially available products in the EU are listed in and .
The most advanced class of polymer therapeutics contain the PEG–protein conjugates intended for parenteral administration in the treatment of viral infections and tumors (). These conjugates are designed to overcome some of the limitations of the peptide-, protein-, and antibody-based drugs, including a short plasma half-life, poor stability, and immunogenicity. Pegylation of interferon α-2a (Pegasys®; Roche Pharma AG, Grenzach-Wyhlen, Germany) and interferon α-2b (PegIntron®; Schering-Plough Labo NV, Heist-Op-Den-Berg, Belgium) significantly improved the therapy of liver diseases. Pegylated interferons have been found to be very effective against viral hepatitis. The combination of these compounds with ribavirin has shown high success in the treatment of hepatitis B and C.Citation51 Pegylated interferons are not liver specific, but the prolonged circulation time of the pegylated compound allows prolonged uptake by the hepatocyte, providing enhanced therapeutic efficiency. However, the uptake in non-target cells is not prevented.
Sevelamer (Renagel® [Genzyme Ltd, Oxford, UK]/Renvela® [Genzyme Ireland Ltd, Co, Waterford City, Ireland]) is the only polymeric drug for oral administration approved in the EU. This non-calcium phosphate binder reduces coronary artery and aortic calcification as compared with calcium-containing phosphate binders. It also exerts an effect on inflammatory biomarkers such as C-reactive protein, and lowers low-density lipoprotein cholesterol in patients with chronic kidney disease. Additionally, it can possibly decrease retinopathy in diabetic patients with hypertension.Citation52
Nanoemulsions
One of the interesting classes of nanotherapeutics suitable for the entrapment of hydrophobic drugs is the oil-in-water nanoemulsion, in which oil nanodroplets are dispersed within an aqueous continuous phase and stabilized with surfactant molecules ().Citation34 Nanoemulsions can be prepared by high-pressure homogenization, microfluidization, ultrasonication, and spontaneous emulsification. Advantages of nanoemulsions include increased drug loading and enhanced bioavailability. Therapeutic nanoemulsion-based products in the form of self-emulsifying drug-delivery systems (SEDDS) formulated in gelatin capsules are particularly interesting because nanoemulsions form when the formulation reaches the gastrointestinal tract. However, only few formulations have been commercialized due to limitations related to usage of surfactants and cosolvents, and a possibility of drug precipitation upon aqueous dilution in vivo resulting in unpredictable oral bioavailability.Citation53
Examples of commercialized SEDDS formulations include cyclosporine (Neoral®; Novartis AG, Basel, Switzerland) and ritonavir (Norvir®; Aesica Queenborough Ltd, Queenborough, UK) (). Neoral® improves cyclosporine bioavailability and reduces its pharmacokinetics/pharmacodynamics inter- and intra-patient variability, offering more predictable and more extensive drug absorption than the standard Sandimmune® (Novartis, AG Basel, Switzerland) formulation.Citation54
Nanoparticles
Polymeric nanoparticles are nanosized solid particles that consist of natural or synthetic polymers. Two types of nanoparticles can be distinguished: 1) nanospheres, which are matrix systems in which drug is uniformly dispersed; and 2) nanocapsules, which are reservoir systems in which drug is located in the core surrounded by a polymer membrane. Drugs are physically entrapped within the nanoparticles, chemically conjugated or adsorbed to the constitutive polymers of the nanoparticle. Polymeric nanoparticles are able to control drug release either by diffusion through polymer matrix or by its degradation. They have been investigated as drug-delivery systems for the site-specific targeting of tumors and for the transport of drugs across biological barriers, particularly the blood–brain barrier.
Although nanoparticles have been studied for approximately 40 years, the anticancer drug Abraxane® (Celgene Europe Ltd, Uxbridge, UK) (paclitaxel stabilized by albumin) is the only polymeric nanoparticle-based product on the market ().Citation55 The albumin–paclitaxel formulation consists of 130 nm diameter nanoparticles and is used in colloidal suspensions.Citation20 This product is commercially available in the EU (). Formulating paclitaxel for systemic administration proved to be a very difficult challenge due to its high hydrophobicity. The first paclitaxel formulation to come to market was Taxol® (Corden Pharma Latina S.p.A, Sermoneta, Italy), in which paclitaxel was solubilized in a Cremophor®EL (BASF Corp, Ludwigshafen, Germany)/ethanol mixture. However, Cremophor®EL can be highly toxic, frequently causing acute hypersensitivity reactions and limiting the allowable dosage for the patient. Abraxane® utilizes the serum protein albumin to solubilize and carry the drug in circulation. The formulation is Cremophor® EL free, and clinical trials have indeed indicated a reduction in acute toxicity in patients.Citation56 In the case of Abraxane®, lower toxicity allows paclitaxel administration at higher doses and infusion rates than in the case of Taxol®. In addition, no premedication is needed to diminish acute side effects.
Nanosized colloidal iron-based formulations
Parenteral iron formulations are colloidal systems with a complex structure that consists of a polynuclear iron (III)-hydroxide core surrounded by carbohydrate polymer coatings ().Citation57,Citation58 These formulations are used when iron supplements given orally cannot be used or fail to provide therapeutic effect. Parenteral iron formulations have been widely used since the 1930s to treat iron-deficiency anemia in patients with chronic kidney disease. Carbohydrates such as sucrose, carboxymaltose, and dextran are used as polymer coatings of the polynuclear iron (III)-hydroxide core. The average particle size in the formulation is 20–30 nm. Therapeutic products that are commercially available in the EU are listed in .
Despite structural similarities between the different iron formulations, the products differ significantly in their physicochemical properties such as particle size, zeta potential, free and labile iron content, and release of iron in serum.Citation57 Their efficacy is directly related to the amount of iron administered, but product differences determine pharmacological and biologic differences between the different iron formulations. These include clearance after injection, iron release in vitro, early evidence of iron bioactivity in vivo, and maximum tolerated dose and rate of infusion, as well as effects on oxidative markers, propensity for inducing hypophosphatemia (Ferinject®; Vifor France SA, Neuilly-sur-Seine, France/Injectafer®; Luitpold Pharmaceuticals, Inc., New York, NY, USA), and propensity to cause transient proteinuria (Venofer®; Vifor France SA) or hepatic damage (Ferrlecit®; Aventis Pharma Dagenham, UK/Sanofi-Aventis SpA, Anagni, Italy) following administration (reviewed in Jahn et alCitation57).
The efficacy of parenteral iron formulations for treating anemia has been consistently proved in a variety of clinical settings, with a very low rate of severe adverse effects. All parenteral iron medicines have a small risk of causing allergic reactions, which can be life-threatening if not treated promptly. Iron dextran complexes may cause well known dextran-induced anaphylactic reactions, which are significantly more frequent with high molecular weight iron dextran than with low molecular weight iron dextran. The new parenteral iron formulations (Monofer® [Pharmacosmos A/S, Holbaek, Denmark] and Ferinject®/Injectafer®) are all based on carbohydrates with reduced immunogenic properties.Citation57
The dose depends on the stability of the iron complex. The newest product, iron isomaltoside 1,000 (Monofer®) is characterized by a special matrix-like structure with interchanging iron molecules and linear isomaltoside 1,000 oligomers.Citation57 The resulting matrix enables a controlled and slow release of bioavailable iron to iron binding proteins with little risk of free iron toxicity.Citation57,Citation59 Therefore, Monofer® can be administered safely as a rapid high-dose intravenous infusion or bolus injection, offering considerable dose flexibility, including the possibility of providing full iron repletion in a single infusion, the so-called one-dose iron repletion.
Polymeric micelles
Polymeric micelles are formed upon the self-assembly of amphiphilic polymers, with the hydrophobic portion of the polymer toward the core and the hydrophilic segments on the outer shell ().Citation13 The micelles are in the size range of 20–80 nm, suitable for the entrapment of hydrophobic drugs. PEG is the shell-forming polymer of choice, because it is nontoxic and already approved by the US Food and Drug Administration (FDA) for use in drug products. Additionally, PEG limits micelle interactions with other micelles (which could lead to aggregation) and proteins (opsonins). Several aspects of polymeric micelle-based delivery systems need to be addressed for further utilization in the clinical setting. One of the key issues is their inability to maintain their structural integrity before reaching the destination site. Thus, it is critical to obtain a more comprehensive understanding of the fate of both amphiphilic polymers and polymeric micelles after administration into the body.Citation38
Polymeric micelles have been most extensively explored for use in anticancer treatments; some are currently in preclinical or clinical development.Citation20 Genexol®-PM (Samyang Biopharmaceuticals, Seoul, South Korea) is a polymeric micelle-based formulation of paclitaxel. Compared with Taxol®, Genexol®-PM displayed similar cytotoxicity against various human cancer cells, including breast, colon, ovarian and non-small cell lung cancer cells.Citation60 Due to its superior efficacies and less adverse reactions, Genexol®-PM has been approved in South Korea for breast and non-small cell lung cancers and is currently undergoing Phase III trials in the US.Citation15,Citation20 Genexol®-PM will probably be registered in the EU under the brand name Cynvilog™.Citation61
Polymeric micelle nanotherapeutics have not yet been approved in the EU. Recently, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) drafted a reflection paper on the pharmaceutical development and preclinical and early clinical studies of polymeric micelle medicinal products designed to affect the pharmacokinetics, stability, and distribution of incorporated or conjugated drug in vivo.Citation62 However, because of the complexity of polymeric micelle products and limited research, their general advice is to seek product-specific scientific consults when developing such nanotherapeutic products.
Toward nanosimilars
To date, a notable number of different nanotherapeutics have gained marketing authorization by the EMA. Liposomal formulations, iron-based preparations, and drug nanocrystals in oral dosage forms are first-generation nanotherapeutics, whose effectiveness and safety have been substantiated for long-term clinical use.Citation63 In the process of their approval, the applications were assessed under a conventional regulatory framework using established principles of benefit/risk analysis.
At this point, it is particularly challenging to develop and evaluate “follow-on” nanotherapeutic products that could be authorized after the innovator product patent expiration. This specifically applies to nanotherapeutics that reach the systemic circulation and thereby determine the pharmacokinetics, biodistribution, and therapeutic performance. Any variations in the manufacturing process and the formulation may result in a generic product with different physicochemical properties (eg, size, size distribution, surface properties, drug loading and release profile, aggregation status, and stability), which could lead to a different biopharmaceutical profile with a significant impact on patient safety and efficacy. In particular, different physicochemical properties can result in different ratios of free to nanoparticle-incorporated/associated drug, pharmacological effects, specific cell–nanotherapeutic interactions, distributions, target organ uptake, immunological effects, and toxicities. To assess these differences, approaches more complex than the simple plasma concentration measurement are required. It is generally considered that the regulatory approach established for similar biological medicinal products (biosimilars) should be adopted for “follow-on” nanotherapeutic products (nanosimilars) because such an approach includes the stepwise comparison of their quality, safety, and efficacy.
The overall experience with the development of nanosimilars has been limited. Although being the oldest nanotherapeutic platform, no “follow-on” liposomal products have yet been approved in the EU. The CHMP drafted a reflection paper with general recommendations intended to assist in the generation of relevant quality, preclinical and clinical data to support the marketing authorization of parenteral liposomal products developed with reference to an innovator liposomal product.Citation64
Significant pitfalls have surfaced during the development of nanosimilar colloidal iron-based formulations with respect to the innovator product.Citation58 Differences in tissue distribution and toxicological profiles have been observed among nanoparticle iron formulations that have different carbohydrate coatings. When differences in the toxicological profiles were observed for nanoparticle iron formulations with the same coating, they were ascribed to the differences in the manufacturing process. The CHMP drafted a reflection paper on the preclinical studies for generic nanoparticle iron medicinal product applications, describing the factors related to the particle characteristics influencing pharmacokinetic parameters and consequently efficacy and toxicity.Citation65 These factors mostly refer to the nanoparticle stability and distribution in relevant compartments, including plasma, the reticuloendothelial system, and pharmacological (eg, bone marrow) and toxicological (eg, kidneys, liver, lungs, and heart) target tissues.
In addition to nanosimilars, recent advances in nanoscience will soon lead to the development of new, more complex nanotherapeutics, presenting a particular regulatory challenge. The assessment of existing nanotherapeutics has provided valuable experience in certain evaluation aspects of emerging (next-generation) nanotherapeutics. However, further scientific research is required to provide an appropriate evaluation of their quality, safety, and efficacy.
Nanotherapeutic regulatory issues
The EMA is a regulatory agency whose main responsibility is to evaluate and supervise medicines for use in the EU for protecting and promoting public (and animal) health. Additionally, the EMA constantly monitors the safety of medicines and acts in cases of safety concerns and changes in the benefit–risk balance of the medicinal product. The EMA also encourages pharmaceutical innovation and research and provides scientific advice and protocol assistance in the development of new medicinal products.
Nanotherapeutic products are currently regulated within a conventional regulatory framework. However, as already mentioned, additional expert evaluations are necessary to confirm the quality, safety, and efficacy of nanotherapeutics because of their complexity.Citation66,Citation67
Recent EMA activities aiming to provide regulatory guidance (ie, implementing the needs of nanotherapeutic-specific properties) and assistance (ie, scientific advice on the appropriate tests and studies) in developing high-quality, effective, and safe nanotherapeutics have been reviewed in the literature.Citation63 Recommendations from the CHMP have led to the approval of a number of medicines based on nanotechnology (see and ).
In addition, the EMA plays a central role in the development and authorization of “orphan medicines,” ie, therapeutics used in the treatment of rare diseases. In this respect, the European Commission has granted orphan status for several nanotherapeutics under development, such as doxorubicin poly(isohexylcyanoacrylate) nanoparticles for the treatment of hepatocellular carcinoma, nanobody directed toward the human A1 domain of the von Willebrand factor for the treatment of thrombotic thrombocytopenic purpura and nanoliposomal irinotecan for the treatment of pancreatic cancer.Citation63
In 2009, the CHMP established an ad hoc expert group meeting on nanomedicines. This group of selected experts from academia and the European regulatory network support the agency’s activities by providing specialist input on new scientific knowledge and reviewing the guidelines on nanomedicine development. The agency also consults this group during discussions with international partners on issues concerning nanomedicines.
The need for sharing and discussing the global academic, industrial, and regulatory experience and perspectives in the field of nanomedicines has been recognized by European and international experts and the medical regulatory agencies of the EU, US, Japan, and Canada. In 2010, the EMA hosted the first international workshop on nanomedicines, which gathered more than 200 participants from all over the world. The discussion was focused on existing and emerging nanomedicines and covered a number of specific aspects, such as the characterization, biodistribution, and the interactions of nanomedicines with biological systems.
The main goal of these interactive communications is to direct the development of nanomedicines toward timely and effective clinical translation. Apart from the activities at the EMA, there are other initiatives aiming to strengthen the links between academia and industry and to promote the research and application of nanomedicines. One of these initiatives, the European Society for Nanomedicine, was founded in 2007. The European Technology Platform on Nanomedicine (ETPN), an initiative led by industry and supported by the European Commission, addresses the application of nanotechnology to achieve breakthroughs in health care and to intensify innovation in nanobiotechnologies. An example of the collaboration between the EU and the US is the Joint Workshop, a group focused on nanosafety research that is open to scientists, policy makers, regulators, administrators, decision makers (from academia, research institutes, and industry), and authorities from the EU and the US. The first Joint Workshop was held in 2011 in Washington. The second Joint Workshop (in 2012) was hosted by the Finnish Institute of Occupational Health.
The CHMP established a multidisciplinary expert group on nanomedicines in 2011 and drafted a series of aforementioned reflection papers aiming to develop scientific and regulatory guidance within the area of specific nanomedicine development. This refers to the development and evaluation of first-generation nanosimilars (ie, nanosimilars developed with reference to the first-generation nanotherapeutics) and emerging (next-generation) nanotherapeutics. This is of particular importance, since nanotherapeutics may exhibit a complex mechanism of action combining mechanical, chemical, pharmacological, and immunological properties.Citation68 Therefore, additional specialized expertise, together with the adaptation of existing methodologies and the development of new methods, may be required for the evaluation of their quality, safety, efficacy, and risk management.Citation66 For the regulatory decision making, it is imperative to define critical product attributes predictive of product performance in vivo (eg, size specifications, shape, and surface characteristics such as area, chemistry, porosity, and coating).Citation68–Citation70 Therefore, both the EMA and the FDA require similar documentation that will prove quality, safety, and efficacy of the nanomedicinal product. Since advances in emerging technologies may be unpredictable and rapid, the FDA also uses a case-by-case approach which is iterative, adaptive, and flexible.Citation71 Particular policies for each product area, both substantive and procedural, will be defined separately when ready. Manufacturers are advised to consult with the FDA early in their development process to facilitate a mutual understanding of the scientific and regulatory issues for their nanotechnology products.Citation72 The FDA has issued a draft guidanceCitation73 on considerations for identifying products containing nanomaterials, with the intention of providing greater regulatory clarity to industry.
Regarding the duration of approval procedure, there are no major differences. In the EU, medicinal products are authorized following a 210-day procedure, which can be shortened to 150 days when a marketing authorization application is submitted for a product which is of major public health interest, in particular from the viewpoint of therapeutic innovation (so-called accelerated assessment procedure).Citation74 In the US, in addition to the standard review that is applied to a medicinal product that offers, at most, only minor improvement over existing marketed therapies, with a set goal of 10 months for completing the review, there is the priority review designation given to products that offer major advances in treatment, or provide a treatment where none existed, with the set goal of 6 months.Citation75
Nanotherapeutic research and development
In early 2009, the European Commission, together with the ETPN, suggested that research and development activities should be focused on identifying translatable trends in research, understanding their expected impact on applications, productions, and markets, and enabling the fine-tuning and targeting of research funding on areas with greater impact on health benefits and commercialization. This focus is especially important in light of recent reduced public funding and the resulting need for public/private funding of research.
Successful translation of research from academia to production lines has been identified as one of the major challenges in nanotherapeutic development. Strategies to foster and initiate this translation have yet to be developed to help European research institutions and industries remain competitive in global markets.Citation19 A quick and successful translation of emerging nanotherapeutics is expected to adapt the established quality-by-design approach.Citation17 The quality-by-design approach, in the field of nanotherapeutic development, promotes the idea that control over the quality, efficacy, and safety should be incorporated into the formulation development. This approach includes clear definitions of the desired performance (ie, the expected specifications of the target formulation), nanoparticle design (ie, the nanoparticle attributes providing efficacy and safety), manufacturing design (ie, establishing the process parameters ensuring reproducibility of nanoparticle properties), and therapy design (ie, the treatment modalities providing efficacy and safety of the therapeutic application). A process of developing an optimal formulation is influenced by a complicated matrix of interlinked or independent input and output parameters, which include critical process parameters, critical product quality attributes, and clinical properties such as safety and efficacy.
The proactive European approach in nanomedicine research is visible through EU funding opportunities. Research in nanomedicine within the Seventh Framework Programme for Research and Technological Development (2007–2013) is supported by the nanotechnology, materials, and production, as well as health themes (Cooperation program), The Marie Curie Actions (People program) and the European Research Council (Ideas program).Citation76 According to the data reported in the literature, the investments in nanomedicine projects during the first four calls of the Seventh Framework Programme have been significantly higher than the investments in nanomedicine projects during the entire Sixth Framework Programme. According to the same report, similar or even higher levels of funding are anticipated in the fields of nanodiagnostics, targeted nanotherapeutics, regenerative medicine, and ethical, legal, and societal aspects.Citation76,Citation77 Based on excellent academic research and innovative SMEs, nanomedicine will actively contribute to the next Framework Programme Horizon 2020 (ie, the Contribution of Nanomedicine to Horizon 2020).
Nanotherapeutic translation and commercialization
The final goal of the research and development of a nanotherapeutic product is its successful translation from bench to bedside. However, there are significant obstacles and challenges in bringing nanotherapeutic products to the market, including: 1) lack of quality control; 2) separation from undesired nanostructures (eg, by products and starting materials); 3) scalability issues; 4) enhancing the production rate; 5) reproducibility from batch to batch with respect to particle size distribution, charge, porosity, and mass; 6) high fabrication costs; 7) lack of knowledge regarding the interaction between nanosystems and living cells (eg, the issues of biocompatibility and toxicity); 8) nanotherapeutic optimization for maximum therapeutic potential; 9) relative scarcity of venture funds; 10) the pharmaceutical industry’s reluctance to invest in nanotherapeutics; 11) relative unpredictability of the EMA with respect to a lack of regulatory and safety guidelines pertaining to nanotherapeutics; and 12) the media focus on the negative aspects of nanomaterials, often without clear scientific evidence.Citation41
The potential health benefits of nanotherapeutic products can be realized only if such products are available and commercially viable. Therefore, the regulatory requirements must be sufficiently rigid to ensure the safety and quality of nanotherapeutics, but the potential negative impact of over-regulation on the introduction of innovative products to the market must also be considered. Many innovative nanotherapeutics focused on rare diseases will not enter the market because the costs for achieving regulatory approval are too high compared with expected sales.Citation4
In the past, health innovations were evaluated on their efficacy and improved patient quality of life. Currently, health care costs must also be considered. Nanotherapeutic products, which are more complex in structure and more expensive than conventional alternatives, are designed to provide an overall reduction in health care costs (). This reduction in health care costs is likely to be obtained by increasing the nanotherapeutic efficacy, reducing the length of in-patient stay, reducing personal health care costs, and the effective treatment of expensive major diseases.Citation78 However, a well-founded assessment of the overall cost-effectiveness of nanotherapeutics requires analysis of many more marketed products.Citation4
Although nanotherapeutics have not yet been mass-marketed, they hold significant potential in offering a pool of novel products, resulting in the growth of the pharmaceutical market and improved health benefits.Citation79 The nanomedicine-related drug market is directly affected by the pharmaceutical regulatory environment, health care policies, demographics, and the wider economic environment. Companies specializing in nanomedicine have employed specific strategies to meet the challenges of this highly competitive market. BCC Research published a report on the global nanomedicine market analyzing in detail the current size and growth of the pharmaceutical market, reviewing major nanomedicine technologies and their practical applications, discussing the nature and structure of the nanomedicine industry, profiling the leading companies, and providing sales forecasts for the nanomedicine market.Citation80 The therapeutic categories covered in the report include cancer, diseases of the central nervous system, infections, and cardiovascular diseases. The country markets analyzed were the US, Japan, Germany, France, the UK, Italy, and Spain.
According to the BCC Research report, the market value of the worldwide nanomedicine industry was US$43.2 billion and US$50.1 billion in 2010 and 2011, respectively, and is estimated to reach US$96.9 billion by the year 2016. The market for central nervous system products was valued at US$11.7 billion and US$14.0 billion in years 2010 and 2011, respectively. The market value is expected to reach US$29.5 billion by the year 2016. The market for anticancer products was valued at US$4.7 billion in the year 2010 and US$5.5 billion in the year 2011, and it is anticipated to reach US$12.7 billion by the year 2016.Citation80
The ETPN and the NANOMED2020 project published the Contribution of Nanomedicine to Horizon 2020, the next European Framework Programme for Research and Innovation.Citation81 This strategic document provides key recommendations for the European Commission and the EU Member States to create a favorable ecosystem for the successful deployment of nanomedicine in Europe, leading to the efficient translation of nanotechnology from a key enabling technology into new and innovative medical products.
The implementation of these recommendations in Horizon 2020 represents a unique opportunity for Europe to promote innovation in the health care and nanomedicine fields and to contribute to the re-localization of pharmaceutical research and to the re-industrialization of Europe. As stated on the ETPN website,Citation82 with nearly 250 “nano”-products used or tested in humans worldwide, representing approximately 10% of pharmaceutical sales and a US$130.9 billion market by 2016, nanomedicine will have a real impact on the European economy.
Considering the research efforts and the increasing investments in nanotherapeutics, the EU health care systems will likely be confronted with an increasing number of new nanomedicine products. The incorporation of nanotherapeutic products into national drug reimbursement strategies will have a strong impact on their availability throughout health care systems. Many factors, such as safety, efficacy, clinical outcomes, and pharmacoeconomic evidence, as well as ethical, legal, and societal issues, will affect the decision-making processes related to the health insurance coverage of particular nanotherapeutic products. Thus, there is much to learn from the literature on health technology assessment methods/criteria/institutionsCitation83,Citation84 and their roles in the uptake of a new drug therapy in PolandCitation85,Citation86 or Germany.Citation87 In addition, some countries have established “horizon scanning systems” to support their decision-making processes using reliable data about new health technologies.Citation88
The challenges and future trends
The application of nanotechnology to medicine resulted in a notable number of nanotherapeutic products on the market. The vast majority of marketed nanotherapeutics are intended for the treatment of tumor diseases, employing passive targeting based on the leaky vasculature and reduced lymphatic drainage in tumor tissue. One of the main streams of current research efforts are directed toward the development of new-generation nanotherapeutics ensuring active targeting through interaction between a surface ligand and certain receptors overexpressed on the surface of targeted cells. However, surface modification with ligands introduces additional production, regulatory, cost-effectiveness, and polydispersity challenges.Citation20 Several issues need to be addressed within the timeframe of 5–10 years in order to achieve safe application of emerging nanomaterials in the clinical setting.Citation70,Citation89 Standardized, validated in vitro assays for nanosafety testing, including a set of reference materials, are urgently needed to screen for potential hazards early in the development of nanotherapeutics. Moreover, in vitro/ex vivo models relevant for the specific routes of administration of nanotherapeutics are critical both from a safety and pharmaceutical application point of view.Citation70,Citation90 A thorough understanding of the absorption, distribution, metabolism, and excretion of emerging nanomaterials in vivo, and their relationship with critical product attributes, is essential for the development of in silico modelling approaches to predict the biological and toxicological responses of nanotherapeutics. It is necessary to establish a paradigm for the understanding of factors (eg, the biocorona, or the coating of the nanomaterial surface with biomolecules in vivo) that dictate nanomaterial interactions with living systems.Citation70 This will be possible only by providing interdisciplinary training and creating interdisciplinary research teams for the development of nanotherapeutics from a clinical, biological, engineering, and toxicological point of view.
When the research plan covering all aspects of nanotherapeutic development and assessment is thought out, the first challenge for academia is related to the difficulty of finding appropriate funding to finance the proof-of-concept. Challenges occurring during the research and development phase are related to the availability of a contract manufacturing organization to manufacture the first batches of the nanotherapeutic product in accordance with the medical requirements for good manufacturing practice. There are also difficulties linked with the acquirement of appropriate knowledge on clinical trials and identification of a clinical research organization competent in developing the specific preclinical studies required before going to the first-in-man trials.Citation81
Successful translation of scientific inventions into nanotherapeutic products implies constant upgrading of industrial infrastructures. This includes a nanocharacterization laboratory (for physical, chemical, and biological characterization of nanomaterials intended for medical use – for resolving in a timely manner the quality problems related to the manufacturing process and product stability), a pilot line for good manufacturing practices of batches (scale up laboratory preparation of nanomaterials according to industrial and regulatory standards for early clinical trials), a network of preclinical centers of excellence, and a coordination of nanomedicine effort with clinical organizations.Citation81 Compared with “conventional” formulations, the control of materials in the nanosize range often presents greater scientific and technical challenges and therefore requires novel approaches to chemistry, manufacturing, and controls. In addition, nanotherapeutic product quality assurance includes the adequate characterization method validation to control the reproducibility of the critical attributes such as particle size, shape, and surface characteristics including area, chemistry, porosity, and coating parameters.Citation68
From the EU regulatory perspective, the main challenges refer to applicability and relevance of current methods to assess characterization and biodistribution of emerging nanotherapeutics, particularly related to their in vivo behavior (eg, interactions with biological systems and impact on the immune system). The main concerns are related to the assessment of the comparability with existing nanoformulations, adequacy of tools for risk assessment, classification of converging technologies, adequacy of regulatory frameworks, and regulators’ expertise.
Concluding remarks
The application of nanotechnology to medicine has already led to significant progress in the treatments of a number of diseases. However, further development of nanotherapeutics with new or improved features to address unmet medical needs and to enable the implementation of personalized medicine is still required.
Currently, the priorities of the field of nanotherapeutics are to direct further research efforts toward translatable and competitive product development and to ensure a well designed regulatory environment. The key to success is an interdisciplinary and open-minded approach complemented with the partnership and knowledge exchange between academia, industry, and regulatory agencies. From this perspective, the rapid and successful translation of emerging nanotherapeutics in the future can be derived from the established quality-by-design approach in the development of nanotherapeutics.Citation17
Europe currently holds a leading position with regards to scientific research but has failed to translate this strength into commercially viable products. However, numerous recent initiatives in the field of nanotherapeutics are aimed at pushing forward the best translatable concepts toward funding and clinical proof-of-concept. Such an approach is expected to generate new interests in nanomedicinal technology with macro-impacts on patient benefits and treatment options and on the EU economy.
Acknowledgments
This work was supported by a project entitled “New Approach in Antimicrobial Therapy: biocompatible, biofilm-penetrating nanosystems” funded by the University of Zagreb.
Disclosure
The authors report no conflicts of interest in this work.
References
- MurthySKNanoparticles in modern medicine: state of the art and future challengesInt J Nanomedicine20072212914117722542
- MuraSCouvreurPNanotheranostics for personalized medicineAdv Drug Deliv Rev201264131394141622728642
- RizzoLYTheekBStormGKiesslingFLammersTRecent progress in nanomedicine: therapeutic, diagnostic and theranostic applicationsCurr Opin Biotechnol20132461159116623578464
- WagnerWHuüsingBGaisserSBockANanomedicine: drivers for development and possible impactsCentre ECJR, Studies IfPT, trans: European Commission Joint Research Centre, Institute for Prospective Technological Studies2006
- WebsterTJProjections for nanomedicine into the next decade: but is it all about pharmaceuticals?Int J Nanomedicine200831i
- HuberFXMcArthurNHeimannLEvaluation of a novel nanocrystalline hydroxyapatite paste Ostim in comparison to Alpha-BSM - more bone ingrowth inside the implanted material with Ostim compared to Alpha BSMBMC Musculoskelet Disord20091016420028538
- PepićIHafnerALovrićJPirkićBFilipović-GrčićJA nonionic surfactant/chitosan micelle system in an innovative eye drop formulationJ Pharm Sci201099104317432520310026
- JunghannsJUMuüllerRHNanocrystal technology, drug delivery and clinical applicationsInt J Nanomedicine20083329530918990939
- ZhangXXEdenHSChenXPeptides in cancer nanomedicine: drug carriers, targeting ligands and protease substratesJ Control Release2012159121322056916
- CrielaardBJLammersTSchiffelersRMStormGDrug targeting systems for inflammatory disease: one for all, all for oneJ Control Release2012161222523422226771
- HafnerALovrićJVoinovichDFilipović-GrčićJMelatonin-loaded lecithin/chitosan nanoparticles: physicochemical characterisation and permeability through Caco-2 cell monolayersInt J Pharm2009381220521319596430
- PavelićZŠkalko-BasnetNFilipović-GrčićJMartinacAJalšenjakIDevelopment and in vitro evaluation of a liposomal vaginal delivery system for acyclovirJ Control Release20051061–2344315979189
- PepićILovrićJFilipović-GrčićJHow do polymeric micelles cross epithelial barriers?Eur J Pharm Sci2013501425523619286
- HafnerALovrićJPepićIFilipović-GrčićJLecithin/chitosan nanoparticles for transdermal delivery of melatoninJ Microencapsul201128880781522117177
- LammersTKiesslingFHenninkWEStormGDrug targeting to tumors: principles, pitfalls and (pre-) clinical progressJ Control Release2012161217518721945285
- PetrosRADeSimoneJMStrategies in the design of nanoparticles for therapeutic applicationsNat Rev Drug Discov20109861562720616808
- DuncanRGasparRNanomedicine(s) under the microscopeMol Pharm2011862101214121974749
- Filipović-GrčićJMrharAJungingerHThematic Issue on Emerging nanopharmaceuticals for nonparenteral application routesEur J Pharm Sci2013501123747658
- European Commission/ETP NanomedicineRoadmaps in Nanomedicine Towards 2020: Joint European Commission/ETP Nanomedicine expert reportEuropean Commission/ETP Nanomedicine2009 Available from: http://www.etp-nanomedicine.eu/public/press-documents/publications/etpn-publications/091022_ETPN_Report_2009.pdfAccessed December 17, 2013
- SvensonSClinical translation of nanomedicinesCurr Opin Solid State Mater Sci2012166287294
- BarenholzYDoxil®-the first FDA-approved nano-drug: lessons learnedJ Control Release2012160211713422484195
- JabirNRTabrezSAshrafGMShakilSDamanhouriGAKamalMANanotechnology-based approaches in anticancer researchInt J Nanomedicine201274391440822927757
- ChangHIYehMKClinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacyInt J Nanomedicine20127496022275822
- TorchilinVPRecent advances with liposomes as pharmaceutical carriersNat Rev Drug Discov20054214516015688077
- DavisMEChenZGShinDMNanoparticle therapeutics: an emerging treatment modality for cancerNat Rev Drug Discov20087977178218758474
- HofheinzRDGnad-VogtSUBeyerUHochhausALiposomal encapsulated anti-cancer drugsAnticancer Drugs200516769170716027517
- SwainSMWhaleyFSEwerMSCongestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trialsCancer200397112869287912767102
- RahmanAMYusufSWEwerMSAnthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulationInt J Nanomedicine20072456758318203425
- FukuiHKoikeTNakagawaTComparison of LNS-AmB, a novel low-dose formulation of amphotericin B with lipid nano-sphere (LNS), with commercial lipid-based formulationsInt J Pharm20032671–210111214602388
- HamillRJAmphotericin B formulations: a comparative review of efficacy and toxicityDrugs201373991993423729001
- AngstMSDroverDRPharmacology of drugs formulated with DepoFoam: a sustained release drug delivery system for parenteral administration using multivesicular liposome technologyClin Pharmacokinet200645121153117617112293
- SeniorJHMedical applications of multivesicular lipid-based particles: DepoFoam™ encapsulated drugsLasicDDPapahadjopoulosDMedical Applications of LiposomesAmsterdam, The NetherlandsElsevier1998733750
- FramptonJEMifamurtide: a review of its use in the treatment of osteosarcomaPaediatr Drugs201012314115320481644
- ChenHKhemtongCYangXChangXGaoJNanonization strategies for poorly water-soluble drugsDrug Discov Today2011167–835436020206289
- KeckCMMuüllerRHDrug nanocrystals of poorly soluble drugs produced by high pressure homogenisationEur J Pharm Biopharm200662131616129588
- XiaDCuiFPiaoHEffect of crystal size on the in vitro dissolution and oral absorption of nitrendipine in ratsPharm Res20102791965197620585842
- DesaiPRDateAAPatravaleVBOvercoming poor oral bioavailability using nanoparticle formulations – opportunities and limitationsDrug Discov Today Technol201292e87e95
- LuYParkKPolymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugsInt J Pharm201345319821422944304
- TziomalosKAthyrosVGFenofibrate: a novel formulation (Triglide) in the treatment of lipid disorders: a reviewInt J Nanomedicine20061212914717722529
- FlorenceATAttwoodDPhysicochemical Principles of PharmacyFourth editionPharmaceutical PressLondon, UK200622
- BawaRNanoparticle-based therapeutics in humans: a surveyNanotechnol Law Bus200852137155
- ShegokarRMuüllerRHNanocrystals: industrially feasible multifunctional formulation technology for poorly soluble activesInt J Pharm20103991–212913920674732
- BroughCWilliamsRO3rdAmorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug deliveryInt J Pharm2013453115716623751341
- LingHLuomaJTHillemanDA review of currently available fenofibrate and fenofibric acid formulationsCardiol Res2013424755
- YooJWIrvineDJDischerDEMitragotriSBio-inspired, bioengineered and biomimetic drug delivery carriersNat Rev Drug Discov201110752153521720407
- UsonisVBakasenasVValentelisRKatilieneGVidzenieneDHerzogCAntibody titres after primary and booster vaccination of infants and young children with a virosomal hepatitis A vaccine (Epaxal)Vaccine200321314588459214575771
- BovierPAEpaxal: a virosomal vaccine to prevent hepatitis A infectionExpert Rev Vaccines2008781141115018844588
- HerzogCHartmannKKunziVEleven years of Inflexal V-a virosomal adjuvanted influenza vaccineVaccine200927334381438719450630
- DuncanRPolymer therapeutics as nanomedicines: new perspectivesCurr Opin Biotechnol201122449250121676609
- GasparRDuncanRPolymeric carriers: preclinical safety and the regulatory implications for design and development of polymer therapeuticsAdv Drug Deliv Rev200961131220123119682513
- PoelstraKPrakashJBeljaarsLDrug targeting to the diseased liverJ Control Release2012161218819722370583
- DracaNLazicRSimicPDumic-CuleILueticATGabricNPotential beneficial role of sevelamer hydrochloride in diabetic retinopathyMed Hypotheses201380443143523357670
- NarangASDelmarreDGaoDStable drug encapsulation in micelles and microemulsionsInt J Pharm20073451–292517945446
- LawrenceMJReesGDMicroemulsion-based media as novel drug delivery systemsAdv Drug Deliv Rev20004518912111104900
- MieleESpinelliGPMieleETomaoFTomaoSAlbumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancerInt J Nanomedicine200949910519516888
- StirlandDLNicholsJWMiuraSBaeYHMind the gap: a survey of how cancer drug carriers are susceptible to the gap between research and practiceJ Control Release201317231045106424096014
- JahnMRAndreasenHBFuttererSA comparative study of the physicochemical properties of iron isomaltoside 1000 (Monofer), a new intravenous iron preparation and its clinical implicationsEur J Pharm Biopharm201178348049121439379
- BorchardGFluhmannBMuhlebachSNanoparticle iron medicinal products – Requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studiesRegul Toxicol Pharmacol201264232432822951348
- ZagerRAJohnsonACHansonSYWasseHParenteral iron formulations: a comparative toxicologic analysis and mechanisms of cell injuryAm J Kidney Dis20024019010312087566
- YokoyamaMClinical applications of polymeric micelle carrier systems in chemotherapy and image diagnosis of solid tumorsJ Exp Clin Med201134151158
- sorrentotherapeutics.com [homepage on the Internet]Cynviloq™ Available from: http://sorrentotherapeutics.com/cynviloq/Accessed November 17, 2013
- European Medicines Agency/Committee for Medicinal Products for Human UseJoint MHLW/EMA reflection Paper on the development of Block Copolymer Micelle Medicinal ProductsLondonEuropean Medicines Agency2013 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500138390.pdfAccessed December 17, 2013
- EhmannFSakai-KatoKDuncanRNext-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicinesNanomedicine (Lond)20138584985623656268
- European Medicines AgencyReflection Paper on the Data Requirements for Intravenous Liposomal Products Developed with Reference to an Innovator Liposomal ProductLondonEuropean Medicines Agency2013 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdfAccessed December 17, 2013
- European Medicines AgencyReflection Paper on Non-Clinical Studies for Generic Nanoparticle Iron Medicinal Product ApplicationsLondonEuropean Medicines Agency2011 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105048.pdfAccessed December 17, 2013
- European Medicines Agency/Committee for Medicinal Products for Human UseReflection Paper on Nanotechnology-Based Medicinal Products for Human UseLondonEuropean Medicines Agency2006 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2010/01/WC500069728.pdfAccessed December 17, 2013
- KettigerHSchipanskiAWickPHuwylerJEngineered nanomaterial uptake and tissue distribution: from cell to organismInt J Nanomedicine201383255326924023514
- VamvakasSMartinalboJPitaRIsaacMOn the edge of new technologies (advanced therapies, nanomedicines)Drug Discov Today Technol201181e1e42
- MaynardADDon’t define nanomaterialsNature201147573543121734686
- NystromAMFadeelBSafety assessment of nanomaterials: implications for nanomedicineJ Control Release2012161240340822306428
- HamburgMAScience and regulation. FDA’s approach to regulation of products of nanotechnologyScience2012336607929930022517845
- fda.gov [homepage on the Internet]FDA’s approach to regulation of nanotechnology productsSilver Spring, MDUS Food and Drug Administration2012 Available from: http://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/ucm301114.htmAccessed November 17, 2013
- fda.gov [homepage on the Internet]Considering whether an FDA-regulated product involves the application of nanotechnology: guidance for industrySilver Spring, MDUS Food and Drug Administration2011 Available from: http://www.fda.gov/RegulatoryInformation/Guidances/ucm257698.htmAccessed November 17, 2013
- European CommissionVOLUME 2A, Procedures for Marketing Authorization. Chapter 4, centralised procedureLondonEuropean Medicines Agency2006 Available from: http://ec.europa.eu/health/files/eudralex/vol-2/a/chap4rev200604_en.pdfAccessed December 17, 2013
- fda.gov [homepage on the Internet]Frequently asked questions about the FDA drug approval processSilver Spring, MDUS Food and Drug Administration2011 Available from: http://www.fda.gov/Drugs/ResourcesForYou/SpecialFeatures/ucm279676.htmAccessed November 17, 2013
- GabellieriCFrimaHNanomedicine in the European Commission policy for nanotechnologyNanomedicine20117551952021802394
- FrimaHJGabellieriCNilssonMIDrug delivery research in the European Union’s Seventh Framework Programme for ResearchJ Control Release2012161240941522333557
- GandjourAChernyakNA new prize system for drug innovationHealth Policy20111022–317017721724290
- PautlerMBrennerSNanomedicine: promises and challenges for the future of public healthInt J Nanomedicine2010580380921042425
- BCC Research [homepage on the Internet]Nanotechnology in medical applications: the global marketWellesley, MABCC Research LLC2010 Available from: http://www.bccresearch.com/market-research/healthcare/nanotechnology-medical-applications-hlc069a.htmlAccessed August 20, 2013
- ETP NanomedicineNANOMEDICINE 2020 Contribution of Nanomedicine to Horizon 2020: white paper to the Horizon 2020 Framework Programme for Research and Innovation – recommendations from the nanomedicine communityETP Nanomedicine2013 Available from: http://www.etp-nanomedicine.eu/public/press-documents/publications/etpn-publications/etpn-white-paper-H2020Accessed December 17, 2013
- http://www.etp-nanomedicine.eu.[homepage on the Internet] Available from: http://www.etp-nanomedicine.euAccessed 17 November 2013
- GolanOHansenPKaplanGTalOHealth technology prioritization: which criteria for prioritizing new technologies and what are their relative weights?Health Policy20111022–312613521071107
- Velasco GarridoMGerhardusARottingenJABusseRDeveloping health technology assessment to address health care system needsHealth Policy201094319620219889471
- KolasaKSchubertSMancaAHermanowskiTA review of Health Technology Assessment (HTA) recommendations for drug therapies issued between 2007 and 2009 and their impact on policy making processes in PolandHealth Policy20111022–314515121641074
- OzieranskiPMcKeeMKingLThe politics of health technology assessment in PolandHealth Policy20121082–317819323127957
- PerlethMBusseRSchwartzFWRegulation of health-related technologies in GermanyHealth Policy199946210512610346284
- WildCLangerTEmerging health technologies: informing and supporting health policy earlyHealth Policy200887216017118295925
- MaynardADAitkenRJButzTSafe handling of nanotechnologyNature2006444711726726917108940
- PepićILovrićJCetina-ČizmekBReichlSFilipović-GrčićJToward the practical implementation of eye-related bioavailability prediction modelsDrug Discov Today2014191314423948143