
Abstract
Since the Great Oxidation Event, about 2.4 billion years ago, the Earth is immersed in an oxidizing atmosphere. Thus, it has been proposed that excess oxygen, originally a waste product of photosynthetic cyanobacteria, induced oxidative stress and the production of reactive oxygen species (ROS), which have since acted as fundamental drivers of biologic evolution and eukaryogenesis. Indeed, throughout an organism’s lifespan, ROS affect directly (as mutagens) or indirectly (as messengers and regulators) all structural and functional components of cells, and many aspects of cell biology. Whether left unchecked by protective antioxidant systems, excess ROS not only cause genomic mutations but also induce irreversible oxidative modification of proteins (protein oxidation and peroxidation), lipids and glycans (advanced lipoxidation and glycation end products), impairing their function and promoting disease or cell death. Conversely, low-level local ROS play an important role both as redox-signaling molecules in a wide spectrum of pathways involved in the maintenance of cellular homeostasis (MAPK/ERK, PTK/PTP, PI3K-AKT-mTOR), and regulating key transcription factors (NFκB/IκB, Nrf2/KEAP1, AP-1, p53, HIF-1). Consequently, ROS can shape a variety of cellular functions, including proliferation, differentiation, migration and apoptosis. In this review, we will give a brief overview of the relevance of ROS in both physiological and pathological processes, particularly inflammation and aging. In-depth knowledge of the molecular mechanisms of ROS actuation and their influence under steady-state and stressful conditions will pave the way for the development of novel therapeutic interventions. This will mitigate the harmful outcomes of ROS in the onset and progression of a variety of chronic inflammatory and age-related diseases.
Introduction
The detection of free radicals (chemical entities possessing highly reactive unpaired electrons) in biological systemsCitation1 gave rise to the free radical theory,Citation2 which was supported by the discovery of superoxide dismutase (SOD)Citation3 and by pioneering concepts such as oxidative stress and antioxidants.Citation4 All these findings have placed aerobic metabolism and reactive oxygen species (ROS) as the most accepted cause of aging and numerous inflammatory diseases. ROS comprise both free radical and non-free radical oxygen intermediates (peroxides), like superoxide radicals (O2•-), hydrogen peroxide (H2O2), hydroxyl radicals (OH•), and singlet oxygen (1O2).Citation5 These molecules are generated by plasma membrane proteins, such as the growing family of NADPH oxidases,Citation6 by lipid metabolism within the peroxisomes,Citation7 and by the activity of various cytosolic enzymes such as cyclooxygenases.Citation8 Although all these sources contribute to the overall oxidative burden, the vast majority of cellular ROS (approximately 90%) come from mitochondria due to oxidative phosphorylation.Citation9
Oxidative stress, defined by an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of oxidation-reduction (redox) reactions and/or molecular damageCitation10 introduces the redox concept, that in living cells operates in fundamental processes, collectively termed “redox signaling” and “redox control”.Citation11 Thus, the original free radical theory has been substituted by the “redox hypothesis”, which postulates that oxidative stress occurs by disruption of thiol redox circuits. This is fundamental in cell signaling and physiological regulation without macromolecular damage. Indeed, thiols in proteins can function as transducing elements linking chemistry (ROS) and biologic structure.Citation12,Citation13
Conversely, elevated levels of ROS may lead to more extensive and irreparable cell damage through oxidation of DNA, RNA, carbohydrates, proteins and lipids, resulting ultimately in cell death through apoptosis or necrosis.Citation14 There are a high variety and range of pro-oxidant and anti-oxidant enzymes and compounds, prompting a classification of sub-forms of oxidative stress: nutritional, dietary, postprandial, radiation-induced and physiological. At the molecular level, oxidative stress can be photooxidative, nitrosative or reductive. Finally, different scales can be considered ranging from physiological oxidative stress to excessive and toxic oxidative burden.Citation10
In pathological situations and exposure to stressful environmental insults, ROS are produced in excessive amounts.Citation15 These pathological effects are usually mediated by ion channel opening, lipid peroxidation, protein modifications, and DNA oxidation. The pro-inflammatory molecules generated in stressful situations through ROS cause inflammation, which plays a major role in aging and the development of a variety of diseases.Citation16 These include vascular disorders (coronary heart disease, hypertension, metabolic syndrome, atherosclerosis),Citation17 autoimmune diseases (rheumatoid arthritis, inflammatory bowel disease),Citation18 neurodegenerative diseases (Alzheimer’s, Parkinson’s, age-related macular degeneration),Citation19 and respiratory diseases (asthma, chronic obstructive pulmonary disease, acute lung injury, cystic fibrosis).Citation20
This review will focus on the role of ROS at the physiological and physiopathological levels. Particularly, we will emphasize their relevance in the immune-inflammatory processes, highlighting the most important molecules/pathways involved. Finally, we will stress the role of ROS in senescence and aging, and the status of the underlying antioxidant therapies.
ROS Production and Compartmentalization
Up to 1–2% of consumed oxygen by a cell can turn into oxygen radicals, which can lead to the production of ROS. The main source of ROS in vivo is through aerobic breathing. Nevertheless, ROS are also produced by peroxisomal β-oxidation of fatty acids, microsomal cytochrome P450, metabolism of xenobiotic compounds, stimulation of phagocytosis by pathogens or lipopolysaccharides, arginine metabolism, and tissue-specific cellular enzymes.Citation21,Citation22 Intracellular ROS generation and their local redox status are important for understanding cell pathophysiology. Some subcellular compartments are more oxidizing (such as endoplasmic reticulum (ER), lysosomes, or peroxisomes) whereas others are more reducing (mitochondria, nuclei). Thus, ROS levels may fluctuate between subcellular compartments and may lead to beneficial effects or pathology.Citation23
Mitochondria are considered the most redox-active compartment in the cell, accounting for more than 90% of oxygen utilization.Citation9 Although most of the oxygen undergoes complete reduction to water at the level of cytochrome oxidase, partial reduction accompanied by ROS generation can occur as well.Citation24 The most common ROS is O2•-, which is converted by mitochondrial SOD into H2O2, which in turn transmute into powerful oxidants like OH• radicals. This occurs primarily through a Fenton reaction, that can oxidize a high number of important biomolecules. SODs, metalloenzymes found in all kingdoms of life, can be classified into four groups according to the metal cofactor present in their active site: copper-zinc-SOD (Cu-ZnSOD), iron SOD (FeSOD), manganese SOD (MnSOD) and nickel SOD (NiSOD).Citation25 From these, three isoforms are ubiquitously expressed in humans, all other mammals and most chordates: cytoplasmatic Cu-ZnSOD or SOD1, mitochondrial MnSOD or SOD2, and extracellular Cu-ZnSOD (EC-SOD) or SOD3.Citation26,Citation27 SOD1 is the most abundant and is constitutively expressed, although its overall expression in the lung is lower compared to several other organ systems. Its functional importance is most likely related to the regulation of cytosolic O2•- levels.Citation28 SOD2 is inducible by oxidative stress, hyperoxia, environmental pollutants and by inflammatory cytokines.Citation29 SOD3 expression is cell- and tissue-specific, being highest in lung. Because SOD3 represents the only O2•- scavenging enzyme within the extracellular environment, it is likely of major importance in protection against exogenous and environmental stress.Citation30
The ER tubular network has a unique oxidizing environment, and under stress conditions, redox-signaling mediators play key roles in ROS generation and dictate the fate of protein folding and secretion.Citation31 The microsomal cytochrome P450-dependent monooxygenase system, responsible for the oxidative metabolism of xenobiotics, is one of the major producers of ROS in the liver cell.Citation32
The peroxisomes contain peroxisomal oxidases and xanthine oxidase, that generate ROS and nitric oxide (NO).Citation7 But catalase is the major oxidoreductase responsible for H2O2 metabolism. Under physiological conditions, H2O2 diffusion is prevented through its rapid conversion to O2. Interestingly, recycling endosomes, late endosomes, and lysosomes are as oxidizing as the ER.Citation33 The lysosomal electron transport chain, which promotes proton translocation to maintain an optimal pH for the acidic hydrolases, generates OH•radicals.Citation34
The main source of ROS in the plasma membrane corresponds to O2•- production by the membrane-bound NADPH oxidases, which help to kill bacterial intruders.Citation35 The NADPH oxidase proteins constitute the only enzyme family with the sole function of producing ROS.Citation36
Regarding the exogenous sources of free radicals, ROS, oxidative stress and environment hold a complex relationship which impact on human health and disease.Citation37 Environmental oxidative stress derives from different artificial and natural sources such as cigarette smoke, traffic exhaust emissions, solar radiation, nutrition, radiotherapy, cosmetic devices or industrial pollution, and is induced by different mechanisms.Citation38 Some of the influences of the environment on organic life can promote or generate oxidative stress. In general, the action of radiation includes the induction of ROS, namely O2−, H2O2, and OH•.Citation39
Functions and Mechanism of Action of ROS at the Physiological Level
At physiological level, besides their role destructing pathogens in the immune defense against external insultsCitation40 or the synthesis of cellular structures like protein complexes,Citation41 ROS function as redox messengers (second messengers) (). Cells can generate ROS constitutively and exogenously, and use them for intracellular signaling and for stimulating redox-sensitive signaling pathways to modify the cellular content of the cytoprotective regulatory proteins.Citation42,Citation43 Thus, ROS control pro-inflammatory signaling, pro-fibrotic signaling, cell proliferation, apoptosis and a range of other biological processes without triggering a requirement for macromolecular damage.Citation11 In fact, the ROS-mediated redox messenger activity is thought to be larger than the ROS-mediated macromolecular damage activity.Citation44
Figure 1 Functions of ROS at physiological level. Besides their direct functions in immune defense and cellular structure, ROS can act as second messengers. They oxidize the three main sulfur switches and indirectly influence pro-fibrotic signaling, autophagy, mitoptosis, pro-inflammatory signaling, apoptosis and cell proliferation. These processes are mediated by kinases, phosphatases and transcription factors, among others.
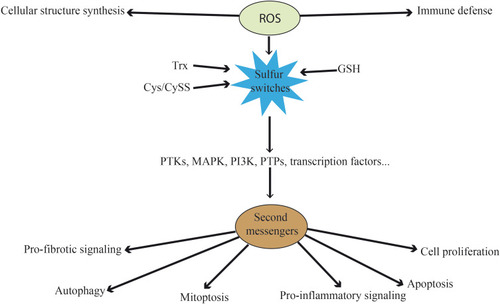
The redox state in the cytosol is normally achieved by the “redox-buffering” capacity of intracellular thiols, such as glutathione (GSH) and thioredoxin (Trx). These are the main redox nodes controlling physiologically relevant processes. Cysteine (Cys)/cystine (CySS) inclusion as a redox control node apart from GSH and Trx1 significantly expands the redox range. These three central couples of proteins are maintained at stable and non-equilibrium values in different organelles. Their function entails controlling the redox state of oxidizable thiols (“sulfur switches”) in proteins.Citation45,Citation46 The high ratios of reduced to oxidized forms are maintained by the activity of GSH reductase and Trx reductase. These sulfur switches are used for the maintenance of protein structure, protein trafficking, and for the regulation of enzyme, cell signaling, receptor, transcription factor and transporter activities.Citation47
Most redox signaling research has focused, on the one hand, on H2O2 and other ROS as oxidants for signaling proteins. On the other hand, it has centered on oxidation of sulfur-containing side chains of cysteine and methionine by ROS, which can result in higher oxidation states of sulfur. While ROS such as OH• may cause irreversible damage with low specificity to macromolecules, the main targets of a mild oxidant such as H2O2 are the thiol groups of protein cysteine residues.Citation48 In fact, O2•-, OH•, and 1O2 are not second messengers because they have no specificity in their interaction with effectors in signaling pathways. H2O2, instead, has an interesting chemistry that provides specificity for the oxidation of thiols and enables its function as a second messenger.Citation49 Accordingly, signaling enzymes and proteins containing cysteine residues have been proposed as potential targets for ROS.Citation50 The sulfur from the cysteine in these proteins can be reversibly or irreversibly oxidized to sulfenic acid (-SOH), sulfinic acid (-SO2H), disulfide bond (-SSR) or sulfonic acid (-SO3H)Citation51, and these were initially thought to be the markers of oxidative damage. Formation of sulfinic and sulfonic acid is irreversible and, therefore, they are not involved in the signaling reaction. Conversely, disulfide bonds and protein sulfenic acids can easily be reduced by systems such as Trx and peroxiredoxin (Prx) and are considered mediators of redox signaling.Citation52
Consequently, ROS formed in the mitochondria and the cytosol influence the redox state of cysteine residues in proteins. This constitutes a regulatory mechanism determining protein conformation and function. ROS are also critical for the establishment of protein–protein and protein–DNA interactions involving many aspects of the signal transduction pathway.
As a result of exposure to different stimuli, intracellular ROS increase and can lead to oxidation of cysteine residues in cytoplasmic proteins such as kinases and phosphatases, ultimately affecting signal transduction processes.Citation53 Overall, H2O2 can induce the phosphorylation of tyrosine residues from numerous cell proteins depending on the regulation of the redox status. This regulation is determined by the content of thiol compounds in the cell, primarily by the content of GSH. Through this mechanism, several proteins, notably kinases, are activated (eg, protein-tyrosine kinases (PTKs)). They promote the effects of growth factors, cytokines, and hormonesCitation54 or induce downstream signaling pathways involving protein kinases of the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) cascades.Citation42
Conversely, the inhibition of phosphatases by oxidants is mediated through oxidation of the reactive cysteine residue in the active center and inhibition of its catalytic activity (eg, protein tyrosine phosphatases (PTPs)Citation55 capable of dephosphorylating PTKs). Thus, phosphatases are directly oxidized by ROS resulting in their inhibition and ultimately in the sustained activation of signaling pathways. This plays a key role in cell proliferation and survival in response to growth factor, hormone, and cytokine stimulation.
Furthermore, ROS can directly modulate the activation of key signaling molecules such as transcription factors. They act as master switch systems in higher organisms, significantly determining the gene expression profile and cellular response to oxidative stress, as detailed in the next sections.
Other functions of ROS include their role in mitoptosisCitation56 and autophagy.Citation57 Moreover, there is a cross-talk between ROS and Ca2+.Citation42 Multiple evidences show that intracellular Ca2+ modulates ROS generation and clearance processes and thereby shift the redox state from oxidized to reduced, and vice versa.Citation58,Citation59
ROS in Inflammation
Imbalanced ROS are key players in the inflammatory process.Citation60 Oxidative stress-mediated signaling mechanisms are involved in inflammation and tissue injury. We acknowledge that many factors including activator protein 1 (AP-1), Redox factor-1 (Ref-1), p53 or hypoxia-inducible factor 1 (HIF-1) have been described to hold key effector roles in redox signaling. Nevertheless, in the following sections, we will focus on the involvement of ROS in a few fundamental aspects of the immune-inflammatory response. First, we will remark on the molecular mechanisms and relevance of ROS generation in inflammatory phagocytes. Indeed, NADPH oxidase-derived ROS has been associated with pathogen killing. Moreover, mitochondria-derived ROS hold also a relevant role in activating the inflammasomes, cytosolic multiprotein oligomers of the innate immune system driving the secretion of mature cytokines and pyroptosis, a pro-inflammatory form of programmed cell death. Furthermore, ROS range of actions encompasses the direct activation of the redox sensor nuclear factor erythroid 2-related factor (Nfr2). This ubiquitous transcription factor activates and modulates antioxidant, drug metabolism, anti-inflammatory detoxification and radical scavenging functions. Also, worth mentioning is its interplay with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), another omnipresent redox-regulated transcription factor that controls cytokine production and cell survival. In fact, ROS overproduction impacts key pathways leading the inflammatory response such as the tumor necrosis factor receptor-alpha (TNF-α)-tumor necrosis factor receptor-1 (TNFR1) pathway, which balances cell survival, apoptosis and necroptosis through regulation of NF-κB and, in turn, controls ROS signaling in immune cells. Finally, we will highlight the consequences of oxidative stress induced by polymorphonuclear cells (PMNs), stimulating the opening of inter-endothelial junctions and facilitating the migration of inflammatory cells across the endothelial barrier.
NADPH Oxidase-Derived ROS in Inflammatory Phagocytic Cells
NADPH oxidases (NOX) were first identified in phagocytes, acknowledging their role inducing respiratory or oxidative burst and bacterial killing.Citation61 The oxidative burst is the first line of defense against environmental pathogens as a part of the innate immune system. It involves neutrophils and macrophages,Citation62 and is characterized by the rapid production of large amounts of intracellular ROS and the activation of proteases that degrade phagocytosed microbes.Citation63 Non-mitochondrial ROS production during the oxidative burst results from the tightly regulated activation of NOX proteins. NOX1, NOX2 and NOX4, the major NOX isoforms expressed in the vascular system, are strongly implicated in inflammation-induced vascular injury.Citation64 Concomitantly, phagocytes such as granulocytes, neutrophils, monocytes and macrophages produce ROS using NOX2.Citation65
The generation of ROS begins with the rapid uptake of oxygen, activation of NOX and the production of the superoxide free radical anion, O2−•. O2−• is then rapidly converted to H2O2 by SOD. These free radicals can destroy microorganisms or other foreign matter. In addition, O2−• and H2O2 react to provide OH•.Citation66 Neutrophil cytoplasmic granules contain O2−• molecules generated by NOX2 and the enzyme myeloperoxidase (MPO), which react with the ubiquitous ion chloride generating hypochlorous acid, a potent oxidant and antimicrobial agent.Citation67 Following phagocytosis, antigen presentation allows a more specific and targeted response against the pathogen. ROS play a role in this specific response triggering the amplification of intracellular signal transduction cascades in T lymphocytes.Citation68 Moreover, ROS regulate macrophages adjusting its intracellular redox state. This has direct consequences in the release of prostaglandins and of pro-inflammatory cytokines such as several interleukins (IL-6, IL-12), necessary to control the ratio of type 1 to type 2 helper T cells.Citation69
Microbes with common pathogen-associated molecular patterns (PAMPs) are recognized by specific pattern recognition receptors, complement components or growth factors, triggering NOX2 complex activation.Citation70 Analogously, exposure of a phagocyte to pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), gamma-interferon (IFN-γ), and/or IL-1β induces NOX2 complex formation. This significantly increases the ROS levels within the cell.Citation71
The NOX2 complex is composed of five subunits: NOX2 (gp91phox) and the four phagocytic oxidases: p67phox, p47phox, p40phox and p22phox. To assemble the NOX complex under pathogen attack, the cytosolic p67phox, p47phox and p40phox subunits converge and recruit a small GTPase (Ras-related C3 botulinum toxin substrate 1 (RAC1) in monocytes or RAC2 in neutrophils), that colocalize with p22phox and NOX2 to the membrane.Citation72 The NOX2 complex converts extracellular O2 into O2.−, which is converted to H2O2 by SOD3 to generate ROS. These species penetrate the phagocyte membrane and act intracellularly to promote pathogen phagocytosis.Citation65
TNF-NOX2 signaling is believed to account for chronic inflammation and its associated tissue damage. ROS regulation is very important because it engages in a positive feedback loop in which the production of ROS is induced by TNF and, in turn, ROS trigger TNF expression through p38, NF-κB and c-Jun N-terminal kinases (JNK).Citation73
Riboflavin kinase (RFK) is important for ROS generation because it interacts with TNFR1 when coupled to the NOX2 complex, activated by pathogen stimulation.Citation74 Consequently, ROS are produced through a pathway involving Vav1, Rac2 and proline-rich tyrosine kinase-2 (Pyk2), which enhances the adherence of macrophages and neutrophils to endothelial cells.
NOX-generated ROS in macrophages induce the translocation of microtubule-associated protein 1 light chain 3 beta (MAP1A/MAP1BLC3B) to the phagosomal membrane, fusing lysosomes and clearing phagosomes in LC3-associated phagocytosis (LAP).Citation75 TLR4 signaling promotes the recruitment of mitochondria to phagosomes to enhance ROS production and kill bacteria.Citation76 This increases ROS production which, in turn, activates NF-κB in T cells.
Mitochondria-Derived ROS in Inflammation
Mitochondrial ROS (MtROS), a byproduct of mitochondrial respiration and metabolic enzyme activity, participate in key molecular functions and signal transduction pathways ensuing in inflammatory cells: 1) cytokine release induced by LPS, 2) NF-κB activation via inositol 1,4,5-trisphosphate receptor (IP3R), 3) Ca2+ signaling induced by thrombin, and 4) AP-1 activation induced by lysophosphatidylcholine (LPC), which triggers endothelial activation.Citation77
Another important role of MtROS is the regulation of the inflammasomes.Citation78 These high molecular weight complexes activate inflammatory caspases (caspase-1 and 11) and cytokines (IL-1β and IL-18) in macrophages.Citation79 At least four different prototypes of inflammasomes (Nucleotide-binding oligomerization domain, Leucine-rich Repeat and Pyrin domain-containing, NLRP, also known as NALP) have been recognized: NALP1, NALP3 (also known as NLRP3, the best-characterized form), NLR Family CARD domain containing 4 (NLRC4 or ice protease-activating factor, IPAF), and absent in melanoma 2 (AIM2).Citation80 MtROS are involved in macrophage activation during inflammation and become primary regulators of NLRP3 inflammasome activation. Thus, in macrophages stimulated by LPS, MtROS regulate IL-1β transcription (inflammasome priming) and the maturation and secretion of IL-1β (inflammasome activation).Citation81 On the other hand, thioredoxin-interacting protein (TXNIP), a negative regulator of Trx and a proapoptotic factor regulated by ROS, mediates the activity of MtROS and NOX4. TXNIP also influences inflammasome activation induced by glucose.Citation82 Furthermore, an increased level of ROS in chronic inflammatory states inhibits mitophagy (the removal of malfunctioning mitochondria through autophagy)Citation83 and fosters inflammasome activation.Citation84
Master Switches and Pathways Regulated by ROS in Inflammation: NF-κB, Nrf2 and TNF-α -TNFR1 as Paradigms
Overall, ROS can activate NF-κB, a major redox-regulated transcription factor, in response to inflammatory agonists ().Citation85 The translocation of NF-κB to the nucleus takes place in response to H2O2 through nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor-alpha (IκBα), tyrosine phosphorylation (Tyr42), phosphorylation of the serine/threonine PEST domain plus subsequent degradation via calpain and p65 phosphorylation.Citation86 In the nucleus, NF-κB initiates the transcription of pro-inflammatory molecules, including cytokines (IL-1, IL-6, TNF-α), cyclooxygenase-2 (COX-2), vascular adhesion molecules and inducible nitric oxide synthase (iNOS). ROS-induced NF-κB is inhibited by SOD2 overexpression,Citation87 and the NOX family of proteins influence, and are influenced by, NF-κB activity.Citation88 Specific inflammatory agonists use ROS as part of their signaling cascades. For example, LPS induces the activation of NF-κB through NOX4.Citation89
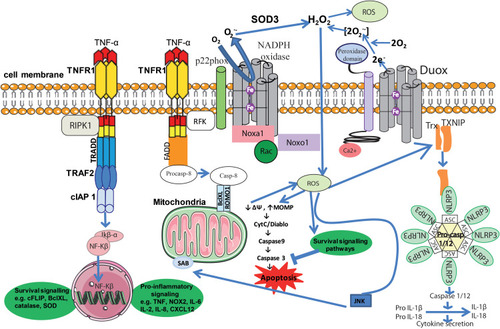
Figure 2 Inflammation and ROS. Elevated ROS production as a result of inflammatory signaling can mediate canonical NF-κB activation and downstream inflammatory gene induction, proteasome activity, antioxidant gene transcription, inflammasome activation, and cytokine secretion. The intricate balance between cell death and cell survival is largely modulated by intracellular ROS generation. In general, high intracellular ROS generation causes cell death by activation of cell death pathways (mitochondrial-dependent and -independent), whereas low levels of ROS act as signaling molecules that facilitate cell survival. Death receptors such as TNFR1 cause cell death by inducing high intracellular ROS production. The binding of TNF-α to TNFR1 recruits proteins such as TRADD and TRAF2 to activate TNFR1 signaling. The associated TRADD further recruits FADD and pro-caspase-8, and this whole signaling complex known as DISC is endocytosed, resulting in caspase-8 activation. Similar to TNFR1, the extent of caspase-8 activation determines whether a cell will follow a mitochondrial-dependent pathway or an independent pathway. Low caspase-8 activation follows a mitochondrial-dependent amplification loop which causes Cyt-C release. The released Cyt-C then subsequently binds Apaf-1 and caspase-9 and activates effector caspases such as caspase-3, which causes cell death. In the event of significant caspase-8 activation, it directly activates caspase-3 independent of mitochondria. High intracellular ROS induce sustained JNK activation and causes mitochondrial Cyt-C-dependent cell death. TNFR1 signaling also activates the NF-κB transcription factor by recruiting TRAF-2 and RIPK1 proteins. NF-κB activation is involved in the transcription of anti-apoptotic signaling proteins such as cIAP-2 and Bcl-xL. On the other hand, TNF can activate the NOX1 (neutrophilic) or NOX2 (epithelial) complex. The first step of NOX1/2 activation is the interaction of the cytosolic domain of TNFR1 with RFK and p22phox. The activated NOX complex converts extracellular O2 into O2−•, which extracellular SOD3 then converts into extracellular H2O2. The generated H2O2 passes freely through the plasma membrane and acts as a major source of intracellular ROS. Furthermore, TNF-induced formation of complex II leads to interactions between activated caspase-8, ROMO1 and Bcl-XL in the outer mitochondrial membrane, which reduce mitochondrial membrane potential, triggering MOMP and the production of MtROS. Conversely, if the levels of caspase-8 are high, TNF can activate directly caspase-3 independent of mitochondria, leading to apoptosis. Additionally, Ca2+-activated DUOX produces even more H2O2, which joins the cycle. Another important role of MtROS is the regulation of the inflammasome. MtROS are involved in immune cell activation during inflammation and are primary regulators of NLRP3 inflammasome activation. These high-molecular-weight complexes activate inflammatory caspases (caspase-1 and −11) and cytokines (IL-1β and IL-18). Also illustrated are ROS activating NF-κB directly or indirectly by inhibiting IKK phosphatases, and JNK stimulating mitochondrial ROS production through SAB.
Nrf2 is a redox-sensitive transcription factor that regulates redox homeostasis, holding anti-oxidant, anti-inflammatory and detoxification responses.Citation90 Nrf2 resides in the cytoplasm at a low basal level, but when induced by oxidative stress translocates to the nucleusCitation91 where it binds to the antioxidant responsive element (ARE) sequence, necessary for ROS detoxification. Nrf2 is composed of seven functional domains (Neh1-7) that regulate its transcriptional activity and stability.Citation92 The Neh1 is responsible for the translocation of Kelch-like ECH-associated protein 1 (KEAP1) into the nucleus.Citation93 The activity of the Nrf2-ARE signaling pathway is controlled by sequestration and degradation of Nrf2 into the cytoplasm by KEAP1.Citation94 Indeed, Neh2 is a negative regulatory domain that promotes Nrf2 degradation.Citation95 Neh3 modulates the transcriptional activation of ARE genes,Citation96 and Nhe4 and 5 facilitate Nrf2 transcription.Citation97 Neh6 and 7 control Nrf2 degradation.Citation98
There is a crosstalk between Nrf2 and NF-κB signaling pathways under stress or pathophysiological conditions.Citation62,Citation64,Citation99 Studies using animal modelsCitation100 or different cell types, such as microglial cells and monocytes,Citation101,Citation102 suggest that Nrf2 up-regulation decreases pro-inflammatory and immune responses that are regulated by NF-κB. Thus, although not completely well understood, Nrf2 and NF-κB influence each other to control anti-oxidant and inflammatory responses.Citation103 It has also been shown that Nrf2 overexpression inhibits RAC1-dependent activation of NF-κB.Citation104 Interestingly, novel therapeutic approaches to prevent or reduce brain injury are being focused to restore Nrf2 activity while preventing NF-κB activation. Thus, several phytochemicals such as curcumin, sulforaphane and metformin have shown a dual effect as Nrf2 activators and NF-κB inhibitors.Citation63
TNF-α is a pro-inflammatory cytokine with important functions in mammalian immunity and cellular homeostasis.Citation105 This master multi-faceted regulator plays an important role in the control of cell survival, apoptosis, necroptosis and intercellular communication through two main pathways: 1) induction of apoptosis through ROS/JNK, and 2) reduction of the apoptotic signal through NF-κB activation and triggering antioxidant gene expression.Citation106,Citation107 Dysregulation of these processes elicits inflammatory diseases and cancer. TNF can present in soluble (sTNF) or membrane-bound (mTNF) form. Its receptor TNFR1, is ubiquitously expressed on almost all cell types and can be activated by both TNF forms.Citation108 TNFR1 signaling under resting conditions is inhibited by association with a 60-kD cytoplasmic protein known as silencer of death domains (SODD). When TNF binds to TNFR1 the corresponding complex, TNF-TNFR1, recruits the adaptor protein tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD) and the receptor-interacting serine/threonine-protein kinase 1 (RIPK1),Citation109 initiating the assembly of membrane-bound TNFR complex I. That is, binding of TNF receptor-associated factor 2 (TRAF2) to cellular inhibitor of apoptosis proteins (cIAP1 and cIAP2).Citation110,Citation111 cIAP1 and cIAP2 facilitate the recruitment of the linear polyubiquitin chain assembly complex (LUBAC) to complex ICitation106,Citation112 and prevent inflammationCitation113 and cell death. This is achieved through the recruitment of TGFβ-activated kinase 1 (TAK1) and TAK1 binding protein 2/3 (TAB2/3), phosphorylation of IκBα and activation of NF-κB.Citation114
RIPK1 acts as a molecular switch between cell survival and cell death, depending on its state of ubiquitination. When RIPK1 in complex I is deubiquitinated by cylindromatosis (CYLD) and TNFR1 is activated by TNF-α, RIPK1 dissociates from the membrane-bound TNFR1 signaling core. Then, it assembles in the cytosol with TRADD and Fas-associated death domain (FADD), and the collective death-inducing signaling complex (DISC) is internalized in an endosome. The release of endosomal DISC to the cytosol further recruits initiator procaspase-8, resulting in its activation through complex IIa. This leads to apoptosis depending on the presence of cellular FLICE inhibitory protein (cFLIPL).Citation115 Complex IIb/necrosome is formed when dissociated and non-ubiquitinated RIPK1 assembles with the same molecular components of complex IIa, except TRADD, with further addition of RIPK3.Citation106
ROS are important regulators of TNF-TNFR signaling, leading to cell survival or death. The generally accepted hypothesis is that TNF-induced ROS suppress NF-κB activation decreasing survival signaling and increasing cell death.Citation116 Nevertheless, several evidences indicate that mitochondrial ROS can promote TNF-mediated NF-κB activation.Citation117 Thus, ROS can positively control NF-κB signaling inhibiting the phosphatases involved in IκBα phosphorylation, triggering its degradation and allowing NF-κB activation.Citation118
TNF-induced ROS signaling initiates with the activation of NOX1/2 (NOX2 concerning phagocytic cells and NOX1 non-phagocytic cells) through the interaction of TNFR1 with RFK and p22phox, which generates H2O2 via extracellular SOD3. This H2O2 traverses the extracellular membrane and is the main source of intracellular ROS. Moreover, TNF-induced formation of complex II (which contains procaspase-8) leads to interactions between BclXL and ROS modulator 1 (ROMO1) in the outer mitochondria membrane. This reduces the mitochondrial membrane potential (ΔΨ), which produces more MtROS through mitochondrial outer membrane permeability (MOMP). Thus, MtROS can contribute to apoptosis releasing mitochondria-derived pro-apoptotic factors such as cytochrome C (Cyt-C).Citation119,Citation120 High levels of ROS activate the JNK pathway by inhibiting MAPK phosphatases, while JNK stimulates MtROS production through SH3 homology associated BTK binding protein (SAB), an outer mitochondrial membrane protein.Citation121 Increased MOMP can be induced by excessive Ca2+ entry and increased oxidative stress, resulting in mitochondrial membrane depolarization. Cyt-C then binds to apoptosis activation factor-1 (Apaf-1) and recruits initiator pro-caspase-9, which undergoes auto-activation. This whole complex is collectively known as “apoptosome”.Citation122 Furthermore, caspase-9 activates effector caspases such as caspase-3. Second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO) also facilitates the activation of effector caspases by removing the blockage by an inhibitor of apoptosis proteins (IAPs).Citation123 Apoptosis-inducing factor (AIF), on activation, induces DNA condensation and cleavage.Citation124 EndoG-mediated DNA fragmentation is the final event in the apoptosis process. The mitochondrial release of Cyt-C is considered the major determinant of cell death, and Smac/DIABLO, which activates caspase-9 and -3, executes apoptosis.Citation115
Indeed, TNF-induced ROS production is important in the crosstalk between the NF-κB-induced cell survival pathway and the JNK-induced cell death pathway.Citation125,Citation126 SAB activates JNK and thus increases MtROS generation, sustaining JNK activation in a self-amplifying loop. Moreover, the quantity of activated caspase-8 will determine whether a cell follows a mitochondrial-dependent (low levels) or -independent (high levels) pathway. Furthermore, dual oxidase (DUOX), the major NOX isoform expressed preferentially in the lungsCitation127 is activated by Ca2+, the concentration of which is enhanced by ROS, and further increases H2O2 production, fostering the self-amplifying cycle.
Oxidative Stress Regulates the Opening of Endothelial Junctions and Immune Cell Migration Across the Endothelial Barrier
Cell adhesion molecules (CAMs) are expressed on activated endothelial cells and leukocytes. They mediate the migration of inflammatory cells through the endothelium of postcapillary venules. Neutrophils, the first cells to reach the inflamed scene, cross the endothelial barrier mainly through endothelial junctions.Citation128 The extravasation process in response to inflammatory stimuli is regulated by oxidative stress induced by the own extravasating leukocytes.Citation129,Citation130 This can occur directly through CAMs activation: intracellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), E-selectin and P-selectinCitation131–Citation133 (), including integrins that bind to ICAM-1.Citation134 Alternatively, neutrophil extravasation may involve indirect activation through a transcription-dependent mechanism involving redox-sensitive transcription factors (ie, NF-κB and AP-1). Thus, oxidative stress induced by leukocytes at the site of inflammation plays a crucial role disturbing endothelial junction assembly. Both adherens junctions (AJs) and tight junctions (TJs) initiate their disassembly, leading to gap formation between cells.Citation135
Figure 3 Role of ROS in neutrophil migration through endothelial cells. Integrins bind to ICAM-1 expressed in activated endothelial cells and arrest neutrophils on the endothelial surface. This leads to the activation of Cdc42 and Rac, which induce intracellular ROS generation through NOX. Increased ROS are involved in activation of different kinases, including c-Src and PKC, and thus induce phosphorylation and destabilization of endothelial junction proteins such as adherens junction. Moreover, ROS enhance the expression of P-selectin on the endothelium. Furthermore, ROS are known to activate NF-κB, inducing the expression of cell adhesion molecules such as ICAM-1, VCAM-1 and E-selectin which, in turn, enhance neutrophil binding on the endothelium. Acute exposure to pro-inflammatory factors triggers NOX-mediated ROS, which results in an increase of [Ca2+] by TRPC/TRPM ion channels. Increased [Ca2+] activates calmodulin and several downstream kinases including myosin light chain kinase (MLCK), which contribute to the disassembly of endothelial junction proteins.
![Figure 3 Role of ROS in neutrophil migration through endothelial cells. Integrins bind to ICAM-1 expressed in activated endothelial cells and arrest neutrophils on the endothelial surface. This leads to the activation of Cdc42 and Rac, which induce intracellular ROS generation through NOX. Increased ROS are involved in activation of different kinases, including c-Src and PKC, and thus induce phosphorylation and destabilization of endothelial junction proteins such as adherens junction. Moreover, ROS enhance the expression of P-selectin on the endothelium. Furthermore, ROS are known to activate NF-κB, inducing the expression of cell adhesion molecules such as ICAM-1, VCAM-1 and E-selectin which, in turn, enhance neutrophil binding on the endothelium. Acute exposure to pro-inflammatory factors triggers NOX-mediated ROS, which results in an increase of [Ca2+] by TRPC/TRPM ion channels. Increased [Ca2+] activates calmodulin and several downstream kinases including myosin light chain kinase (MLCK), which contribute to the disassembly of endothelial junction proteins.](/cms/asset/94aed9fd-65a7-4958-8b98-681077ae8b46/djir_a_12198534_f0003_c.jpg)
ROS-mediated regulation of intracellular free Ca2+ concentration is also a major mechanism of increased vascular permeability, the hallmark of inflammation. An increase in intracellular Ca2+ was associated with enhanced H2O2 generation.Citation136 Moreover, the major Ca2+ driveway in endothelial cells is through store-operated Ca2+ channels (SOC channels: TRPC1 and TRPC4), receptor-operated Ca2+ channels (ROC channels: TRPC3, TRPC6 and TRPC7), and transient receptor potential melastatin channels (TRPM), presumably regulated by ROS.Citation137
Other enzymes that contribute to the opening of endothelial junctions include: 1) myosin light chain (MLC), which is activated by ROS and contributes to actin reorganization in endothelial cells;Citation138 2) Cell division control protein 42 (Cdc42), a member of the Rho GTPase family involved in ethanol-induced NADPH oxidase activation and ROS generation, rearranging actin filaments in endothelial cells;Citation139 3) protein kinase C (PKC) isoforms, activated by H2O2, targets for multiple tyrosine phosphorylation events by various tyrosine kinases, including the Src family kinases;Citation140 and 4) toll-like receptors (TLRs), a subset of the pathogen-associated pattern recognition receptors family expressed in a wide range of immune cells the most relevant of which is TLR4, receptor for LPS signaling.Citation141
ROS and Aging
The multifactorial and inexorable phenomenon of aging deteriorates human homeostasis at multiple levels, reducing gradually the ability to resist stress, damage, and illness.Citation142 Aging mechanisms have been classified into two main categories: 1) genetically programmed by developmental processes (cell senescence, neuroendocrine alterations, immunological alterations), and 2) caused by random damage, that is, accumulation of somatic mutations and oxidative stress. Here we will focus on the “free radical theory of aging” and its evolved “mitochondrial free radical theory of aging”, highlighting the ROS ability to induce cell senescence in aging processes. Nevertheless, ROS generation might not be the initial trigger of the aging process. In fact, several studies present an alternative point of view to Harman’s hypothesis,Citation2 whose free radical theory remains inconclusive. These reports suggest that: 1) oxidative damage is just one type of damage, 2) biological imperfectness drives the aging process, and 3) cumulative damage defines lifespan.Citation143,Citation144 Accordingly, antioxidant strategies against aging will be briefly considered.
The Free Radical Theory of Aging
One of the preferential causal theories of aging is the “free radical damage theory”, firstly proposed by Denham Harman in the 1950s to explain aging focusing on ROS generation. Free radicals cause oxidative damage to the macromolecular components of the cell, affecting homeostasis (cell death, inflammation) and shaping the organism’s pathophysiological network. Indeed, multiple studies have reported age-related oxidative modifications to a large variety of proteins. These include structural proteins, enzymes, and proteins important in signal transduction pathways.Citation145–Citation149 Furthermore, telomerase shortening is affected by oxidative damage.Citation150,Citation151 As stated previously, ROS such as OH•, O2•−, and H2O2 are extremely reactive and are the major cause of damage to proteins, lipids, and DNA. Oxidative damage to these biomolecules seems to depend on H2O2 and a reduced transition metal.Citation152 Therefore, molecules that contain transition metals, such as aconitase, are likely to undergo oxidative damage.Citation153
Any perturbation of the redox balance during aging can activate the redox-sensitive transcription factors and generate numerous pro-inflammatory mediators. In addition to the apoptotic and senescence pathways, redox modification of transcription factors can regulate cellular proliferation, differentiation, senescence, death, and aging. For instance, NF-κB altered signaling has been causally linked to aging and diverse pathological conditions.Citation154 Macrophages, central effectors of the innate immune system, are strongly involved in inflammation and generate reactive species during age-associated diseases.Citation155 However, there is still a lack of evidence to conclusively determine that inflammation is one of the causal factors influencing biological aging and longevity in mammalian species.
Mitochondria play a significant role in the free radical theory of aging.Citation156 According to the endosymbiotic theoryCitation157, aging started 1 billion years ago with mitochondria. Because mitochondria are mostly responsible for the production of ROS, in the 1970s the free radical theory of aging was extended into the “mitochondrial free radical theory of aging”.Citation158 It states that aging is produced by the toxicity of ROS through a malicious cycle in which ROS injury to mitochondrial elements precedes the generation of more ROS. The mitochondrial activity declines with age because of replication errors of mitochondrial DNA (mtDNA) and toxic adducts on mitochondrial proteins.Citation159 These adducts are harmful to the function of proteins, being promoted by high levels of acetyl-CoA and succinyl-CoA, which acylate the ε-amino groups of lysines. Sirtuins like SIRT3, SIRT4 and SIRT5, which are enriched in mitochondria and possess deacylase activity, catalyze the removal of the adducts.Citation160 This decline in mitochondrial function includes a decreased activity of the respiratory chain and ATP synthase, Krebs cycle fluxes, oxidative alterations of cardiolipin, disrupted regulation, and defective mtDNA regulation and activity.Citation161 This results in an increase in the production of O2•− and H2O2 in mitochondria, causing oxidative damage to mtDNA (more sensitive than nuclear DNA) and mitochondrial membrane lipids.Citation162 Consequently, changes in mitochondrial membrane permeability ensue, which culminates in the release of apoptogenic factors mediating cell apoptosis through the previously described pathway.Citation163
The cellular senescence process, which influences somatic, tumor and stem cellsCitation164 is considered an aging hallmark. It drives the cell through longevity, by hampering tumorigenesis and cell death, and is involved in many age-related diseases.
The DNA damage caused by ROS as mutating agents induce and maintain the cell senescence process.Citation165 Moreover, attention has also been focused on the ROS involvement as signaling molecules in cell senescence induction without DNA damage, through Ras, p53, p21 or p16.Citation166 Furthermore, ROS contribution at the epigenetic level seems to modulate cell senescence.Citation167 Hence, senescent cells have been identified as a novel potential therapeutic target in aging and age-related diseases.Citation168
Therapeutic Strategies Against Aging
Studies in different model organisms indicate that inhibition of oxidative stress contributes to an increase in the life span. For example, administration of Vitamin E was previously shown to extend the life span of different animals.Citation169,Citation170 Nevertheless, despite a vast amount of publications on ROS and the implications of oxidative stress and free radical damage in almost all pathological conditions, no pharmaceutical drugs are yet available or prescribed for antioxidant therapies in the clinical practice. Similarly, the commercial use of many natural antioxidants, mainly in the form of dietary supplements is a fact. Nevertheless, the results of a number of clinical trials suggest that, as yet, there is no sound clinical evidence that such antioxidants can represent effective treatments in any clinical condition.Citation171,Citation172 Notable exceptions are GSH (administered either orally or by inhalation),Citation173,Citation174 although questioned,Citation175 and perhaps β-carotene at high doses.Citation176,Citation177 Indeed, antioxidants have met with limited success because only a small fraction reaches mitochondria.Citation178 To circumvent this limitation, antioxidants are being conjugated with lipophilic cations so that they can accumulate in mitochondria.Citation179 Alternatively, human clinical trials regarding dietary supplementation have reported to reduce ROS injury related to aging.Citation180 Thus, vitamin D supplementation can reduce the risk of hip and other fractures in housebound elderly.Citation181 Moreover, functional foods and nutraceuticals possessing antioxidant activity, such as supplemented blueberry extract, might slow the aging process.Citation182
Human aging could also be influenced by the concentration and activity of enzymes involved in the antioxidant defense system. Several reports suggest that the levels of SOD, catalase, and glutathione peroxidase from a variety of tissues (liver, brain, kidney, heart, etc.) are reduced in senescent animals.Citation183 In this respect, SOD has demonstrated great physiological relevance and therapeutic potential in various pathological conditions such as cancer, inflammatory diseases, cystic fibrosis, ischemia, aging, rheumatoid arthritis, neurodegenerative diseases and diabetes.Citation184 Accordingly, SOD has proved effective in preclinical studies.Citation185 SOD is also considered an anti-aging enzyme. In animal models such as Drosophila, a reduction in SOD activity accelerated loss of olfactory behavior on aging.Citation186 Furthermore, SOD mimetics, synthetic compounds that have an equivalent function as SOD, attenuate aging.Citation187 On the other hand, energy restriction diet, moderate reduction of nutrient availability without malnutrition, significantly increased the expression of SOD1 and SOD2 and extended lifespan by 16% in fruit flies.Citation182
Although contrasting reports and inconclusive studies have emerged, as stated previously, it can be postulated that the increase in oxidative stress and damage to cellular constituents associated with aging could be due to a decline in antioxidant defense systems. For many years, coenzyme Q 10 (CoQ10) has been considered a key factor in the progression of aging-associated complications.Citation188 However, although several studies have supported the safety and the potential of CoQ10 reducing oxidative stress biomarkers, adequate large-scale clinical trials are needed to confirm the beneficial effects of CoQ10 supplementation.
Concluding Remarks
A double-edged sword of ROS in health and pathology is being increasingly appreciated, although the underlying mechanisms are still incompletely understood. In physiological processes environmental and metabolic ROS levels are tightly regulated and play an essential role as signaling molecules, contributing to cell and tissue homeostasis. Conversely, excess of ROS generation and/or impaired antioxidant activity under acute and chronic oxidative stress associate with metabolic “reprogramming”, inflammation, tissue damage/dysfunction and toxicity, leading to senescence or death. Emerging “omics” technologies together with single-cell analyses and novel imaging tools will allow specific, spatiotemporal, comprehensive and quantitative dissection of the whole spectrum of pathways and processes induced by ROS at whole tissue or organism level. In this scenario, further advances in personalized redox medicine may combine dietary/nutritional and pharmacological strategies (anti-inflammatory drugs, activators of mitophagy, uncouplers, hTERT activators, antioxidants). Whether successful, these approaches might enable to tip the balance equilibrating the redox state of the tissues towards homeostasis, while maintaining the ability of immune cells to clear infections (eg, pathogen killing through the respiratory burst of neutrophils). This will surely become highly beneficial in a variety of age-related pathologies and ultimately in the health of the elderly population.
Acknowledgments
We thank CERCA Programme/Generalitat de Catalunya for institutional support. J.C. is supported by a grant from the “Associació Catalana de Fibrosi Quística”. J.M.A. has received support from the Ministerio de Economía y Competitividad (Madrid, Spain) (grants FIS-ISCIII PI13/01490 and PI16/00377, co-funded by FEDER funds/European Regional Development Fund (ERDF)-a way to build Europe-), and from the Generalitat de Catalunya (grant 2014S GR 541). J.M.A. is sponsored by the “Researchers Consolidation Program” from the SNS-Dpt. Salut Generalitat de Catalunya (Exp. CES06/012).
Disclosure
The authors report no conflicts of interest in this work.
References
- Commoner B, Townsend J, Pake GE. Free radicals in biological materials. Nature. 1954;174:689–691. doi:10.1038/174689a013213980
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi:10.1093/geronj/11.3.29813332224
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244:6049–6055.5389100
- Sies H. Oxidative Stress. Academic Press; 1985.
- Pizzino G, Irrera N, Cucinotta M, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. doi:10.1155/2017/841676328819546
- Buvelot H, Jaquet V, Krause KH. Mammalian NADPH oxidases. Methods Mol Biol. 2019;1982:17–36.31172464
- Del Río LA, López-Huertas E. ROS generation in peroxisomes and its role in cell signaling. Plant Cell Physiol. 2016;57:1364–1376.27081099
- Martínez-Revelles S, Avendaño MS, García-Redondo AB, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxidants Redox Signal. 2013;18:51–65. doi:10.1089/ars.2011.4335
- Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–181.19513674
- Sies H. Memories of a senior scientist: biological redox systems and oxidative stress. Cell Mol Life Sci. 2007;64:2181–2188. doi:10.1007/s00018-007-7230-817565441
- Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi:10.1016/j.redox.2015.01.00225588755
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:849–868. doi:10.1152/ajpcell.00283.2008
- Benhar M. Oxidants, antioxidants and thiol Redox switches in the control of regulated cell death pathways. Antioxidants. 2020;9:309. doi:10.3390/antiox9040309
- Ghosh N, Das A, Chaffee S, Roy S, Sen CK. Reactive oxygen species, oxidative damage and cell death In: Chatterjee S, Jungraithmayr W, Bagchi D, editors. Immunity and Inflammation in Health and Disease: Emerging Roles of Nutraceuticals and Functional Foods in Immune Support. Elsevier; 2018:45–55.
- Brieger K, Schiavone S, Miller FJ, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly. 2012;142:w13659.22903797
- Srivastava KK, Kumar R. Stress, oxidative injury and disease. Indian J Clin Biochem. 2015;30:3–10. doi:10.1007/s12291-014-0441-525646036
- Aldosari S, Awad M, Harrington EO, Sellke FW, Abid MR. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants. 2018;7:14. doi:10.3390/antiox7010014
- Glennon-Alty L, Hackett AP, Chapman EA, Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic Biol Med. 2018;125:25–35. doi:10.1016/j.freeradbiomed.2018.03.04929605448
- Umeno A, Biju V, Yoshida Y. In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic Res. 2017;51:413–427. doi:10.1080/10715762.2017.131511428372523
- Paola Rosanna D, Salvatore C. Reactive oxygen species, inflammation, and lung diseases. Curr Pharm Des. 2012;18:3889–3900. doi:10.2174/13816121280208371622632750
- Phaniendra A, Jestadi DB, Periyasamy L, Properties S. Free radicals: Targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11–26.25646037
- Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi:10.1016/j.biocel.2006.07.00116978905
- Di Meo S, Reed TT, Venditti P, Victor VM. Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. 2016;2016:1245049. doi:10.1155/2016/124504927478531
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13.19061483
- Younus H. Therapeutic potentials of superoxide dismutase. Int J Health Sci. 2018;12:88–93.
- Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants Redox Signal. 2011;15:1583–1606. doi:10.1089/ars.2011.3999
- Wang Y, Branicky R, Noë A, Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217:1915–1928. doi:10.1083/jcb.20170800729669742
- Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxidants Redox Signal. 2009;11:1313–1333. doi:10.1089/ars.2008.2363
- Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–1619.12796054
- Nguyen NH, Tran GB, Nguyen CT. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J Mol Med (Berl). 2020;98:59–69. doi:10.1007/s00109-019-01845-231724066
- Zeeshan HMA, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. 2016;17:327. doi:10.3390/ijms1703032726950115
- Cederbaum AI. Cytochrome P450 and oxidative stress in the liver In: Muriel P, editor. Liver Pathophysiology: Therapies and Antioxidants. Elsevier; 2017:401–419.
- Austin CD, Wen X, Gazzard L, et al. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc Natl Acad Sci USA. 2005;102:17987–17992. doi:10.1073/pnas.050903510216322102
- Nohl H, Lysosomal GL. ROS formation. Redox Rep. 2005;10:199–205. doi:10.1179/135100005X7017016259787
- Rada B, Leto T. Oxidative innate immune defenses by Nox/Duox Family NADPH oxidases. Contrib Microbiol. 2008;15:164–187.18511861
- Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. 2014;9:119–145. doi:10.1146/annurev-pathol-012513-10465124050626
- Bardaweel SK, Gul M, Alzweiri M, et al. Reactive oxygen species: the dual role in physiological and pathological conditions of the human body. Eurasian J Med. 2018;50:193–201. doi:10.5152/eurasianjmed.2018.1739730515042
- Samet JM, Wages PA. Oxidative stress from environmental exposures. Curr Opin Toxicol. 2018;7:60–66. doi:10.1016/j.cotox.2017.10.00830079382
- Halliwell B, Gutteridge JMC, eds. Free Radicals in Biology and Medicine. Oxford University Press, Oxford; 2015.
- Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive oxygen species in the immune system. Int Rev Immunol. 2013;32:249–270.23617726
- Reichmann D, Voth W, Jakob U. Maintaining a healthy proteome during oxidative stress. Mol Cell. 2018;69:203–213. doi:10.1016/j.molcel.2017.12.02129351842
- Zhang J, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;2016:4350965.26998193
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi:10.1152/physrev.00018.200111773609
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–R462. doi:10.1016/j.cub.2014.03.03424845678
- Yatmaz S, Seow HJ, Gualano RC, et al. Glutathione peroxidase-1 reduces influenza A virus-induced lung inflammation. Am J Respir Cell Mol Biol. 2013;48:17–26. doi:10.1165/rcmb.2011-0345OC23002098
- Xu J, Li T, Wu H, Xu T. Role of thioredoxin in lung disease. Pulm Pharmacol Ther. 2012;25:154–162. doi:10.1016/j.pupt.2012.01.00222293327
- Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta Proteins Proteomics. 2014;1844:1335–1343. doi:10.1016/j.bbapap.2014.03.007
- García-Santamarina S, Boronat S, Hidalgo E. Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry. 2014;53:2560–2580. doi:10.1021/bi401700f24738931
- Di Marzo N, Chisci E, Giovannoni R. The role of hydrogen peroxide in redox-dependent signaling: homeostatic and pathological responses in mammalian cells. Cells. 2018;7:156. doi:10.3390/cells7100156
- Fu L, Liu K, Sun M, et al. Systematic and quantitative assessment of hydrogen peroxide reactivity with cysteines across human proteomes. Mol Cell Proteomics. 2017;16:1815–1828. doi:10.1074/mcp.RA117.00010828827280
- Chauvin JPR, Pratt DA. On the reactions of thiols, sulfenic acids, and sulfinic acids with hydrogen peroxide. Angew Chemie Int Ed. 2017;56:6255–6259. doi:10.1002/anie.201610402
- Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–326. doi:10.1016/j.freeradbiomed.2011.04.03121605662
- Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965.23461806
- Dustin CM, Heppner DE, Lin MCJ, van der Vliet A. Redox regulation of tyrosine kinase signalling: more than meets the eye. J Biochem. 2020;167:151–163. doi:10.1093/jb/mvz08531599960
- Östman A, Frijhoff J, Sandin Å, Böhmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150:345–356. doi:10.1093/jb/mvr10421856739
- Venditti P, Di Meo S. The role of reactive oxygen species in the life cycle of the mitochondrion. Int J Mol Sci. 2020;21:2173. doi:10.3390/ijms21062173
- Cordani M, Donadelli M, Strippoli R, Bazhin AV, Sánchez-Álvarez M. Interplay between ROS and autophagy in cancer and aging: from molecular mechanisms to novel therapeutic approaches. Oxid Med Cell Longev. 2019;2019:8794612. doi:10.1155/2019/879461231467639
- Lewis A, Hayashi T, Su TP, Betenbaugh MJ. Bcl-2 family in inter-organelle modulation of calcium signaling; Roles in bioenergetics and cell survival. J Bioenerg Biomembr. 2014;46:1–15. doi:10.1007/s10863-013-9527-724078116
- Thompson MD, Mei Y, Weisbrod RM, et al. Glutathione Adducts on Sarcoplasmic/Endoplasmic Reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J. Biol. Chem. 2014;289:19907–19916. doi:10.1074/jbc.M114.55445124920669
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167.23991888
- Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234:1355–1362.13654378
- Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J Biomed Biotechnol. 2012;2012:936486. doi:10.1155/2012/93648622927725
- Geissmann F, Manz MG, Jung S, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi:10.1126/science.117833120133564
- Burtenshaw D, Hakimjavadi R, Redmond EM, Cahill PA. Nox, reactive oxygen species and regulation of vascular cell fate. Antioxidants. 2017;6:90. doi:10.3390/antiox6040090
- Dupré-Crochet S, Erard M, Nüβe O. ROS production in phagocytes: why, when, and where? J Leukoc Biol. 2013;94:657–670. doi:10.1189/jlb.101254423610146
- Collin F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci. 2019;20:2407. doi:10.3390/ijms20102407
- Hurst JK. What really happens in the neutrophil phagosome? Free Radic Biol Med. 2012;53:508–520. doi:10.1016/j.freeradbiomed.2012.05.00822609248
- Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 2018;39:489–502. doi:10.1016/j.it.2018.01.00529452982
- Tan HY, Wang N, Li S, et al. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev. 2016;2016:2795090. doi:10.1155/2016/279509027143992
- Jiang F, Zhang Y, Dusting GJ, Sibley DR. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi:10.1124/pr.110.00298021228261
- Singel KL, Segal BH. NOX2-dependent regulation of inflammation. Clin Sci (Lond). 2016;130:479–490. doi:10.1042/CS2015066026888560
- Rastogi R, Geng X, Li F, Ding Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci. 2017;10:301. doi:10.3389/fncel.2016.0030128119569
- Sandoval R, Lazcano P, Ferrari F, et al. TNF-α increases production of reactive oxygen species through Cdk5 activation in nociceptive neurons. Front. Physiol. 2018;9:65. doi:10.3389/fphys.2018.0006529467671
- Yazdanpanah B, Wiegmann K, Tchikov V, et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi:10.1038/nature0820619641494
- Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin Cell Dev Biol. 2020;101:68–76. doi:10.1016/j.semcdb.2019.04.01631029766
- Wang Y, Sun L, Song Z, et al. Maspin inhibits macrophage phagocytosis and enhances inflammatory cytokine production via activation of NF-κB signaling. Mol Immunol. 2017;82:94–103. doi:10.1016/j.molimm.2016.12.02128064070
- Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902. doi:10.1161/CIRCRESAHA.117.31140129700084
- Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26:99–114. doi:10.1038/s41418-018-0212-630341423
- Hughes MM, O’Neill LAJ. Metabolic regulation of NLRP3. Immunol Rev. 2018;281:88–98. doi:10.1111/imr.1260829247992
- Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res. 2020;30:315–327.32152420
- He Y, Hara H, Mechanism NG. Regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. doi:10.1016/j.tibs.2016.09.00227669650
- Gu C, Liu S, Wang H, Dou H. Role of the thioredoxin interacting protein in diabetic nephropathy and the mechanism of regulating NOD‑like receptor protein 3 inflammatory corpuscle. Int J Mol Med. 2019;43:2440–2450.31017263
- Schofield JH, Schafer ZT. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal. 2020. doi:10.1089/ars.2020.8058
- Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. 2014;5:352.25324778
- Lingappan K. NF-κB in oxidative stress. Curr Opin Toxicol. 2018;7:81–86. doi:10.1016/j.cotox.2017.11.00229862377
- Takada Y, Mukhopadhyay A, Kundu GC, et al. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi:10.1074/jbc.M21238920012711606
- Chen C-J, Fu Y-C, Yu W, Wang W. SIRT3 protects cardiomyocytes from oxidative stress-mediated cell death by activating NF-κB. Biochem Biophys Res Commun. 2013;430:798–803. doi:10.1016/j.bbrc.2012.11.06623201401
- Manea SA, Constantin A, Manda G, Sasson S, Manea A. Regulation of Nox enzymes expression in vascular pathophysiology: focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015;5:358–366. doi:10.1016/j.redox.2015.06.01226133261
- Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-κB in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2010;396:901–907. doi:10.1016/j.bbrc.2010.05.01920457132
- Cuadrado A, Manda G, Hassan A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. 2018;70:348–383. doi:10.1124/pr.117.01475329507103
- Choi S, Krishnan J, Krishnan J, et al. Cigarette smoke and related risk factors in neurological disorders: an update. Biomed Pharmacother. 2017;85:79–86. doi:10.1016/j.biopha.2016.11.11827930990
- Cardozo LFMF, Pedruzzi LM, Stenvinkel P, et al. Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie. 2013;95:1525–1533. doi:10.1016/j.biochi.2013.04.01223643732
- Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. 2018;1865:721–733. doi:10.1016/j.bbamcr.2018.02.01029499228
- Sivandzade F, Prasad S, Bhalerao A, Cucullo L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019;21:101059. doi:10.1016/j.redox.2018.11.017
- Plafker KS, Nguyen L, Barneche M, et al. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J Biol Chem. 2010;285:23064–23074. doi:10.1074/jbc.M110.12191320484052
- Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2 - an update. Free Radic Biol Med. 2014;66:36–44. doi:10.1016/j.freeradbiomed.2013.02.00823434765
- Xiang MJ, Namani A, Wu SJ, Wang XL. Nrf2: bane or blessing in cancer? J Cancer Res Clin Oncol. 2014;140:1251–1259.24599821
- Chowdhry S, Zhang Y, McMahon M, et al. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi:10.1038/onc.2012.38822964642
- Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans. 2015;43:621–626. doi:10.1042/BST2015001426551702
- Zhou H, Wang Y, You Q, Jiang Z. Recent progress in the development of small molecule Nrf2 activators: a patent review (2017-present). Expert Opin Ther Pat. 2020;30:209–225. doi:10.1080/13543776.2020.171536531922884
- Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-Induced Expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–6737. doi:10.4049/jimmunol.181.10.673018981090
- Rojo AI, Innamorato NG, Martín-Moreno AM, et al. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598. doi:10.1002/glia.2094719908287
- Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxidants Redox Signal. 2010;13:1649–1663. doi:10.1089/ars.2010.3216
- Sanlioglu S, Williams CM, Samavati L, et al. Lipopolysaccharide Induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. J Biol Chem. 2001;276:30188–30198. doi:10.1074/jbc.M10206120011402028
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi:10.1016/S0962-8924(01)02064-511514191
- Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15:362–374.26008591
- Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Hindawi Publ Corp. 2015;2015:610813.
- Fritsch J, Zingler P, Särchen V, Heck AL, Schütze S. Role of ubiquitination and proteolysis in the regulation of pro- and anti-apoptotic TNF-R1 signaling. Biochim Biophys Acta Mol Cell Res. 2017;1864:2138–2146. doi:10.1016/j.bbamcr.2017.07.01728765050
- Füllsack S, Rosenthal A, Wajant H, Siegmund D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019;10:122. doi:10.1038/s41419-019-1396-530741924
- Park YC, et al. A novel mechanism of TRAF signaling revealed by structural and functional analyses of the TRADD-TRAF2 interaction. Cell. 2000;101:777–787.10892748
- Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 2011;278:862–876. doi:10.1111/j.1742-4658.2011.08015.x21232017
- Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi:10.1016/j.molcel.2009.10.01320005846
- Gerlach B, Cordier SM, Schmukle AC, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi:10.1038/nature0981621455173
- Zinngrebe J, Montinaro A, Peltzer N, Walczak H. Ubiquitin in the immune system. EMBO Rep. 2014;15:28–45. doi:10.1002/embr.20133802524375678
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi:10.1016/j.freeradbiomed.2009.12.02220045723
- Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26:249–261. doi:10.1016/j.tcb.2015.12.00226791157
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115.21187859
- Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi:10.1038/nri342323618831
- Win S, Than TA, Fernandez-Checa JC, Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014;5:e989. doi:10.1038/cddis.2013.52224407242
- Kim JJ, Lee SB, Park JK, Yoo YD. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010;17:1420–1434. doi:10.1038/cdd.2010.1920203691
- Nathan C, Ding A. SnapShot: reactive Oxygen Intermediates (ROI). Cell. 2010;140:951–951.e2. doi:10.1016/j.cell.2010.03.00820303882
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi:10.1126/science.109932015286356
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi:10.1007/s10495-006-0525-717136322
- Candé C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734.12432061
- Reuther-Madrid JY, et al. The p65/RelA Subunit of NF- B suppresses the sustained, antiapoptotic activity of jun kinase induced by tumor necrosis factor. Mol Cell Biol. 2002;22:8175–8183.12417721
- Tang F, et al. The Absence of NF- B-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol. 2002;22:8571–8579. doi:10.1128/MCB.22.24.8571-8579.200212446776
- Yang HT, Huang YH, Yang GW. Mini review: immunologic functions of dual oxidases in mucosal systems of vertebrates. Brazilian J Biol. 2019. doi:10.1590/1519-6984.208749
- Jyoti SO, Söhnlein O. outstanding investigator award of the European Society of Cardiology council for basic cardiovascular sciences. Cardiovasc Res. 2017;113(2017):e47.29088376
- Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi:10.1038/nri390826471775
- Zuchtriegel G, Uhl B, Puhr-Westerheide D, et al. Platelets guide leukocytes to their sites of extravasation. PLoS Biol. 2016;14:e1002459. doi:10.1371/journal.pbio.100245927152726
- Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells: essential roles of a variant NF-κB site and p65 homodimers. Journal of Biological Chemistry. 1995;270(2):933–943. doi:10.1074/jbc.270.2.933
- Lo SK, Janakidevi K, Lai L, Malik AB. Hydrogen peroxide-induced increase in endothelial adhesiveness is dependent on ICAM-1 activation. Am J Physiol Lung Cell Mol Physiol. 1993;264:L406–12. doi:10.1152/ajplung.1993.264.4.L406
- Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol. 1999;66:876–888. doi:10.1002/jlb.66.6.87610614768
- Evans R, Patzak I, Svensson L, et al. Integrins in immunity. J Cell Sci. 2009;122:215–225. doi:10.1242/jcs.01911719118214
- Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci. 2008;13:7210–7226. doi:10.2741/322318508729
- Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–271. doi:10.1016/j.redox.2015.08.01026296072
- Song MY, Makino A, Yuan JXJ. Role of reactive oxygen species and redox in regulating the function of transient receptor potential channels. Antioxidants Redox Signal. 2011;15:1549–1565. doi:10.1089/ars.2010.3648
- Usatyuk PV, et al. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. J Biol Chem. 2012;287:9360–9375. doi:10.1074/jbc.M111.29454622219181
- Wang X, Ke Z, Chen G, et al. Cdc42-dependent activation of NADPH oxidase is involved in Ethanol-Induced neuronal oxidative stress. PLoS One. 2012;7(5):e38075. doi:10.1371/journal.pone.003807522662267
- Haidari M, Zhang W, Willerson JT, Dixon RA. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-β-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc Diabetol. 2014;13:105. doi:10.1186/1475-2840-13-10525927959
- Park HS, Jung HY, Park EY, et al. Cutting edge: direct interaction of TLR4 with NAD(P)H Oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol. 2004;173:3589–3593. doi:10.4049/jimmunol.173.6.358915356101
- Catic A. Cellular metabolism and aging In: Ottinger MA, editor. Progress in Molecular Biology and Translational Science. Metabolic Aspects of Aging. Vol. 155 Elsevier B.V.; 2018:85–107.29653684
- Huang Y, Hong H, Li M, et al. Age-dependent oxidative DNA damage does not correlate with reduced proliferation of cardiomyocytes in humans. PLoS One. 2017;12(1):e0170351. doi:10.1371/journal.pone.017035128099512
- Gladyshev VN. The free radical theory of aging is dead. Long live the damage theory! Antioxidants Redox Signal. 2014;20:727–731. doi:10.1089/ars.2013.5228
- I S-B, et al. Role of oxidative, nitrative, and chlorinative protein modifications in aging and age-related diseases. Oxid Med Cell Longev. 2018;2018:3267898.30159111
- Rinnerthaler M, Bischof J, Streubel MK, Trost A, Richter K. Oxidative stress in aging human skin. Biomolecules. 2015;5:545–589.25906193
- Aitbaev KA, Murkamilov IT, Fomin VV. Molecular mechanisms of aging: the role of oxidative stress and epigenetic modifications. Adv Gerontol. 2019;9:417–425.
- Chakravarti B, Chakravarti DN. Oxidative modification of proteins: age-related changes. Gerontology. 2007;53(3):128–139. doi:10.1159/00009786517164550
- Goto S, Radak Z. Implications of oxidative damage to proteins and DNA in aging and its intervention by caloric restriction and exercise. J Sport Heal Sci. 2013;2:75–80.
- Reichert S, Stier A. Does oxidative stress shorten telomeres in vivo? A review. Biol Lett. 2017;13:20170463. doi:10.1098/rsbl.2017.046329212750
- Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev. 2019;177:37–45. doi:10.1016/j.mad.2018.03.01329604323
- Rodrigo-Moreno A, Poschenrieder C, Shabala S. Transition metals: a double edge sward in ROS generation and signaling. Plant Signal Behav. 2013;8:e23425. doi:10.4161/psb.2342523333964
- Lushchak OV, Piroddi M, Galli F, Lushchak VI. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014;19:8–15. doi:10.1179/1351000213Y.000000007324266943
- Osorio FG, Soria-Valles C, Santiago-Fernández O, Freije JMP, López-Otín C. NF-κB signaling as a driver of ageing Int Rev Cell MolBiol. 2016;326:133–174. doi:10.1016/bs.ircmb.2016.04.003
- Linehan E, Fitzgerald D. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol. 2015;5:14–24. doi:10.1556/EuJMI-D-14-00035
- Giorgi C, et al. Mitochondria and reactive oxygen species in aging and age-related diseases. Int Rev Cell Mol Biol. 2018;340:209–344.30072092
- Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14:225–274. doi:10.1016/0022-5193(67)90079-3
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi:10.1111/j.1532-5415.1972.tb00787.x5016631
- Payne BAI, Chinnery PF. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta - Bioenerg. 2015;1847:1347–1353. doi:10.1016/j.bbabio.2015.05.022
- Lee SH, Lee JH, Lee HY, Min KJ. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52:24–34. doi:10.5483/BMBRep.2019.52.1.29030526767
- Emelyanova L, Preston C, Gupta A, et al. Effect of aging on mitochondrial energetics in the human atria. J Gerontol A Biol Sci Med Sci. 2018;73:608–616. doi:10.1093/gerona/glx16028958065
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi:10.1152/physrev.00026.201324987008
- Rottenberg H, Hoek JB. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017;16:943–955.28758328
- McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77. doi:10.1083/jcb.20170809229114066
- Van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi:10.1038/nature1319324848057
- Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and and novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longev. 2016;2016:3565127. doi:10.1155/2016/356512727247702
- Romá-Mateo C, Seco-Cervera M, Ibáñez-Cabellos JS, et al. Oxidative stress and the epigenetics of cell senescence: insights from progeroid syndromes. Curr Pharm Des. 2019;24:4755–4770. doi:10.2174/1381612824666190114164117
- Amaya-Montoya M, Pérez-Londoño A, Guatibonza-García V, Vargas-Villanueva A, Mendivil CO. Cellular senescence as a therapeutic target for age-related diseases: a review. Adv Ther. 2020;37:1407–1424. doi:10.1007/s12325-020-01287-032185730
- Harrington LA, Harley CB. Effect of vitamin E on lifespan and reproduction in Caenorhabditis elegans. Mech Ageing Dev. 1988;43:71–78. doi:10.1016/0047-6374(88)90098-X3374177
- Navarro A, Gómez C, Sánchez-Pino M-J, et al. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1392–1399. doi:10.1152/ajpregu.00834.200416020519
- Lezo A, Biasi F, Massarenti P, et al. Oxidative stress in stable cystic fibrosis patients: do we need higher antioxidant plasma levels? J Cyst Fibros. 2013;12:35–41. doi:10.1016/j.jcf.2012.06.00222781546
- Shamseer L, Adams D, Brown N, Johnson JA, Vohra S. Antioxidant micronutrients for lung disease in cystic fibrosis. Cochrane Database Syst Rev. 2010;12:CD007020.
- Calabrese C, et al. Randomized, single blind, controlled trial of inhaled glutathione vs placebo in patients with cystic fibrosis. J Cyst Fibros. 2015;14:203–210. doi:10.1016/j.jcf.2014.09.01425458463
- Hector A, Griese M, Hartl D. Oxidative stress in cystic fibrosis lung disease: an early event, but worth targeting? Eur Respir J. 2014;44:17–19. doi:10.1183/09031936.0003811424982050
- Griese M, Kappler M, Eismann C, et al. Inhalation treatment with glutathione in patients with cystic fibrosis: a randomized clinical trial. Am J Respir Crit Care Med. 2013;188:83–89. doi:10.1164/rccm.201303-0427OC23631796
- de Vries JJV, Chang AB, Bonifant CM, Shevill E, Marchant JM. Vitamin A and beta (β)-carotene supplementation for cystic fibrosis. Cochrane Database Syst Rev. 2018;2018:CD006751.
- González Jiménez D, Díaz Martín JJ, Arias Llorente RP, Bousoño García C. Oxidative stress in cystic fibrosis In: Wat D, editor. Cystic Fibrosis in the Light of New Research. InTech; 2015. doi:10.5772/60661
- Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxidants Redox Signal. 2015;22:686–729. doi:10.1089/ars.2014.5952
- Plecitá-Hlavatá L, Engstová H, Ježek J, et al. Potential of mitochondria-targeted antioxidants to prevent oxidative stress in pancreatic β -cells. Oxid Med Cell Longev. 2019;2019:1826303. doi:10.1155/2019/182630331249641
- Liu Z, et al. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. 2018;9:477. doi:10.3389/fphys.2018.0047729867535
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001;22:477–501. doi:10.1210/edrv.22.4.043711493580
- Peng C, Wang X, Chen J, et al. Biology of ageing and role of dietary antioxidants. Biomed Res Int. 2014;2014:831841. doi:10.1155/2014/83184124804252
- Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–1249. doi:10.1152/ajpregu.00226.200415271654
- Noor R, Mittal S, Iqbal J. Superoxide dismutase - applications and relevance to human diseases. Med Sci Monit. 2002;8:RA210–215.12218958
- Salvemini D, Riley DP. Nonpeptidyl mimetics of superoxide dismutase in clinical therapies for diseases. Cell Mol Life Sci. 2000;57:1489–1492. doi:10.1007/PL0000063211092442
- Paul A, Belton A, Nag S, et al. Reduced mitochondrial SOD displays mortality characteristics reminiscent of natural aging. Mech Ageing Dev. 2007;128:706–716. doi:10.1016/j.mad.2007.10.01318078670
- Extracellular Superoxide LE. Dismutase (EC-SOD) quenches free radicals and attenuates age-related cognitive decline: opportunities for novel drug development in aging. Curr Alzheimer Res. 2005;2:191–196. doi:10.2174/156720505358571015974918
- De Barcelos IP, Haas RH. Coq10 and aging. Biology (Basel). 2019;8:28.