Abstract
Neurodegenerative diseases are a growing public health challenge, and amyotrophic lateral sclerosis (ALS) remains a fatal incurable disease. The advent of stem cell therapy has opened new horizons for both researchers and ALS patients, desperately looking for a treatment. ALS must be considered a systemic disease affecting many cell phenotypes besides motor neurons, even outside the central nervous system. Cell replacement therapy needs to address the specific neurobiological issues of ALS to safely and efficiently reach clinical settings. Moreover, the enormous potential of induced pluripotent cells directly derived from patients for modeling and understanding the pathological mechanisms, in correlation with the discoveries of new genes and animal models, provides new opportunities that need to be integrated with previously described transplantation strategies. Finally, a careful evaluation of preclinical data in conjunction with wary patient choice in clinical trials needs to be established in order to generate meaningful results.
Introduction
Amyotrophic lateral sclerosis (ALS) still remains, more than a century since first description, a fatal and untreatable disease. The use of pluripotent or multipotent cells for the restoration of damaged neuronal networks is one of the factors that holds promise from a translational medicine perspective and a relevant part of the strongest forces driving research in stem cell (SC) biology applied to neurodegenerative diseases. Novel specific neuropathological findings in ALS suggest a more generalized involvement outside the nervous system that needs to be fully evaluated before strategies for replacement or rescue of damaged neurons can be further developed.
Amyotrophic lateral sclerosis and the frontotemporal involvement
ALS is a fatal disease caused by the progressive loss of motor neurons (MNs) in both the brain and the spinal cord leading to paralysis of voluntary muscles and death within 2–5 years from clinical onset.Citation1 Most cases of ALS are classified as sporadic ALS (sALS), albeit approximately 7%–10% are inherited in a dominant mode (familial ALS [fALS]).Citation2 Although several hypotheses have been proposed to explain the specific MN involvement, leading to their progressive degeneration, the underlying mechanisms remain elusive.Citation3
ALS has been considered to be the prototypical pyramidal motor system neurodegenerative disease for decades. In terms of neuropathology, degeneration of the upper and lower motor neuron with MN cytoplasmic inclusions immunoreactive for ubiquitin (U) and degeneration of the corticospinal tract were considered to be diagnostic for ALS.Citation4 Recently, researchers have begun to recognize an important connection between frontotemporal dementia (FTD) and ALS or Lou Gehrig’s disease (). FTD is a syndrome of progressive changes in behavior, language, and cognition due to loss of function of neurons in both the frontal and temporal lobes. Usually, FTD has relatively little effect on the parts of the nervous system that control movement, and so many FTD patients remain physically strong and relatively agile until late in the illness. However, in approximately 10%–15% of patients with FTD, the disease also involves the nerve cells controlling voluntary movement, the MNs. When this occurs, the syndrome is called FTD with motor neuron disease (FTD-MND). TDP-43 (ubiquitinated TAR DNA-binding protein) is a multifunctional DNA/RNA-binding factor that has been implicated in the regulation of neuronal plasticity.Citation5 The notion that pathological TDP-43 is involved in neurological diseases was proposed when it was discovered by Neumann et alCitation6 since this protein has been identified as the major constituent of pathological inclusions in FTD with U (FTD-U, now known as FTD-TDP), FTD-MND, and ALS. Therefore, a common pathogenesis linked to TDP-43 abnormalities in these disorders has been suggested and further confirmed.Citation5,Citation7 This scheme reflects the considerable overlap of clinicopathological features between all neurodegenerative diseases:Citation8 ALS, FTD-MND, and FTD-U may be situated at different points along a continuous and broad spectrum of a multisystemic degenerationCitation9,Citation10 (). Recent findings of mutations in the TARDBP gene, encoding for TDP-43, in cases of autosomal-dominant fALS and rare sALS patients further corroborate the significance of pathological TPD-43 as being mechanistically implicated in the disease process.Citation11 Patients with both FTD-MND and TARDBP mutations have been reported.Citation12 Recently, it has been demonstrated that elevated expression of TDP-43 in mouse forebrain causes neuropathological patterns similar to FTD-U, mimicking its specific behavior phenotype.Citation13 Moreover, both TDP-43 and Cu/Zn superoxide dismutase-1 (SOD-1) proteins modulate sequestration of neuroflament mRNAs in the aggregates characteristic in ALS MN degeneration.Citation14
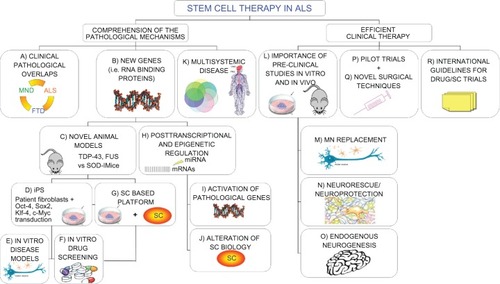
Figure 1 Novel “holistic” approach to ALS therapy. Clinical overlaps between ALS and other neurodegenerative diseases could unravel common molecular/pathological mechanisms (A). Moreover, new insights on causative genetic mutations (B) and the development of novel animal models (C) widen our knowledge of the possible therapeutic targets in the pathological pathways. In the meantime, recent iPS technology (D) provides patient-derived specimens as disease modeling and cell assays to dissect pathological mechanisms and specific cell contribution (E). The development of SC-based therapies is also directly exploitable for new drug screening (F and G). The discovery of the importance of epigenetic regulation in the pathological processes is paralleled by a relevant role in SC biology. Any alteration in this complex network could alter SC dynamic cross talk to the diseased surroundings, thus precluding possible therapeutic effects (H–J). The complex nature requires a multifaceted strategy, able to efficiently contrast widespread degeneration in all tissue districts (K), which should be carefully evaluated in accurate preclinical studies (L). Efficient therapeutic treatments are required both to replace MNs (M) and provide an healthy environment for them (N), capable also of enhancing endogenous repair (O). These laboratory studies will lead to successful clinical trials (P), based on novel surgical techniques (Q), able to slow the disease progression. Consensus international guidelines for drug/SC trials will guarantee the conscientious translation of basic SC research into appropriate treatment applications for patients aiming to create optimized efficient protocols able to slow down (neuro)degeneration (R).
Mutations in a gene encoding another DNA/RNA-binding protein with striking structural and functional similarities to TDP-43 named FUS (fused in sarcoma) or TLS (translocation in liposarcoma) have been recently reported to trigger degeneration of MNsCitation15–Citation17 and be responsible for FTD.Citation18 Although this gene was initially identified as a component of a fusion pro-oncogene resulting from a chromosomal translocation seen in liposarcomas, it belongs similarly to a subfamily of RNA-binding proteins, involved in MN (patho) physiological biology/metabolism (). Interestingly, FUS/TLS protein interacts with RNA, single-stranded DNA, and double-stranded DNA, and is involved in unique functions in the mRNA processing and transport, transcriptional regulation, and maintenance of genomic stability.Citation19 It has been also reported the selective presence of FUS+ inclusions in an elderly patient apparently not mutated in the gene but affected by MND.Citation20
The emerging scenario of multiple regional involvements due to the TDP-43/FUS neuropathological inclusions in the central nervous system (CNS) of ALS patients entails a significant impact on the therapeutic strategies applicable to them and, particularly, on the SC approach, since RNA-binding proteins appear as key regulators of signaling networks responsible for neuronal development and homeostasis,Citation21 as well as neural SC biology.Citation22 Therefore, healthy transplanted cells cross talk with the surroundings may also be compromised by the abnormal cellular RNA metabolism, able to trigger MN degeneration, thus impeding any therapeutic outcomes. FUS and TDP-43 harbor also a “prion domain” very similar to the specific one present in several yeast prion proteins prone to pathological misfolding transmissible within or between healthy cells or species. In this case, no SC strategy could maintain positive therapeutic outcomes in the long term, without a supportive treatment able to prevent the spread of the disease.Citation23 Moreover, mere cell substitution appears insufficient to contrast all the alterations in the interrelated complex pathways activated by ALS, as described in the following paragraphs.
Stem cells and new pharmacotherapeutic strategies: approaching novel genes, disease modeling, and drug candidates
Additional interesting hints derive from the discovery of the novel pathological genes TARDBPCitation6,Citation24 and FUS/TLSCitation15 since they also have led to the derivation of new animal models, alternatives to the classic transgenic (tg) SOD-1, for deciphering the mechanisms responsible for the motor system neurodegeneration with large implications for SC therapies in the CNSCitation25,Citation26 (). In particular, it appears that TDP-43 plays a pivotal role in many forms of MND, and this protein, being implicated in some forms of dementia, exerts a contributory role in a wider number of neurodegenerative diseases.Citation27 Moreover, a careful examination of the pathological SCs in these new animal models, as well as of the surrounding niche, could reveal abnormalities that may influence reparative mechanisms, as already suggested in tgSOD-1 NPs,Citation28 wobbler mice,Citation29 and in the bone marrow of sporadic ALS patients.Citation30
Furthermore, remarkable pieces of information are expected from the recent generation of iPS cells (induced pluripotent SCs, obtained by transducing cells with 4 transcription factors: Oct-4, Sox2, Klf-4, and c-Myc; ), deriving from somatic tissues of an elderly ALS patient, which could be successfully differentiated toward well-characterized MNs.Citation31 iPS cells possess the same advantages as traditional SCs due to their ability to produce differentiated affected cells, such as neurons.Citation32 However, the use of oncogenes and retrovirus in the current iPS cell establishment protocol raises safety concerns since their progeny show high teratoma-forming propensities that actually restricts their potential use in cell therapy.Citation33 Nevertheless, neurodegenerative disease-specific iPS may be used for assays with cell specimens uncollectible from live patients (such as MNs, glial cells, and so on) to dissect their distinct peculiar influence on pathological events () and to define new drug targetsCitation34 (). Although reprogramming adult patient cells would be particularly interesting for both fALS and sALS, so far no ALS iPS-derived MNs appear to be affected by the disease. Conversely, both iPS cells from spinal muscular atrophyCitation35 (SMA) and familiar dysautonomia,Citation36 characterized by childhood onset, recapitulate all the pathological selective deficits. Therefore, it appears that the complex interactions between genetic and environmental features combined to senescence processes,Citation32 which are completely lacking during iPS-derived MN differentiation, may play a key role in ALS late-onset degeneration. Additional long-term studies would be necessary to recreate the correct pathophysiological conditions before validation of this model for drug screening or as a disease model for ALS.
A precise dissection of the synergistic effects of SC implantation on the ALS surroundings is limited by the absence of a primate model and by the still developing unique SC identification upon grafting.Citation37 Additional studies to clarify the complex stem/niche interactions could be derived from in vitro cocultures as demonstrated by the pioneering experiments of both Di Giorgio and Nagai. Human embryonic SC (ES) – derived MNs selectively die whenever cultivated in the presence of tgSOD-1 MNs or glial cells through a Bax-dependent mechanism triggered by toxic soluble factors.Citation38–Citation40 These studies may conduct to the development of standardized SC-based platforms to study cell–cell interactions and to identify novel pharmacotherapeutic targets ().
Innovative perspectives are derived also by the novel findings on the importance of posttranscriptional and epigenetic regulation in neurodevelopment, as well as in neurodegenerative diseases.Citation41 Recent evidence suggests that altered RNA metabolism may play a role in ALS pathogenesisCitation16,Citation19,Citation42 (). Micro-RNAs (miRNAs) are small RNA molecules highly conserved and able to regulate the expression of genes by binding to the 3’-untranslated regions of target mRNAs. miRNAs have emerged as critical regulators of gene expression, and they have been implicated in the control of virtually all the biological processes, including the biology of SCs.Citation43 Recently, it has been described how the deficit of a specific miRNA expressed in skeletal muscle (miR-206) of tgSOD-1 mice led to compromised reinnervation of neuromuscular junction with an accelerated disease progression due to an increase of denervated muscle fibers.Citation44 Moreover, recently a widespread modification in the total miRNA compartment after TDP-43 knockdown in culture cells has been reported, thus suggesting a direct link between the altered protein levels to specific candidate genes, crucial for TDP-43 pathogenic role in FTD and ALS.Citation45 Concordantly, it has been recently suggested that SC biology is also strictly controlled by some differentially expressed miRNAs that act in concerted actions to regulate self-renewal, differentiation, and division.Citation46,Citation47 As a consequence, mere cell substitution by SC progeny alone could not recover a widespread compromised transcriptional regulation responsible for disease-related gene activation. Altogether, these novel findings support the idea that a combination of approaches, related to different pathological mechanisms, is needed for effective disease modeling and, consequently, cell therapies. Conversely, SC transplantation supporting the MN surroundings in conjunction with miRNAs or RNA interference using small inhibitory RNA (siRNA) to downregulate specific pathology-involved genes (such as TDP-43 and GluR2)Citation48,Citation49 or RNAs may become, therefore, an innovative pharmacotherapeutic strategy to efficiently contrast both the MN degeneration and the environmental glial activation.
ALS viewed as a systemic disease implies new therapeutic strategies
The precise cause of ALS is still unknown, but several mutated genes have been found to predispose to ALS and recently other potential contributors to the complex pathological mechanism have been proposed.Citation50 Both sALS and fALS share similar clinical hallmarks, and some common genetic alterations, such as mutations in the SOD1 or TARDBP genes, are also reported in sALS, thus suggesting common pathological pathways.Citation51 Moreover, the picture arising from different studies on ALS animal models and patients, leads to the idea that multiple cell types, even outside the motor system, result as pathophysiologically affected. Beside cognitive and personality changes, indicating frontotemporal involvement, abnormalities have been described in the fibril organization compartment of the skin,Citation52–Citation54 as well as in the spinal cord collagen content of ALS patients.Citation55 Moreover, metalloproteinases (MMPs) involvements in different cellular districts have been reported as peculiar finding in ALS.Citation56,Citation57 Widespread variations in several MMPs and their tissue inhibitors have been demonstrated in serum and cerebrospinal fluid of ALS patients,Citation58–Citation60 around atrophic myofibers,Citation61 in mesenchymal SCs,Citation30 and in postmortem brain/spinal cord tissues in sALS cases.Citation62 Alterations of MMPs were also retrieved in tgSOD-1 mice spinal cord,Citation63 affecting both neuronal and glial cells.Citation63 Moreover, early administration of MMP synthetic inhibitors extends survival in tg mice,Citation64 while mutating tgSOD-1 mice knockout for a specific MMP (MMP9) show exacerbation of disease progression.Citation65 Since MMPs are actively responsible for all cellular signaling cascades in a variety of physiological and pathological processes,Citation66 their widespread variations appear to involve all cell lineages in both the sporadic and familial forms of this neurodegenerative disorder. ALS patients are also hypermetabolic and present systemic alterations of lipidemia,Citation67 while abnormal lipid clearance characterizes the pathological end stages of the tgSOD-1 mice.Citation68 Finally, it has been recently reported that mutant tgSOD-1 protein contributes to mitochondrial toxicity also in muscle tissue.Citation69 Interestingly, it has been recently suggested that the primary pathogenic event in tgSOD-1 mice involves muscle hypermetabolism which in turn causes neuromuscular junction destruction, followed by axonal degeneration, and finally MN loss.Citation70
Altogether, this evidence supports the idea of a “multisystemic” disease, affecting multiple cell types, either neuronal or nonneuronal (). Notably, ALS involvement in SCs outside the motor system could represent a subject for consideration in autotransplantation of ALS patients,Citation71–Citation75 as discussed by our group in a recent article.Citation30 Major metabolic pathways (such as MMP regulation, SC biology, mitochondrial toxicity, and metabolic rate), contemporaneously affected in different cellular districts, require an appropriate therapeutic strategy able to contrast all these pathological symptoms, instead of focusing mainly on MN degeneration. Therefore, outcomes of SC transplantation may be greatly improved by a global tactic directed toward MN substitution, recovery of the surroundings and rescue effect on suffering neurons (neurorescue effect), as well as action on different pathological mechanisms outside the CNS. Today, patients suffering from ALS may have a hope to improve their quality of life by slowing down or even healing their condition through SC technology combined with pharmacological approaches. This innovative concept deeply affects the traditional approach of SC therapy based on the need for MN replacement or protection to obtain clinical recovery in ALS patients: new cellular targets appear critical to slowing down MN degeneration, as well as more widespread symptoms, due to the broad organ involvement.
MN replacement and the importance of surroundings for SC progeny in ALS
The easiest clinical strategy in treating ALS should consist of the graft of SC-derived spinal MN precursors/neuroblasts to replace damaged or dead spinal neurons. Several interesting data have been already derived from in vitro and in vivo preclinical studies (). MNs have been generated from different lineages of SCs including mouse and human ES in vitro.Citation76–Citation80 These ES-derived MNs correctly innervate muscle fibers in vitro,Citation81 as well as in motor-injured adult rats.Citation82,Citation83 Motor functions have been re-established by grafted embryonic cortical neurons,Citation84 whereas fetal neural progenitors (NPs) may well survive and proliferate both in vitro and in vivo after transplantation.Citation85 Furthermore, they could be engineered to produce neutrophines, such as glial cell-derived neurotrophic factor, to protect host MNs during the pathological progression,Citation86,Citation87 while transfection of fully differentiated MNs would be difficult and less effective. However, both ES and fetal NP cells, as well as their progeny, possess advantages and limitations that ought to be considered. Actually, ES cells display unlimited growth in culture, an undifferentiated state and great differentiation potential associated with high risk to form teratomas. Conversely, fetal NP cells exhibit long-term stability in culture and multiple differentiation potential in conjunction with absence of tumor formation, but both ethical and immunological constraints limit their use in humans. Moreover, ALS hostile inflammatory and oxidative environment could seriously challenge fully differentiated endogenous MNs, independent of their initial origin (ES or NPs). Therefore, the harsh surroundings may seriously hamper incorporation of grafted MNS into the host neural circuitry, target axonal growth, and reinnervate pathological muscle fibers.Citation88
Surprisingly, both ES-derived MNsCitation89,Citation90 and NP-derived MNsCitation69,Citation91–Citation93 transplanted in mice affected by SMA survived, integrated, and sprouted axonal terminals appropriately, thus ameliorating animal behavior, functional end points, and lifespan. These papersCitation89,Citation91 constitute the first important proof of principle that functional restoration of the motor circuit with SC-derived MNs is feasible and therapeutic through an overall neuroprotective effect associated with the decrease of proinflammatory molecules (). These milestone experiments also demonstrate how the complex axonal growth processes toward the skeletal muscle target could be driven and modulated by pharmacological treatment, even in adult mice. Nevertheless, many issues still need to be clarified before any clinical application: possible toxicity in human, surviving, migratory and differentiative potential of grafted cells in the degenerate environment, and their capacity to induce efficient synaptogenesis are still not fully analyzed and described.Citation94 Moreover, it is improbable that this approach could be soon translated in clinical trials since the restoration of the human motor circuit from MNs toward the appropriate muscle target will require considerable time (months or years) in a disease characterized by a mean survival of 36 months. Presently, it is unclear if transplanted SC-derived MNs may endure in the long term, once exposed to the harsh microenvironment of the spinal cord in ALS patients.Citation88 Moreover, the progressive nature of ALS, characterized by time-increased dysfunctional astrocytes and overactivated microglia, may gradually damage grafted MNs, spreading pathological hallmarks from diseased to healthy cells, as demonstrated in transplanted parkinsonian patients.Citation95 Actually, the importance of the surroundings during MN degeneration in ALS has been extensively proved,Citation96–Citation98 showing that disease onset can be delayed and survival increased in tgSOD-1 mice by providing genetically noncompromised supporting cells.Citation99 Therefore, ALS appears to be even more convincingly a noncell autonomous process wherein different cell types (such as astrocytes and microglia) play a key role in the disease progression.Citation100,Citation101 On the other hand, SCs and their MN progeny may provide large quantities of affected MNs to define earlier pathological events relevant to disease initiation.Citation102,Citation103 As elegantly discussed in the recent review by Thonhoff et al,Citation88 these specimens can also be used for studying glia-mediated toxin mechanisms and test potential therapies in ALS.Citation38,Citation104 In fact, ES-derived MNs expressing mutant tgSOD-1 recapitulate the main pathological hallmarks (reduced cell survival and shortened axonal processes) and may be exploited to elucidate ALS pathophysiology or to arrest degeneration using cell-based assaysCitation39,Citation105 (). Moreover, riddance of immature myeloid precursor cells, abnormally proliferating during late pathological phases in tgSOD-1 mice, has enhanced how this process does not primarily affect MN degeneration,Citation106 thus shifting research toward other potential targets such as inhibition of host immune or inflammatory response.Citation107 As a matter of fact, bone marrow transplant reconstituted T-cell compartment, prolonged survival, and restored glial activation within tgSOD-1 mice lacking immune modulation. These results demonstrate that the absence of immune recruitment accelerates disease progression and death, while functional CD4+ T cells provide supportive neuroprotection by modulating the trophic/cytotoxic balance of glia.Citation108 SC choice, cell dose, and delivery route (as recently reviewed)Citation107 are fundamental for a successful therapeutic strategy since subtle variations may cause divergent results.Citation109,Citation110 Nevertheless, several factors may influence enduring positive outcomes, other than structural integration in the mice motor circuitryCitation111 or migration to patient injury sites.Citation112 Due to its systemic and multifactorial nature, contrasting ALS entails multiple approaches able to contemporaneously act on different pathological mechanisms. As a matter of fact, effective growth release by engineered grafted SCs may be helpful to prevent MN loss, with an overall neurorescue effect able to slow down neuronal degeneration, thus avoiding pathological spreading to the surroundings. Nevertheless, additional strategies, such as SC differentiation toward glial cells, are required to preserve neuromuscular connectionsCitation113,Citation114 and to ameliorate pathological symptoms in ALS mice.Citation115 Combining growth factor administration and SC therapy to increase healthy astroglial cell numbers also appears as a successful strategy to modulate disease progression by detoxification of the MN environment.Citation114 Altogether, these new results point out the importance of multiple strategies able to counteract MN degeneration and positively influence the surrounding niche ().
Therefore, additional studies on the properties of SC progeny, in combination with pioneering cellular and molecular techniques, are required to provide a trophic environment for endogenous neural cells able also to contrast the reactive astrogliosis and microglial activation.
Innovative SC therapies beyond replacement: neuroprotection by nonneural surrounding cells and activation of endogenous repair
An interesting perspective comes from the observation that NP-derived neurons support host MNs in the tgSOD-1 rat model with an advanced degree of structural integration in the motor circuit, although in the absence of any replacement or new axonal innervations into target muscle.Citation111 Moreover, recently it has been demonstrated that selective reduction of human SOD-1 levels in mice microglia or astrocytes, using a CRE-lox system, extends disease progression and host MN survival.Citation98,Citation99 Therefore, replacing nearby supporting cells appears a feasible strategy to preserve remaining MN functionality and activity. Transplantation of glial-restricted cell precursors by focal delivery into the ventral horn of the spinal cord extends both survival and disease duration, as well as attenuating MN loss.Citation116 Interestingly, the observed neuroprotection appears partially due to normalization of the astrocyte glutamate transporter GLT1 levels, which is reduced in ALS patients and animal models.Citation117,Citation118 Concordantly, human adipose-derived SCs have the capability to increase their cytokine release when cocultured with tgSOD-1 astrocytes, which in turn show enhanced GLT1 expression and reduced caspase-3 activation, at least in vitro.Citation119 Multiple neuroprotective effects including partial preservation of spinal cord GLT1 levels, modulation of growth factors, and microglial reduction have been recently reported after wild-type bone marrow-derived murine c-kit+ cells were systemically engrafted into tgSOD-1 mice.Citation120 Microglia appears as another potential target for cell transplantation in ALS since wild-type bone marrow grafted into tgSOD-1 mice, both irradiatedCitation121 or lacking CNS microglia/peripheral immune cells,Citation122 augments healthy microglial content with reduction of MN loss and increased animal survival. Replacement with normal allogenic hematopoietic SCs by intra-bone marrow-bone marrow transplantation in irradiated mice also improves the neural surroundings by delaying the disease progression, in contrast to the autologous tgSOD-1 graft.Citation123 Unfortunately, hematopoietic SC transplantation in irradiated sporadic ALS patients has been demonstrated to be ineffective for improving clinical symptoms or survival, although implanted SCs correctly engraft injury sites, as demonstrated by autoptic samples.Citation112 Conversely, grafts in frontal motor cortex of autologous multipotent hematopoietic CD-133 SCs, collected following mobilization, appear to be able to prolong ALS patient survival in a recent study,Citation124 although no suggested mechanisms of action are provided. Similarly, two different open-label studies using bone marrow-derived hematopoietic progenitor SCsCitation125 and autologous mesenchymal SCsCitation72–Citation74 report clinical benefits demonstrating that SC therapy is a safe, effective, and promising treatment for ALS patients still requiring additional evidence and standardization before being extensively applied in clinical practice.Citation94
Although the mammalian CNS shows a very limited capacity to regenerate after injury, endogenous precursors/SCs may provide a potential source of new neurons in the adult brain and spinal cord. As a matter of fact, endogenous precursors can differentiate into highly complex long-projection corticospinal MNs and send new projections to spinal cord targets in the healthy, adult mammalian brain, following synchronous apoptotic degeneration.Citation126 Alternative therapeutic strategies based on the modulation of adult neurogenesis have been already proposed and appeared promising for other neurodegenerative diseases, such as Parkinson disease (PD).Citation94 Few data on this specific issue have been published in ALS models, but there is convincing evidence of widespread regenerative response, mainly toward glia, in the spinal cord of tgSOD-1 mice.Citation127 Moreover, a temporal and regional plasticity of NPs in the dorsal horns of the spinal cord, as well as in motor cortex and lateral ventricle, in conjunction with their differentiation into neuron-like cells in response to MN loss, has been described during the disease onset or progression stages.Citation128 These data suggest that, when stimulated by the neurodegenerative process, the adult spinal cord possesses, at least, limited ability for regeneration, which could be potentially exploited for therapeutic purposes (Figure 10). However, this endogenous response appears impossible in contrasting widespread degeneration, but may be strategically supported by allogenic SC grafts, as recently demonstrated in a PD model where reciprocal influences between implanted cells and endogenous NPs exert multiple neurorescue effects on several brain regions.Citation129
Collectively, these data highlight how many potential protective strategies, alternative to MN replacement, may be explored for future applications to ALS therapy after careful testing in appropriate preclinical studies, clarifying the underlying mechanisms of action.
Caveats and the importance of patient choice in clinical SC therapy
The collected preclinical data demonstrate the feasibility of SC application to ALS animal models speeding the route towards clinical trials, but several hurdles limit a direct translation into new therapies, as clearly discussed by Lindvall and Kokaia.Citation94 Retrieval of inconsistent behavioral improvements after SC grafting in animal models have not been paralleled by adequate comprehension of the underlying regulatory mechanisms exploitable for the development of standardized protocols applicable to patients. Altogether, the collected preclinical data show the feasibility of SC therapy for ALS, but more definitive answers are needed on the biological cascades activated by transplantation, such as the regulation of grafted SC behavior in terms of survival, proliferation and migration, as well as novel functional synaptogenesis, before widespread clinical trials can be contemplated.Citation130
Multiple small pilot trials using a variety of different SC types have been published, but inconsistency on safety procedures, optimal cell dose/source, and delivery route reduce the interpretation of their potential efficiencyCitation107 (as detailed in ). An impressive debate on expensive SC treatment offered worldwide has enlightened a complex reality where both meaningful interpretations and anecdotal reports are strictly linked. Nevertheless, since efficacious therapy is lacking, the severity of ALS might justify the potential risks of intervention in patients to demonstrate clinical feasibility of pioneering SC techniques.Citation131
Table 1 Recent clinical trials with SCs in ALS patients (in chronological order)
A clinical trial aimed to evaluate the safety of human SC implant in ALS patients is currently ongoing in the United States, which follows the preclinical validations of the surgical procedures.Citation132 The SCs used in the study, prepared from cultured neural SCs, have previously been shown to extend the life of rats with ALSCitation133 and reverse the paralysis in rats affected by ischemic spastic paraplegia.Citation134 Direct neuronal differentiation of the grafted cells and release of growth factors to host MNs via graft–host connections have been suggested as the mechanisms responsible for the positive effects observed in both models. The phase 1 trial directed by Dr Glass at Emory University (Georgia, USA.) will enroll up to 12 ALS patients who will receive 5–10 SC injections in the lumbar area of the spinal cord. The patients will be examined at regular intervals after surgery, with final review of the data to come about 24 months later. Depending on the success of this initial trial, a follow-up phase 2 trial or a modified phase 1 trial is expected to implement the surgical and experimental procedures. The clinical trial is recruiting ALS patients (for additional data, refer ).
Intravenous, intrathecal, and more often intraparenchymal administrations of hematopoietic SCs derived from peripheral blood or bone marrow have been tested in a small series of patients.Citation73,Citation112,Citation124,Citation125,Citation135,Citation136 Even if safety and lack of early side effects have been claimed, the majority of these studies did not exhibit solid preclinical evidence as recommended before translation to clinical application.Citation137,Citation138 Clinical efficacy appears unproven and long-term safety needs to be demonstrated (). A large number of ALS patients has been recently reported after intracerebral transplantation with olfactory ensheathing cells,Citation139–Citation141 with disabling side effects reported for a patient who received this therapy in Beijing, China.Citation142 No sham operations have been documented, and the interpretation of the reported data is difficult because it is generated mostly outside the construct of a well-designed clinical study (see ). Finally, novel surgical techniques for efficient SC delivery within the spinal cord have to be developed and tested to maximize safety and to support grafted cell integration in the host circuits, as recently suggested by several reportsCitation73,Citation132,Citation143,Citation144 ().
Guidelines for clinical trials using SC need to be specifically designed for ALS patients after selection of the most appropriate end points to reach clinical significance (): the historical data related to cell transplantation in PD become instrumental in achieving this goal. The neurological community has to reach a consensus on the design of clinical trials and proceed with a long follow-up to define the outcomes.
Conclusion
SC contribution in understanding the relevant pathways involved in ALS pathobiology becomes as essential as the SC application to cell therapy: their exploitation, in combination with advanced molecular techniques (ie, cell engineering, siRNA, and miRNA application), will guarantee optimized and innovative protocols for cell therapy, as well as new drug or pharmacological strategies able to influence the pathological progression of ALS. The emerging evidence of ALS as a system disease affecting, besides the CNS, also several peripheral organs in the affected patients will dictate new approaches to solve an old problem, namely the MN loss that still represents the more vulnerable side of a generalized process. Human iPS technology for generation of large numbers of MNs and other cells could help to further define generalized disease mechanism, offering powerful biological assays for drug screening. These combined efforts could broaden the chance of success, and thus, the journey that started in 1993 with the identification of the first underlying genetic defects in ALS could finally lead to successful therapy in ALS patients.
| Abbreviations | ||
| ALS | = | amyotrophic lateral sclerosis |
| ES | = | embryonic stem cells |
| iPS | = | iPS inducible pluripotent stem cells |
| FTD | = | frontotemporal dementia |
| FTD-U | = | frontotemporal dementia with ubiquitin inclusions |
| MN | = | motor neuron |
| MND | = | motor neuron disease |
| NP | = | neural progenitor |
| SC | = | stem cell |
| SMA | = | spinal muscular atrophy |
| SOD-1 | = | superoxide dismutase |
| U inclusion | = | Ubiquitin-positive inclusion |
Acknowledgments
This study was kindly supported financially by Francesco Caleffi and the Peviani family.
Disclosure
The authors have no conflicts of interest that are directly relevant to the content of this review.
References
- BruijnLIMillerTMClevelandDWUnraveling the mechanisms involved in motor neuron degeneration in ALSAnnu Rev Neurosci20042772374915217349
- KunstCBComplex genetics of amyotrophic lateral sclerosisAm J Hum Genet20047593394715478096
- PasinelliPBrownRHMolecular biology of amyotrophic lateral sclerosis: insights from geneticsNat Rev Neurosci2006771072316924260
- HiranoANeuropathology of ALS: an overviewNeurology199647S63S668858053
- BurattiEBaralleFEMultiple roles of TDP-43 in gene expression, splicing regulation, and human diseaseFront Biosci20081386787817981595
- NeumannMSampathuDMKwongLKUbiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosisScience200631413013317023659
- AraiTHasegawaMAkiyamaHTDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosisBiochem Biophys Res Commun200635160261117084815
- ArmstrongRALantosPLCairnsNJOverlap between neurodegenerative disordersNeuropathology20052511112415875904
- StrongMJThe syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosisAmyotroph Lateral Scler2008932333818752088
- StrongMJGraceGMFreedmanMConsensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosisAmyotroph Lateral Scler20091013114619462523
- CorradoLRattiAGelleraCHigh frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosisHum Mutat20093068869419224587
- BenajibaLLe BerICamuzatATARDBP mutations in motoneuron disease with frontotemporal lobar degenerationAnn Neurol20096547047319350673
- TsaiKJYangCHFangYHElevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-UJ Exp Med20102071661167320660618
- VolkeningKLeystra-LantzCYangWJaffeeHStrongMJTar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS)Brain Res2009130516818219815002
- KwiatkowskiTJJrBoscoDALeclercALMutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosisScience20093231205120819251627
- Lagier-TourenneCClevelandDWRethinking ALS: the FUS about TDP-43Cell20091361001100419303844
- VanceCRogeljBHortobagyiTMutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6Science20093231208121119251628
- TicozziNSilaniVLeClercALAnalysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohortNeurology2009731180118519741215
- YangSWarraichSTNicholsonGABlairIPFused in sarcoma/translocated in liposarcoma: A multifunctional DNA/RNA binding proteinInt J Biochem Cell Biol2010421408141120541619
- FujitaYFujitaSTakatamaMIkedaMOkamotoKNumerous FUS-positive inclusions in an elderly woman with motor neuron diseaseNeuropathology201010.1111/j.1440-1789.2010.01146.x
- BolognaniFPerrone-BizzozeroNIRNA-protein interactions and control of mRNA stability in neuronsJ Neurosci Res20088648148917853436
- Deschenes-FurryJPerrone-BizzozeroNJasminBJThe RNA-binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticityBioessays20062882283316927307
- CushmanMJohnsonBSKingODGitlerADShorterJPrion-like disorders: blurring the divide between transmissibility and infectivityJ Cell Sci20101231191120120356930
- TicozziNLeclercALvan BlitterswijkMMutational analysis of TARDBP in neurodegenerative diseasesNeurobiol Aging200910.1016/j.neurobiolaging.2009.11.018
- LiYRayPRaoEJA Drosophila model for TDP-43 proteinopathyProc Natl Acad Sci U S A1073169317420133767
- ZhouHHuangCChenHtransgenic rat model of neurodegeneration caused by mutation in the TDP genePLoS Genet20106e100088720361056
- SleegersKCrutsMvan BroeckhovenCMolecular Pathways of Frontotemporal Lobar DegenerationAnnu Rev Neurosci201033718820415586
- LiuZMartinLJThe adult neural stem and progenitor cell niche is altered in amyotrophic lateral sclerosis mouse brainJ Comp Neurol200649746848816736475
- DianaVOttolinaABottiFNeural precursor derived astrocytes of wobbler mice induce apoptotic death of motor neurons through reduced glutamate uptakeExp Neurol201010.1016/j.expneurol.2010.06.008
- BossolascoPCovaLCalzarossaCMetalloproteinase alterations in the bone marrow of ALS patientsJ Mol Med20108855356420091292
- DimosJTRodolfaKTNiakanKKInduced pluripotent stem cells generated from patients with ALS can be differentiated into motor neuronsScience20083211218122118669821
- KiskinisEEgganKProgress toward the clinical application of patient-specific pluripotent stem cellsJ Clin Invest2010120515920051636
- MiuraKOkadaYAoiTVariation in the safety of induced pluripotent stem cell linesNat Biotechnol20092774374519590502
- InoueHNeurodegenerative disease-specific induced pluripotent stem cell researchExp Cell Res201010.1016/j.yexcr.2010.04.022
- EbertADYuJRoseFFJrInduced pluripotent stem cells from a spinal muscular atrophy patientNature200945727728019098894
- LeeGPapapetrouEPKimHModelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCsNature200946140240619693009
- VillaCErraticoSRaziniPStem cell tracking by nanotechnologiesInt J Mol Sci2010111070108120480000
- Di GiorgioFPBoultingGLBobrowiczSEgganKCHuman embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutationCell Stem Cell2008363764819041780
- Di GiorgioFPCarrascoMASiaoMCManiatisTEgganKNon-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS modelNat Neurosci20071060861417435754
- NagaiMReDBNagataTAstrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neuronsNat Neurosci20071061562217435755
- WeinbergMSWoodMJShort non-coding RNA biology and neurodegenerative disorders: novel disease targets and therapeuticsHum Mol Genet200918R27R3919297399
- StrongMJThe evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS)J Neurol Sci201028811219840884
- MallannaSKRizzinoAEmerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cellsDev Biol2010344162520478297
- WilliamsAHValdezGMoresiVMicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in miceScience20093261549155420007902
- BurattiEde ContiLStuaniCRomanoMBaralleMBaralleFNuclear factor TDP-43 can affect selected microRNA levelsFEBS J20102772268228120423455
- InuiMMartelloGPiccoloSMicroRNA control of signal transductionNat Rev Mol Cell Biol20101125226320216554
- WangYRussellIChenCMicroRNA and stem cell regulationCurr Opin Mol Ther20091129229819479662
- KawaharaYItoKSunHAizawaHKanazawaIKwakSGlutamate receptors: RNA editing and death of motor neuronsNature200442780114985749
- MackenzieIRBigioEHIncePGPathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutationsAnn Neurol20076142743417469116
- KikuchiHAlmerGYamashitaSSpinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS modelProc Natl Acad Sci U S A20061036025603016595634
- GruzmanAWoodWLAlpertECommon molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosisProc Natl Acad Sci U S A2007104125241252917636119
- OnoSThe skin in amyotrophic lateral sclerosisAmyotroph Lateral Scler Other Motor Neuron Disord2000119119911464952
- OnoSImaiTShimizuNNakayamaMYamanoTTsumuraMSerum markers of type I collagen synthesis and degradation in amyotrophic lateral sclerosisEur Neurol200044495610894996
- ProvincialiLCangiottiATulliDCarboniVCintiSSkin abnormalities and autonomic involvement in the early stage of amyotrophic lateral sclerosisJ Neurol Sci199412654617836947
- OnoSImaiTMunakataSCollagen abnormalities in the spinal cord from patients with amyotrophic lateral sclerosisJ Neurol Sci19981601401479849796
- FangLHuber-AbelFTeuchertMLinking neuron and skin: Matrix metalloproteinases in amyotrophic lateral sclerosis (ALS)J Neurol Sci2009285626619523650
- LudolphACMatrix metalloproteinases – a conceptional alternative for disease-modifying strategies in ALS/MND?Exp Neurol200620127728016808915
- BeucheWYushchenkoMMaderMMaliszewskaMFelgenhauerKWeberFMatrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosisNeuroreport2000113419342211095490
- DemestreMParkin-SmithGPetzoldAPullenAHThe pro and the active form of matrix metalloproteinase-9 is increased in serum of patients with amyotrophic lateral sclerosisJ Neuroimmunol200515914615415652414
- Niebroj-DoboszIJanikPSokolowskaBKwiecinskiHMatrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosisEur J Neurol20101722623119796283
- SchoserBGBlottnerDMatrix metalloproteinases MMP-2, MMP-7 and MMP-9 in denervated human muscleNeuroreport1999102795279710511442
- LimGPBackstromJRCullenMJMillerCAAtkinsonRDTokesZAMatrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patientsJ Neurochem1996672512598666998
- KiaeiMKipianiKCalingasanNYMatrix metalloproteinase-9 regulates TNF-alpha and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosisExp Neurol2007205748117362932
- LorenzlSNarrSAngeleBThe matrix metalloproteinases inhibitor Ro 26-2853 extends survival in transgenic ALS miceExp Neurol200620016617116516196
- DewilMSchurmansCStarckxSOpdenakkerGvan Den BoschLRobberechtWRole of matrix metalloproteinase-9 in a mouse model for amyotrophic lateral sclerosisNeuroreport20051632132415729130
- MalemudCJMatrix metalloproteinases (MMPs) in health and disease: an overviewFront Biosci2006111696170116368548
- DupuisLCorciaPFerganiADyslipidemia is a protective factor in amyotrophic lateral sclerosisNeurology2008701004100918199832
- FerganiAOudartHGonzalez De AguilarJLIncreased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosisJ Lipid Res2007481571158017438338
- CortiSDonadoniCRonchiDAmyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunctionJ Neurol Sci200927617017419000626
- DupuisLLoefflerJPNeuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic modelsCurr Opin Pharmacol2009934134619386549
- MazziniLFagioliFBoccalettiRStem-cell therapy in amyotrophic lateral sclerosisLancet20043641936193715567004
- MazziniLFagioliFBoccalettiRStem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humansAmyotroph Lateral Scler Other Motor Neuron Disord2003415816113129802
- MazziniLFerreroILuparelloVMesenchymal Stem Cell Transplantation In Amyotrophic Lateral Sclerosis: A Phase I Clinical TrialExp Neurol200910.1016/j.expneurol.2009.08.007
- MazziniLMareschiKFerreroIAutologous mesenchymal stem cells: clinical applications in amyotrophic lateral sclerosisNeurol Res20062852352616808883
- MazziniLMareschiKFerreroIStem cell treatment in Amyotrophic Lateral SclerosisJ Neurol Sci2008265788317582439
- LeeHShamyGAElkabetzYDirected differentiation and transplantation of human embryonic stem cell-derived motoneuronsStem Cells2007251931193917478583
- LiXJDuZWZarnowskaEDSpecification of motoneurons from human embryonic stem cellsNat Biotechnol20052321522115685164
- LiXJHuBYJonesSADirected differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small moleculesStem Cells20082688689318238853
- Singh RoyNNakanoTXuingLKangJNedergaardMGoldmanSAEnhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cellsExp Neurol200519622423416198339
- WichterleHLieberamIPorterJAJessellTMDirected differentiation of embryonic stem cells into motor neuronsCell200211038539712176325
- MilesGBYohnDCWichterleHJessellTMRafuseVFBrownstoneRMFunctional properties of motoneurons derived from mouse embryonic stem cellsJ Neurosci2004247848785815356197
- DeshpandeDMKimYSMartinezTRecovery from paralysis in adult rats using embryonic stem cellsAnn Neurol200660324416802299
- HarperJMKrishnanCDarmanJSAxonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult ratsProc Natl Acad Sci U S A20041017123712815118094
- GaillardAPrestozLDumartinBReestablishment of damaged adult motor pathways by grafted embryonic cortical neuronsNat Neurosci2007101294129917828256
- EmgardMHolmbergLSamuelssonEBHuman neural precursor cells continue to proliferate and exhibit low cell death after transplantation to the injured rat spinal cordBrain Res20091278152619376093
- CapowskiEESchneiderBLEbertADLentiviral vector-mediated genetic modification of human neural progenitor cells for ex vivo gene therapyJ Neurosci Methods200716333834917397931
- KleinSMBehrstockSMcHughJGDNF delivery using human neural progenitor cells in a rat model of ALSHum Gene Ther20051650952115871682
- ThonhoffJROjedaLWuPStem cell-derived motor neurons: applications and challenges in amyotrophic lateral sclerosisCurr Stem Cell Res Ther2009417819919492980
- CortiSNizzardoMNardiniMEmbryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in miceBrain201013346548120032086
- YohnDCMilesGBRafuseVFBrownstoneRMTransplanted mouse embryonic stem-cell-derived motoneurons form functional motor units and reduce muscle atrophyJ Neurosci200828124091241819020033
- CortiSLocatelliFPapadimitriouDNeural stem cells LewisX+ CXCR4+ modify disease progression in an amyotrophic lateral sclerosis modelBrain20071301289130517439986
- CortiSNizzardoMNardiniMMotoneuron transplantation rescues the phenotype of SMARD1 (spinal muscular atrophy with respiratory distress type 1)J Neurosci200929117611177119776263
- CortiSNizzardoMNardiniMNeural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophyJ Clin Invest20081183316333018769634
- LindvallOKokaiaZStem cells in human neurodegenerative disorders – time for clinical translation?J Clin Invest2010120294020051634
- BrundinPLiJYHoltonJLLindvallOReveszTResearch in motion: the enigma of Parkinson’s disease pathology spreadNat Rev Neurosci2008974174518769444
- ClementAMNguyenMDRobertsEAWild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS miceScience200330211311714526083
- YamanakaKBoilleeSRobertsEAMutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS miceProc Natl Acad Sci U S A20081057594759918492803
- YamanakaKChunSJBoilleeSAstrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosisNat Neurosci20081125125318246065
- BoilleeSYamanakaKLobsigerCSOnset and progression in inherited ALS determined by motor neurons and microgliaScience20063121389139216741123
- HedlundEHefferanMPMarsalaMIsacsonOCell therapy and stem cells in animal models of motor neuron disordersEur J Neurosci2007261721173717897390
- MarchettoMCMuotriARMuYSmithAMCezarGGGageFHNon-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cellsCell Stem Cell2008364965719041781
- RalphGSRadcliffePADayDMSilencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS modelNat Med20051142943315768029
- RaoulCAbbas-TerkiTBensadounJCLentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALSNat Med20051142342815768028
- EbertADSvendsenCNHuman stem cells and drug screening: opportunities and challengesNat Rev Drug Discov2010936737220339370
- KarumbayaramSKellyTKPaucarAAHuman embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron degeneration linked to ALSDis Model Mech2009218919519259395
- GowingGPhilipsTvan WijmeerschBAblation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutaseJ Neurosci200828102341024418842883
- MazziniLVercelliAFerreroIStem cells in amyotrophic lateral sclerosis: state of the artExpert Opin Biol Ther200991245125819663719
- BeersDRHenkelJSZhaoWWangJAppelSHCD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALSProc Natl Acad Sci U S A2008105155581556318809917
- HabischHJJanowskiMBinderDIntrathecal application of neuroectodermally converted stem cells into a mouse model of ALS: limited intraparenchymal migration and survival narrows therapeutic effectsJ Neural Transm20071141395140617510731
- KimHKimHYChoiMRDose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS miceNeurosci Lett46819019419879334
- XuLRyugoDKPongstapornTJoheKKoliatsosVEHuman neural stem cell grafts in the spinal cord of SOD1 transgenic rats: differentiation and structural integration into the segmental motor circuitryJ Comp Neurol200951429730919326469
- AppelSHEngelhardtJIHenkelJSHematopoietic stem cell transplantation in patients with sporadic amyotrophic lateral sclerosisNeurolog y20087113261334
- SuzukiMMcHughJTorkCGDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALSPLoS One20072e68917668067
- SuzukiMSvendsenCNCombining growth factor and stem cell therapy for amyotrophic lateral sclerosisTrends Neurosci20083119219818329734
- ParkSKimHTYunSGrowth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS miceExp Mol Med20094148750019322031
- LeporeACRauckBDejeaCFocal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron diseaseNat Neurosci2008111294130118931666
- HowlandDSLiuJSheYFocal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS)Proc Natl Acad Sci U S A2002991604160911818550
- MaragakisNJDykes-HobergMRothsteinJDAltered expression of the glutamate transporter EAAT2b in neurological diseaseAnn Neurol20045546947715048885
- GuRHouXPangRHuman adipose-derived stem cells enhance the glutamate uptake function of GLT1 in SOD1(G93A)-bearing astrocytesBiochem Biophys Res Commun201039348148620152807
- CortiSNizzardoMNardiniMSystemic transplantation of c-kit+ cells exerts a therapeutic effect in a model of amyotrophic lateral sclerosisHum Mol Genet201010.1093/hmg/ddq293
- CortiSLocatelliFDonadoniCWild-type bone marrow cells ameliorate the phenotype of SOD1-G93A ALS mice and contribute to CNS, heart and skeletal muscle tissuesBrain20041272518253215469951
- BeersDRHenkelJSXiaoQWild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosisProc Natl Acad Sci U S A2006103160211602617043238
- OhnishiSItoHSuzukiYIntra-bone marrow-bone marrow transplantation slows disease progression and prolongs survival in G93A mutant SOD1 transgenic mice, an animal model mouse for amyotrophic lateral sclerosisBrain Res2009129621622419686706
- MartinezHRGonzalez-GarzaMTMoreno-CuevasJECaroEGutierrez-JimenezESeguraJJStem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patientsCytotherapy200911263419191058
- DedaHInciMCKurekciAETreatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-upCytotherapy200911182519012065
- ChenJMagaviSSMacklisJDNeurogenesis of corticospinal motor neurons extending spinal projections in adult miceProc Natl Acad Sci U S A2004101163571636215534207
- ErieEAShimHSmithALMice deficient in the ALS2 gene exhibit lymphopenia and abnormal hematopoietic functionJ Neuroimmunol200718222623117156857
- ChiLGanLLuoCLienLLiuRTemporal response of neural progenitor cells to disease onset and progression in amyotrophic lateral sclerosis-like transgenic miceStem Cells Dev20071657958817784831
- CovaLArmenteroMTZennaroEMultiple neurogenic and neurorescue effects of human mesenchymal stem cell after transplantation in an experimental model of Parkinson’s diseaseBrain Res20101311122719945443
- Garbuzova-DavisSSanbergPRFeasibility of cell therapy for amyotrophic lateral sclerosisExp Neurol20092163619084005
- GornallJStem cell renegades or pioneers?BMJ2010340c204120444823
- RileyJFedericiTParkJCervical spinal cord therapeutics delivery: preclinical safety validation of a stabilized microinjection platformNeurosurgery200965754761 discussion 761–75219834381
- XuLYanJChenDHuman neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic ratsTransplantation20068286587517038899
- CizkovaDKakinohanaOKucharovaKFunctional recovery in rats with ischemic paraplegia after spinal grafting of human spinal stem cellsNeuroscience200714754656017524565
- CashmanNTanLYKriegerCPilot study of granulocyte colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells in amyotrophic lateral sclerosis (ALS)Muscle Nerve20083762062518335482
- JansonCGRameshTMDuringMJLeonePHeywoodJHuman intrathecal transplantation of peripheral blood stem cells in amyotrophic lateral sclerosisJ Hematother Stem Cell Res20011091391511798518
- BadayanICudkowiczMEIs it too soon for mesenchymal stem cell trials in people with ALS?Amyotroph Lateral Scler2008932132218819027
- SilaniVCovaLCorboMCiammolaAPolliEStem-cell therapy for amyotrophic lateral sclerosisLancet200436420020215246734
- ChenLHuangHZhangJShort-term outcome of olfactory ensheathing cells transplantation for treatment of amyotrophic lateral sclerosisZhongguo Xiu Fu Chong Jian Wai Ke Za Zhi20072196196617933231
- HuangHChenLXiHFetal olfactory ensheathing cells transplantation in amyotrophic lateral sclerosis patients: a controlled pilot studyClin Transplant20082271071818673377
- HuangHChenLXiHOlfactory ensheathing cells transplantation for central nervous system diseases in 1,255 patientsZhongguo Xiu Fu Chong Jian Wai Ke Za Zhi200923142019192871
- ChewSKhandjiAGMontesJMitsumotoHGordonPHOlfactory ensheathing glia injections in Beijing: misleading patients with ALSAmyotroph Lateral Scler2007831431617917850
- BlanquerMPerez-EspejoMAMartinez-LageJFIniestaFMartinezSMoraledaJMA surgical technique of spinal cord cell transplantation in amyotrophic lateral sclerosisJ Neurosci Methods201010.1016/j.jneumeth.2010.06.014
- FeronFPerryCCochraneJAutologous olfactory ensheathing cell transplantation in human spinal cord injuryBrain20051282951296016219671