Abstract
Epigenetic regulation of gene transcription by small molecule inhibitors of histone deacetylases (HDAC) is a novel cancer therapy. Vorinostat (Zolinza™) is the first FDA approved HDAC-inhibitor for treatment of patients with cutaneous T cell lymphoma (CTCL) who have progressive, persistent or recurrent disease on or following two systemic therapies. Vorinostat was active against solid tumors and hematologic malignancies as intravenous and oral preparations in Phase I development. In two Phase II trials, vorinostat was safe and effective at an oral dose of 400 mg/day with an overall response rate of 24%–30% in refractory advanced patients with CTCL including large cell transformation and Sézary syndrome (SS). The common side effects of vorinostat, similar in all studies, included gastro-intestinal symptoms, fatigue, and thrombocytopenia and the most common serious events were thrombosis. Vorinostat, in combination with other agents such as radiation therapy and chemotherapy, have shown synergistic or additive effects in a variety of cancers in clinical trials.
Mycosis fungoides and Sezary syndrome: Pathogenesis and features
Cutaneous T-cell lymphomas (CTCL) are a clinically heterogeneous group of postthymic lymphomas, accounting for the majority of all lymphomas arising in skin. Mycosis fungoides (MF) and Sézary syndrome (SS), the most common variants, are still rare with an annual incidence of 3 to 4 new cases per million or 1200 new cases per year in the United States (CitationGiardi et al 2004; CitationKim et al 2005). The next most common entities are the CD30+ lymphoproliferative disorders: lymphomatoid papulosus and anaplastic large T-cell lymphoma (ALCL). Sub-cutaneous panniculitic T-cell lymphoma and NK-T cell lymphomas are quite rare, and more aggressive. Cutaneous T-cell lymphomas that do not fit into one of these categories are peripheral T-cell lymphomas, unclassified and are described on the basis of immunohistochemical phenotype.
Mycosis fungoides (MF) and Sézary syndrome (SS) are defined by their cutaneous presentation and the accumulation of a T helper memory/effector subset with a CD4+, CD8−, CD45RO+CLA+ phenotype (CitationKazakov et al 2004). In many patients, MF starts as an indolent and chronic dermatitis in the sun shielded areas and a diagnostic biopsy is difficult to obtain for years. In order to diagnose MF, there must be atypical lymphocytes within the epidermis, and the presence of CD4+>>CD8+ cells and clonality are supporting characteristics. In SS, the atypical cells have lost epidermotropism and are found around the dermal vessels (CitationDiwan et al 2005).
Mycosis fungoides is staged using the Tumor Nodes Metastasis classification schema which is used to stage patients and to predict disease prognosis. MF may progress to a leukemic and erythrodermic condition called Sézary syndrome (SS) which requires the presence of >1000/μL of atypical circulating cells. Sézary cells secrete Th2 cytokines, IL-4 and IL-10, causing loss of cellular immunity due to decreased production of Th1 cytokines, interferon gamma and interleukin 2 (IL-2) (CitationKim et al 2005). This results in an atopic state characterized by erythroderma and staphylococcus colonization, peripheral eosinophilia, increased IgE production, and intractable pruritus. Thus, one strategy used to treat MF/SS, is to restore the immune Th2>Th1 imbalance.
Tan et al first hypothesized that T-cell proliferation was initiated by persistence of chronic antigen stimulation (CitationTan et al 1974). Molecular methods have shown that in MF there is emergence of one or more T-cell clones of skin-homing CD4+ cells and that with progression to SS, these appear in the blood and can be detected by flow cytometry (CitationVega et al 2002). If MF/SS is antigen driven, the stimulus would not need to be the same in each patient and could be autoantigens or environmental and once initiated might not resolve with removal of the antigen. That MF/SS patients have class II HLA associations with DR5 and DQB*03 alleles does support genetic predisposition similar to other autoimmune skin diseases (CitationJackow et al 1996).
Infectious agents such as Epstein-Barr virus, cytomegalovirus, HTLV-1, and Staphylococcus aureus have been hypothesized as possible triggers (CitationAxelrod et al 1992; CitationTokura et al 1995; CitationJackow et al 1997; CitationShimakage et al 2001; CitationGiardi et al 2004). The observation that the atypical CD4+ lymphocytes cluster around epidermal Langerhans cells (the Pautrier’s microabcess characteristic of MF) also supports an antigen driven immune response (CitationGiardi et al 2004; CitationDiwan et al 2005). Immature dendritic cells (Langerhans cells) in the epidermis are thought to activate T cells through direct contact and providing the stimulus for their clonal expansion by migrating to the dermis (CitationBerger et al 2002). It is hypothesized that the loss of activation induced cell death following T-cell proliferation through loss of Fas may lead to accumulation of the T-cells in skin resulting in chronic inflammatory lesions (CitationNi et al 2005).
Tumor cells expressing Fas Ligand may eliminate tumor infiltrating, cytotoxic CD8+ T-cells, allowing disease progression to occur (CitationNi et al 2005). Progression of MF/SS is accompanied by clonal dominance of the malignant cells (CitationVega et al 2002) leading to the elaboration of Th2 cytokines (CitationRook et al 1997), impairment of the hosts’ immune response, and further tumor cell growth (CitationGiardi et al 2004; CitationKim et al 2005).
Treatment of early MF
There is a very limited number of FDA-approved therapies available to treat patients with MF/SS. However, agents that are used for eczema, psoriasis, and other forms of lymphoma are used for MF/SS and represent the standard of care. Early MF is characterized by eczematous or psoriasiform dermatitis limited to less than 10% of the body surface. Lesions are often most prominent in sun-shielded areas and will fade with exposure to UVB or UVA light. Early MF lesions are treated with one or more skin directed therapies and are capable of inducing durable complete responses. The first therapy used frequently before MF is diagnosed is the application of topical corticosteroids of increasing potency. For thicker or hypertrophic lesions, the topical retinoid gels or creams may be effective, can reduce the time of clearing with phototherapy, but may cause irritation, especially in intertriginous areas. The response rate of targretin gel 1% was 76% in patients not previously treated and this agent is the only topical therapy approved for MF (CitationBreneman et al 2002). We have found this gel useful for MF of the hands or feet (CitationLain et al 2003) and for aborting lymphomatoid papulosis lesions (CitationKraken et al 2003). In an opened label pilot study, tazarotene gel was active as a steroid adjuvant for treatment of MF lesions (CitationApisarnthanarax et al 2004). The topical inducer of interferon, imiquimod 5%, has produced 50% histologic clearing of MF lesions when applied to lesions of 6 patients for 3 days per week for 12 weeks (CitationDeeths et al 2005).
Treatment of intermediate or refractory early MF
Patients with <10% involvement (IA) who do not respond to first line topical therapies or who have >10% of the body surface area I (IB) or dermatopathic nodes (IIA) often need more extensive therapies such as topical chemotherapy with mustargen, BCNU, or with a combination of skin directed therapy plus one or more biological response modifiers. Agents that are not specifically approved for MF/SS are commonly used as the standard of care: topical steroids and mustargen, phototherapy, interferons, and chemotherapies. For patch disease, narrow band UVB is effective and can be effectively combined with topical steroids or retinoids. Thick plaque lesions or folliculotropic MF lesions are more difficult to clear and are treated with PUVA plus interferon or an oral retinoid (bexarotene, soriatane, or accutane). Total electron beam radiation is reserved for patients who need skin palliation or who have extensive skin involvement and have failed to respond to skin directed therapies, and should be followed with a form of maintenance therapy such as mustargen, PUVA, or oral bexarotene.
Oral bexarotene and intravenous denileukin diftitox have received FDA approval for treatment of the cutaneous manifestations of CTCL and are used in this setting, along with interferon alpha. Bexarotene monotherapy has a response rate of 54% at a dose of 300 mg/m2 in early patients and 45% in more advanced patients (CitationDuvic et al 2001). Its dose limiting toxicity is hypertriglyceridemia, especially in predisposed patients. Use of lower doses of bexarotene is often effective in combination therapy and the triglyceridemia can often be controlled with addition of an HMG-coA reductase and/or a statin (CitationAssaf et al 2006). Alpha interferon is another biological response modifier that has shown significant activity for MF and SS and may be used in combination with PUVA, photopheresis, and retinoids. It is widely used for MF/SS but has some dose limiting toxicities such as fever, fatigue, low white count, anemia, and hepatitis. Vorinostat, a novel histone deacetylase inhibitor, discussed in detail below, has also been approved recently and would also be considered to be in the category of the biological response modifiers, based on its known mechanism of action so far and its potential ability to synergize with radiation or phototherapy.
Denileukin diftitox (Ontak®), a fusion protein composed of IL-2 and diphtheria toxin, is an example of a biologic response modifier as well as one of the newer targeted therapies developed for CTCL. It received FDA approval for cutaneous manifestations of CTCL of all stages based on a randomized two dose arm controlled multicenter trial showing a response rate of 30% (CitationOlsen et al 2001). Patients with tumors (IIB) had significantly better responses at the higher dose of 18 μg/kg/day given for 5 days, every 3 weeks and it is a non-immunosuppressive alternative to chemotherapy for debulking MF tumors. Complete responses were seen in 10% of the patients enrolled (CitationOlsen et al 2001). Capillary leak syndrome is seen in 20%–30% of patients but may be decreased with hydration and acute fusion reactions are blocked by steroid pre-medication. Bexarotene and steroids may increase CD25 expression on T-cells as measured by flow and could suggest synergism (CitationFoss et al 2005). Our study suggested that high expression of CD25+ in >20% of the tumor cells was associated with higher response rates 60% compared to 20% of the low expressing patient and 30% in the pivotal trial. Additional studies have supported the use of this agent for other lymphomas, chronic lymphocytic leukemia, graft versus host disease, psoriasis, and elimination of tumor suppressor T-regs.
Generalized exfoliative erythroderma (EE) may be induced by S. aureus (CitationDeeths et al 2005). MF and SS can present with EE, and the diagnosis of SS depends on having blood involvement fitting the B2 criteria (Atypical circulating lymphs >1000/μL or presence of a clone of CD4+ 26 or 7-cells). Extracorporeal photopheresis was approved in 1987 for the treatment of CTCL patients and significant responses were seen in erythrodermic patients (CitationLim et al 1995). Photopheresis is usually combined with biological response modifiers, especially interferon and retinoids, for responses approaching 60%–70% (CitationMcGinnis et al 2005). Patients with refractory or transformed MF and SS who do not respond to immune stimulating therapy have a poor prognosis and further therapy is usually palliative. There are several active targeted agents used in this setting which include nucleoside analogues (pentostatin, and the experimental agent forodesine), and monoclonal antibody to CD52 Campath H1.
Patients with transformed MF, tumors, or nodal disease may respond to local radiation, denileukin diftitox, nucleoside analogues (gemcitabine, pentostatin), doxil, or combination chemotherapy. Various chemotherapy combinations, while effective for a limited time, can also induce further immunosuppression leading to line induced sepsis or other opportunistic infections. There is a definite need for new therapies to treat patients. The recent FDA-approved HDAC inhibitor, vorinostat, represents a new strategy for targeted therapy of CTCL (CitationMarks et al 2007). As shown in two Phase II trials it has efficacy in patients who have failed prior therapies. This new agent is a biological response modifier but can work when chemotherapy has failed and has the advantage of being orally administered (CitationDuvic et al 2007). Vorinostat is the first HDAC-inhibitor to be approved for a malignancy indication.
Classification of the HDAC and HDAC inhibitors (HDAC-I)
HDAC inhibitors (HDAC-I) are a new class of antineoplastic agents and are currently being evaluated in a number of clinical trials for a wide variety of cancer types, as monotherapy and in combination with other effective modalities. Normal cells have a balance between acetylation and deacetylation of histones mediated through the activity of histone acetyl-transferases (HATs) and histone deacetylases (HDACs) (Citationde Ruijter et al 2003; CitationGregoretti et al 2004). HDACs remove acetyl groups on histones causing chromatin to be compacted resulting in silencing of gene transcription. Cancer cells tend to have over-expression of HDACs and aberrant recruitment HDACs to oncogeneic transcription factors causing hypoacetylation of core nucleosome histones. Cancer cells may also have reduced HAT activity through mutations resulting in silencing of genes such as tumor suppressors. HDAC-inhibitors are small molecules that bind to the active enzymatic sites in class I and II zinc containing HDAC enzymes. Depression of gene transcription by HDAC-I that block removal of acetyl-groups permits DNA transcription to occur. Although these enzymes are called “histone” acetylases and deacetylases, they actually have broad enzymatic effects on many other proteins, and predate histones in evolution (Citationde Ruijter et al 2003; CitationGregoretti et al 2004). Many genes are suppressed as upregulated in expression by HDAC inhibitors.
As shown in , there are four known classes of HDAC- inhibitors based on their chemical structures and the prototypes are sodium n-butyrate, a short chain fatty acid; vorinostat (SAHA) an organic hydroxamic acid; depsipeptide (romidepsin), a bicyclic depsipeptide; and MS-275, a benzamide. HDAC-I prevent histone deacetylases from catalyzing the hydrolysis of acetyl groups from the amino-terminal lysine residues of the nucleosomal core histones (). Athough HDAC-inhibitors can affect specific HDAC enzymes, vorinostat has broad class I and II activity (CitationGregoretti et al 2004).
Figure 1 HDACs and HATs regulate the balance of acetylation. HDAC inhibitors block removal of acetyl groups.
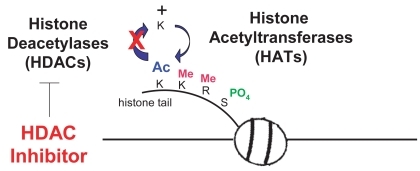
Table 1 Structure of HDAC-inhibitors
Eighteen HDAC enzymes have been described to date. They are divided into three families based on homology to yeast HDAC proteins. HDACs 1, 2, 3, 8 are class I (between 350 and 500 amino acids in length) (Citationde Ruijter et al 2003; CitationGregoretti et al 2004), localized in the nucleus and are inhibited by vorinostat (SAHA) and romidepsin. HDACs 4–7, 9–11 are class II (about 1000 amino acids in length), found in cytoplasm and nucleus, inhibited by vorinostat (SAHA) but not romidepsin. HDAC 11 contains conserved residues that are shared by both class I and class II enzymes and thus is usually classified as a class I enzyme. Recent phylogenetic studies indicate that it represents a separate class of HDACs (class IV) (CitationGregoretti et al 2004; CitationLedent and Vervoort 2006). Class III HDACs are homologous to the yeast “silencing information regulator 2” (Sir2) family of deacetylases and have a unique catalytic mechanism that requires the cofactor nicotinamide adenine dinucleotide (NAD+) (CitationBlander and Guarente 2004). Vorinostat is a non-specific agent inhibiting the activity of class I (types 1, 2, 3), II (type 6) and IV (type 11) HDAC enzymes, specifically (CitationGregoretti et al 2004). Romidepsin preferentially inhibits only class I enzymes, but is also classified as a broad spectrum inhibitor because it does inhibit Class II enzymes, at higher concentrations (CitationFurumai et al 2002).
HDAC-inhibitors – mechanisms of action
Acetylation of histones proteins on lysine tails prevents close interactions with the DNA backbone and promotes a loosening of chromatin structure. This permits access of transcription factors to specific promoter regions increasing transcription of genes. Although HDAC-I only influence 1%–2% of genes, genes controlling differentiation, cell proliferation, and apoptotic cell death are affected. HDAC inhibitors induce the cdk kinase inbitor (p21) causing cell cycle arrest leading to death. HDAC inhibitors target critical cell-cycle regulatory pathways and may induce the expression of various silenced tumor-suppressor genes critical in controlling cancer cell growth (CitationLindemann et al 2004). HDAC inhibitors trigger both caspase-dependent and caspase-independent apoptosis and may be as much as 1000-fold more toxic to malignant T-cells as to nonmalignant cells (CitationZhang et al 2005). The precise pathway that leads to cellular differentiation or apoptosis is not well understood. It has also been hypothesized that HDAC inhibitors may work as anti-angiogenesis agents though decreasing VEGF (CitationHeider et al 2006).
The rationale for clinical use of HDAC inhibitors for cancer is based on their anti-proliferative effects: induction of differentiation, cell cycle growth arrest, and apoptosis, demonstrated for numerous cancer cell-lines in culture and in vivo mouse models (CitationLindemann et al 2004). HDAC-I induces the accumulation of acetylated histones and changes gene expression (CitationLindemann et al 2004; CitationKelly et al 2005; CitationKelly and Marks 2005; CitationO’Connor 2005; CitationDuvic and Zhang 2006). However, histone acetylation is seen in all patients’s cells and is not correlated with clinical response at least in CTCL patients (CitationZhang et al 2005; CitationHeider et al 2006; CitationKelly et al 2005; CitationDuvic and Zhang 2006). HDAC inhibitors have also been shown to block deacetylation of non-histone substrates, ie, transcriptional factors such as p53, transduction mediators, and molecular chaperones all of which may also be important in the anti-cancer effects (CitationButler et al 2000, Citation2002; CitationLindemann et al 2004; CitationKelly and Marks 2005; CitationO’Connor 2005).
In addition to the potential use of HDAC inhibitor monotherapy for cancer, they may also be combined eventually and more effectively with other conventional chemotherapeutics and/or biological agents (CitationCohen et al 1999, Citation2002; CitationHe et al 2001; CitationReddy et al 2004). HDAC inhibitors have already been shown to be additive or synergistic with anthracyclines (CitationMarchion et al 2004), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (CitationButler et al 2006), and all-trans-retinoic acid (CitationZhang et al 2005). They are efficient radiation modifying agents and therefore, have been proposed as possible clinical radiation sensitizers or protectors (CitationMoradei et al 2005).
Vorinostat and romidepsin have shown in vitro and in vivo cytotoxic activity against various tumors which made them attractive to bring into clinical trials (CitationGore and Carducci 2000). In Phase I and II clinical trials, these HDAC-I were active in mycosis fungoides and peripheral T cell lymphoma patients (CitationQuerfeld et al 2005). They received orphan drug designation from the FDA for the treatment of advanced CTCL. Based the similar results in two Phase II trials, vorinostat was approved for the cutaneous manifestations of CTCL in October 2006.
Vorinostat: “SAHA”
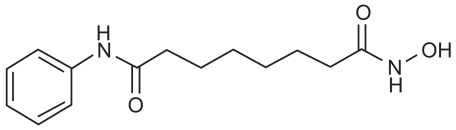
Vorinostat (Zolinza™; Merck, Whitehouse Station, NJ, USA), was previously known as suberoylanilide hydroxamic acid or SAHA. The drug is an orally bioavailable inhibitor of Class I and II HDACs (CitationGregoretti et al 2004). It is described chemically as N-hydroxy-N-′phenyloctanediamide, a small (shown in molecule with the molecular formula C14H20N2O3 ) and a molecular weight of 264.32. It has no chiral centers and is non-hygroscopic. It is very slightly soluble in water and freely soluble in dimethyl sulfoxide with a pH of 6.6 in saturated water. The discovery and pre-clinical development of vorinostat was a collaboration between Breslow, Professor of Chemistry at Columbia, and Marks, Rifkind and Richon, Cell Biology at Sloan-Kettering and Phase I Clinical Trials at Memorial Sloan Kettering with Kelly Scher, and colleagues (CitationMarks and Breslow 2007). Subsequently, Phase I clinical trials were conducted by Aton Pharmaceuticals prior to its being acquired by Merck in April 2004. Vorinostat comes in 100 mg capsules.
Figure 2 Suberolyanilide hydroxamic acid.
Preclinical studies
In preclinical studies, vorinostat was shown to cause accumulation of acetylated histones, induce cell-cycle arrest and apoptosis in a broad range of cancer cell lines, including Sezary cells (CitationZhang et al 2005). Vorinostat also showed antitumor activity in leukemia, lymphoma, and solid-tumor models in vivo (CitationVrana et al 1999; CitationKelly et al 2003, Citation2005; CitationGarcia-Manero et al 2005; CitationSakajiri et al 2005; CitationZhang et al 2005; CitationO’Connor et al 2006).
Marks et al first demonstrated the efficacy of SAHA (vorinostat) in vivo in nude mice bearing a human prostate cancer cell (CWR22) xenograft (CitationMarks et al 2001; CitationMarks and Dokmanovic 2005; CitationMarks and Jiang 2005). The intraperitoneal administration of vorinostat daily for 21 days completely inhibited tumor cell growth at doses that had no detectable toxic effects on normal cells as determined by hematological studies and extensive autopsy examination (CitationButler et al 2000). Subsequently, orally or parenterally administered vorinostat was shown effective in inhibiting tumor growth in a carcinogen-induced mammary tumor in rats, a human neuroblastoma xenograft in mice, a transgenic mouse model of therapy-resistant acute promyelocytic leukemia, and a carcinogen-induced lung cancer in mice, all with little or no toxicity (CitationDesai et al 2003).
Mechanism of vorinostat in CTCL cells
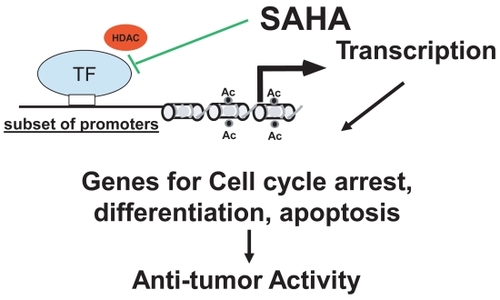
Vorinostat inhibits HDACs of class I (types 1, 2, 3) and II (type 6) at about 50 nM concentrations (CitationRichon et al 1998). It arrests cell growth of a wide variety of transformed cells in culture at 2.5–5.0 μM, including Sezary and MF cell lines (CitationMoradei et al 2005; CitationZhang et al 2005). Class I HDACs are primarily nuclear in localization and ubiquitous. Class II HDAC expression is more tissue restricted; these HDACs shuttle between the nucleus and the cytoplasm, and some are primarily cytoplasmic proteins. At concentrations inducing growth arrest of both normal and transformed cultured cells, vorinostat causes cell death of only the transformed cells (including neuroblastoma, melanoma, leukemia, multiple myeloma and breast, prostate, lung, ovary, colon) suggesting selective and broad anti-cancer effects. As shown in , by inhibiting HDAC activity, vorinostat facilitates transcription of genes governing growth arrest, caspase-dependent apoptotic cell death and/or caspase-independent autophagic cell death of transformed cells (CitationRichon et al 2000; CitationRosato et al 2003; CitationGui et al 2004; CitationGuo et al 2004; CitationLindemann et al 2004; CitationShao et al 2004; CitationZhang et al 2005). Normal cells are up to tenfold more resistant to vorinostat-induced cell death compared with transformed cells (CitationMarks and Jiang 2005; CitationUngerstedt et al 2005; CitationZhang et al 2005). Vorinostat causes the accumulation of acetylated histones in both normal and transformed cells, and histone acetylation, as measured in an assay using peripheral mononuclear cells, has proved to be a predictive marker for HDAC inhibitory activity of the drug in vivo (CitationKelly and Marks 2005).
Figure 3 Proposed mechanism of reaction.
Zhang et al showed that vorinostat at 1–5 μM selectively causes apoptosis of CTCL cell lines and SS/MF patients’ PBL compared to healthy donors’ PBL (CitationZhang et al 2005). This difference in sensitivity to vorinostat-induced apoptosis appears not to be caused by a difference in the ability to inhibit HDAC activity because accumulation of acetylated histones occurs in both tumor and normal cells (CitationKelly et al 2005; CitationMarks and Jiang 2005; CitationUngerstedt et al 2005; CitationZhang et al 2005). Vorinostat induced tumor cell apoptosis at concentrations (1–5 μM) to which normal cells are relatively resistant (CitationZhang et al 2005). This was associated with an accumulation of acetylated histones, increased p21WAF1 and bax, decreased Stat6 and phosphor-Stat6, and activation of caspase 3 (CitationZhang et al 2005). This study agrees with the finding that romidepsin (FK228), also induces apoptosis of Hut78 CTCL cells in vitro (CitationPiekarz et al 2004). The results presented by CitationUngerstedt et al (2005) indicate that vorinostat may cause an accumulation of reactive oxygen species (ROS) and caspase activation in transformed but not normal cells as well as an increase in the level of Trx, a major reducing protein for many targets, in normal cells but not in transformed cells (CitationZhang et al 2005).
In vitro studies of CTCL cell lines revealed that vorinostat at concentrations associated with apoptosis resulted in induction of expression of p21waf1 (CitationZhang et al 2005). P21 is a cdk inhibitor responsible for cell cycle arrest and apoptosis induced by HDAC inhibitors (CitationMei et al 2004; CitationSomech et al 2004). Although upregulation of p21waf1 was demonstrated in response to vorinostat, immunoblot analysis showed that this effect was independent of the tumor suppressor p53. In MF skin lesions and cell lines in vitro, the nuclear to cytoplasmic expression of p-Stat 3 protein shifted (CitationDuvic and Zhang 2006). Stat proteins belong to a family of transcription factors that, once activated, are believed to contribute to oncogenesis by stimulating cell proliferation and preventing apoptosis (CitationSommer et al 2004; CitationTurkson 2004). They have been shown to be constitutively expressed in CTCL cell lines, and therefore are interesting targets of vorinostat action. Vorinostat at concentrations capable of producing apoptosis, decreased the expression of Stat6 and phosphor-Stat-6 (but not Stat3 and phosphor-Stat3) were seen in CTCL cell lines and peripheral blood lymphocytes from SS patients (CitationDuvic and Zhang 2006).
Phase I clinical trials for vorinostat in solid tumors and hematologic malignancies
Preclinical studies in mice and rabbits showed that a 2-hour infusion of vorinostat at the maximal administered dose of 3000 mg/m2/day for 3 days produced no drug related deaths or organ toxicities. On the basis of this and a mouse tumor xenograft model (efficacy at 150 mg/m2/day), Kelly et al at Memorial Sloane Kettering performed the first dose escalation study of intravenous vorinostat given by 2 hour IV infusion in 37 patients with advanced cancer (CitationKelly et al 2003). The starting dose of 75 mg/m2/day was escalated to 900 mg/m2/day with no dose limiting toxicities (CitationHe et al 2001). In part B of the trial, vorinostat was administered for 3 days every 21 days for solid tumors patients (n = 7) and 5 days per week for 1–3 weeks for those patients with hematological malignancies (n = 12). The maximum tolerated dose in hematologic malignancies was 300 mg/m2/day × 5 days per week for 3 weeks. Median duration of response was 6.4 weeks (range, 1.6–40 weeks). The half-life was 21–58 minutes. There was dose related increase in the AUC, and the plasma concentration exceeded 2.5 μM at all dose levels (CitationKelly 2003). Acetylated H3 histones accumulated in peripheral blood mononuclear cells and in tumor biopsies at all doses. The duration of acetylation increased with dose escalation. The drug was generally well tolerated with some fatigue and gastrointestinal symptoms noted.
In 73 patients with hematologic or solid tumors treated with oral vorinostat in Phase I (CitationKelly et al 2005), the maximum tolerated dose was defined as 400 mg daily, or 200 mg BID for continuous daily dosing, or 300 mg BID for 3 consecutive days per week. Dose limiting thrombocytopenia was reported in 87% of the patients with hematologic malignancies compared to only 44% of the patients with solid-tumors, and was more severe in the former group (CitationKelly et al 2005). There was anti-tumor activity in lymphomas and in patients with solid tumors, including thyroid, renal cell, mesothelioma, laryngeal, and urothelial carcinomas.
Thirty-five patients with hematologic malignancies were treated in a Phase I trial with either intravenous (n = 12) or oral vorinostat (n = 23) at continuous doses ranging from 200 mg/daily to 300 mg/twice daily (CitationO’Connor et al 2006). Oral vorinostat showed good oral availability and favorable pharmacokinetics. Of interest, the IV formulation gave a four fold higher C max while oral administration produced significantly higher (22 fold) AUC values. Vorinostat was active in different hematologic malignancies: Hodgkin disease, diffuse large B-cell lymphomas, and 3 patients with CTCL. One MF patient experienced a >4 month partial response. Dose limiting toxicities observed were nausea, vomiting, diarrhea, anorexia, dehydration, fatigue, and myelosuppression. Patients with hematologic malignancies appeared to be more sensitive to thrombocytopenia and myelosuppressive effects (thrombocytopenia, neutropenia, and leukopenia) (CitationO’Connor et al 2006). Thrombocytopenia with vorinostat was accompanied by hypolobular and decreased numbers of megakaryocytes, suggesting an impairment in differentiation, and recovery was observed in 3–7 days.
Dose ranging Phase II trial of oral vorinostat
In light of the favorable preclinical studies and the response of CTCL patients to oral vorinostat in Phase I, a single center, non-randomized, Phase II dose ranging trial was initiated. The purpose of the trial was to determine response rate and duration, safety, and tolerability of oral vorinostat in heavily pretreated patients with refractory CTCL (CitationDuvic et al 2007). Thirty-three patients with refractory or relapsed CTCL (stages IA–IVB) unresponsive to at least one conventional systemic therapy were enrolled into one of three dosing cohorts (CitationDuvic et al 2007). Four patients received treatment in 2 cohorts but were considered only once for efficacy.
The patients were highly pre-treated with a median of 5 prior therapies (range 1–15). Twenty-nine (83%) had advanced stage MF (>IIB) and one third had Sézary Syndrome, defined as erythroderma with blood involvement. Twenty-nine of the 33 patients had received prior chemotherapy, 22 had received bexarotene, and 14 had received denileukin diftitox (). Patients were treated sequentially with one of three oral dosing schedules and the demographics were similar among the three cohorts (). The first cohort received 400 mg daily, the second received 300 mg twice daily (for 3 days with 4 days’ rest), and the third received 300 mg twice daily for 14 days, with 7 days off followed by 200 mg twice daily for maintenance. The MF stages and response for each cohort as well as the dose modification schedule are shown in .
Table 2 Vorinostat in CTCL: Patient demographicsTable Footnotea
Table 3 Phase II oral vorinostat: intent to treat response of 24% (8/33)
Table 4 Oral vorinostat dosing and dose modification schedule
The primary efficacy end point of the study was the complete and partial response rate (CR, PR). However, the study also evaluated the time to response, time to progressive disease, response duration, pruritus relief, and safety. The response to therapy was categorized according to the World Health Organization criteria (CitationMiller et al 1981) as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). The skin involvement was calculated as body surface area (BSA) involvement with patch, plaque, or tumor disease.
Considering unique patients for an intent-to-treat analysis, 8 out of 33 or 24% of the patients achieved a documented PR defined as a 50% reduction in skin score and no increase in other disease sites. There were no complete responses. An additional 11 patients had pruritus relief, stable disease, or both such that 19 (58%) of 33 study patients received clinical benefit from the drug. Responses to vorinostat were observed in a broad spectrum of the study population, including a patient with early IA refractory MF, one patient with advanced tumors with histologic large-cell transformation, and patients with nodal and/or blood involvement (). One elderly patient with large cell lymphoma tumors had very fast response in his existing tumors, but new tumors formed while he was on therapy (). There was no difference in response rate in patients who had received prior bexarotene or not.
Figure 4 Vorinostat in CTCL: resolved large-cell tumor (*Phase 2A).
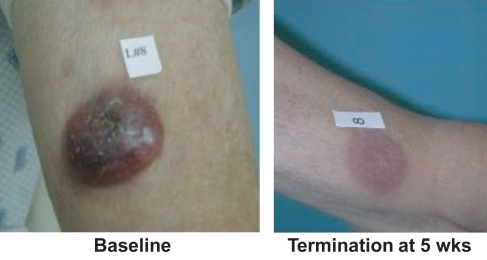
Table 5 Vorinostat in CTCL: Response ratesTable Footnotea
Secondary objectives were to determine the duration of response and to evaluate the safety and tolerability of vorinostat in these patients. The median duration of response overall and in patients with stage IIB or higher CTCL was 15.1 weeks (106 days) and ranged from 9.4 to 19.4 weeks (66–136 days). The median duration of response was numerically lowest in group 2 who received intermittent dosing (9.4 weeks [66 days]) and highest in group 1 treated with 400 mg daily (16.1 weeks [113 days]).
The most common major toxicities that were possibly or probably related to oral vorinostat therapy were fatigue and gastrointestinal symptoms, including diarrhea, altered taste, nausea, and dehydration from not eating/drinking (). Thrombocytopenia was dose limiting in cohort 3 following the higher dose of 300 mg twice daily for 14 days. Overall, the daily dose of 400 mg vorinostat orally provided the most favorable risk-benefit profile and dose of 400 mg daily was selected for evaluation in a second Phase IIb multi-center trial (CitationDuvic et al 2007; CitationOlsen et al 2007).
Table 6 Zolinza™ (vorinostat) clinical or laboratory adverse events occurring in CTCL patients (incidence ≥ 10% of patients)
Phase II B multi-center single arm trial of oral vorinostat in CTCL patients
Phase IIb was conducted as an open-label, multi-center trial of oral vorinostat at 400mg daily in patients with IB-IVA MF/SS (CitationOlsen et al 2007). Two dose modifications were allowed: 300 mg daily or 300 mg daily for 5 days per week. Safety assessments were done with NCI Common Terminology Criteria for Adverse Events v3.0. Patients were eligible with MF/SS Stage IB to IVB that was progressive, persistent, or recurrent on or following more than 2 prior systemic therapies. One must have contained bexarotene unless the patient was intolerant. Patients with ECOG performance status of 0–2, adequate hematologic, hepatic, and renal function were included. Patients with prior use of HDAC inhibitor or anticancer treatment within 3 weeks of study entry were excluded. demonstrates baseline CTCL stage prior to dosing.
Table 7 Zolinza™ (vorinostat) Study 1: Baseline patient characteristics (all patients as treated)
Approximately 30% of patients with treatment refractory advanced MF/SS > Stage IIB had at least a partial response and one patient with tumors had a complete long lasting response ( and ). The median time to response was less than 2 months. Clinically long lasting responses were observed and 15 patients stayed on the drug for over a year. Median response duration and time to progression in advanced patients (IIB or >) were not reached but were estimated to be at least 6.1 and 9.8 months, respectively. Median time to progression in all patients was 4.9 months. Vorinostat provided pruritus relief in 30% of patients including 25% of those who did not meet the criteria for objective cutaneous response.
Figure 5 Vorinostat in CTCL: stage IVA Sézary and keratoderma partial response (*Phase 2A).
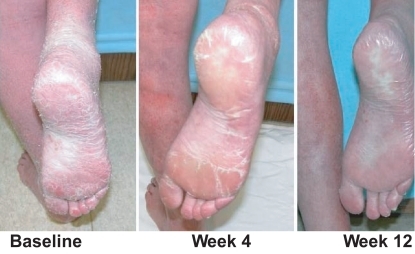
The 400 mg dose was well tolerated since less than 15% of patients (n = 10) required dose reduction. Only 11% of patients had related serious adverse events: thrombosis, anemia, thrombocytopenia, increased creatinine, gastrointestinal hemorrhage, ischemic stroke, syncope, and streptococcal bacteremia. The most common drug related adverse events were gastrointestinal symptoms (diarrhea (49%), nausea (43%), anorrhexia (26%), dysgeusia, dry mouth, vomiting, constipation, anorexia), or fatigue (46%), thrombocytopenia, weight decrease, alopecia, muscle spasms, increase in creatinine, anemia, and chills. Most adverse events were grade 2 or less. Grade 3 events included fatigue (5%), pulmonary embolism (5%), thrombocytopenia (5%) and nausea (4%). There were three deaths in the study, including one sudden death on day 2 (CitationOlsen et al 2007). Based on the Phase I and II studies, on October 6, 2006, the FDA approved a New Drug Application for vorinostat for the treatment of cutaneous manifestions of CTCL in patients who have progressive, persistent, or recurrent disease on or following two prior systemic treatments.
Pharmacokinetics and caveats
Vorinostat is 71% bound to plasma proteins over a range of 0.5 to 50 μg/mL (CitationKelly and Marks 2005). It is metabolized by glucuronidation and hydrolysis followed by B-oxidation. The two serum metabolites are not active and are excreted through metabolism. The mean half-life is about 2.0 hours for vorinostat and the O-glucuronide metabolite. It is not an inhibitor of CYP drug metabolizing enzymes but there is potential for suppression of CYP2C9 and CYP3A at concentrations above 10 μM (CitationDesai et al 2003).
Lab abnormalities seen in patients taking 400 mg in the clinical trials included increased serum glucose in 69%, transient increases in creatinine levels in 46.5%, and proteinuria in 51.4%.
Vorinostat should not be taken by nursing or pregnant women as it could harm the fetus. It was not studied in patients with hepatic or renal impairment. Patients under age 18 were not included in the trials. Patients on vorinostat should be encouraged to maintain adequate hydration to compensate for the GI symptoms.
Other HDAC inhibitors currently in clinical trials for CTCL ()
Romidepsin (depsipeptide)
Depsipeptide, (E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraaza bicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentanone, is a bicyclic pentapeptide isolated from the soil bacterium Chromobacterium violaceum. Romidepsin or depsipeptide (FK228) (Gloucester Pharmaceuticals) is a class I novel HDAC inhibitor currently in Phase II clinical trials and demonstrates clinical activity in patients. Depsipeptide has been shown to induce differentiation, cause G1 and G2/M cell cycle arrest and apoptosis, increase expression of p21 and cyclin E, decrease expression of cyclin D1 and c-myc, and cause hypophosphorylation of Rb (CitationSandor et al 2000; CitationPicardo et al 2004; CitationPiekarz et al 2004; CitationPiekarz and Bates 2004).
Table 8 HDAC-Is in clinical trials for CTCL
In a Phase I trial of intravenous depsipeptide conducted at the National Cancer Institute, 3 patients with cutaneous T-cell lymphoma had a partial response, and 1 patient with peripheral T-cell lymphoma, unspecified, had a complete response. Sézary cells isolated from patients after treatment had increased histone acetylation (CitationPiekarz et al 2001). Clinically, following treatment with depsipeptide, there was a rapid decrease of circulating Sézary cells and improvement of skin erythema and edema.
To date, over 400 cancer patients have received depsipeptide in Phase I and II studies. Ongoing clinical investigations include Phase II studies including a pivotal trial in cutaneous T-cell lymphoma and another in peripheral T-cell lymphomas. Common adverse events are myelotoxicity, asthenia, nausea, vomiting, and cardiac repolarization effects and occasional cardiac dysrhythmias including QTc prolongation. In cutaneous T-cell lymphoma patients, an objective response was observed in 10 of 28 evaluable patients, including 3 patients with complete response (CR) and 7 with partial response (PR), for an overall response rate of 36%.
PXD-101 or bellinostat
PXD-101 (CuraGen) is a low molecular weight HDAC inhibitor with a sulfonamide-hydroxamide structure. A Phase I open label, dose escalation, safety, PK, and PD study of intravenous PXD101 was conducted in patients with advanced cancer. Sequential dose cohorts of 3–6 patients were examined (150, 300, 600, 900, and 1200 mg/m2). An expanded cohort at 1000 mg/m2 was recruited for IV and oral testing. The main drug-related adverse events were fatigue, nausea, vomiting (infusion-related) and phlebitis. Other adverse events included headache, diarrhea, constipation, and dyspnea. No hematological toxicity was identified. Dose limiting toxicities of Grade 3 fatigue at 600 mg/m2, atrial fibrillation (spontaneously reversible) at 1200 mg/m2, Grade 3 diarrhea and lethargy prevented completion of a cycle at 1200 mg/m2. The presumptive maximum tolerated dose was 1000 mg/m2. Four patients had stable disease for 2, 3, 4, 5 cycles respectively. Preliminary data with oral dosing showed good tolerability. Pharmacodynamic analysis showed histone H4 hyperacetylation in peripheral blood mononuclear cells following PXD-101 intravenous administration lasting from 6–24 hours post-dose (CitationSteele and Vidal 2005).
LBH 589
LBH-589 (Novartis) is a novel HDAC Inhibitor, similar to SAHA, but with a longer half-life. There is an ongoing Phase I trial of CTCL patients with advanced stage or who had progressed following prior systemic therapy evaluating oral LBH-589 every Monday, Wednesday, and Friday at either 30 mg or 20 mg in a 28-days treatment cycle (CitationPrince et al 2006). The 30 mg dose was dose limiting. Oral LBH589 was continued until progressive disease (PD) or unacceptable toxicity. Of 10 patients enrolled to date, one was enrolled in the 30 mg cohort and nine in the 20 mg cohort. Two patients attained CRs, 4 had a PR, and 3 had SD before progressing, and one had PD. Therefore, responses were seen in 6 out of 10 patients treated with oral LBH (CitationPrince et al 2006) in the Phase I study. A Phase II clinical trial will evaluate the oral formulation in this disease indication.
Conclusion
Cancer may arise through multiple defects in the expression and/or function of proteins controlling cell proliferation, death, and migration. T-cell malignancies remain challenging to treat due to the essential role of T-cells for host immune function.
Histone deacetylase inhibitors, including vorinostat, have the potential to modulate the transcription of multiple genes and pathways, yet are selective and efficacious as anti-cancer agents. Acetylation of histones, tumor suppressors, and transcription factors are thought to underlie the ability of vorinostat and other HDAC-inhibitors to selectively induce tumor cell apoptosis and growth arrest (CitationBolden et al 2006). HDAC-I should probably be classified as biological response modifiers, along with retinoids and interferons. With different specificity assigned to each of the 18 HDACs, the potential effects of HDAC-I are as yet largely unknown.
Based on preliminary responses of CTCL patients in Phase I trials, we performed a dose ranging study of oral vorinostat in heavily pretreated CTCL patients and observed the most dramatic responses in patients with large cell transformation and Sézary syndrome. This clinical observation is supported by the selectivity of vorinostat for malignant cells in vitro. Oral vorinostat was well tolerated at a dose of 400 mg daily and had a rapid onset of action (CitationOlsen et al 2007). The drug also improved skin, nodal, and blood involvement as well as reducing pruritus. This significantly impacts quality of life in SS and MF patients. The side effects of vorinostat are well tolerated, and quickly reversible. Even in advanced CTCL patients who had limited treatment options, vorinostat was well tolerated, with fatigue and gastrointestinal symptoms being the most common side effects at lower doses. Thrombocytopenia was dose limiting at the highest doses but was reversible.
For the treatment of patients with MF and SS who have failed traditional therapies, vorinostat is an important new therapeutic option with an overall response rate of about 30%. It was active in patients resistant to chemotherapy and has the advantage of being taken orally, avoiding line sepsis that can accompany chemotherapy. Advanced MF patients often have hyper-coagulable states and caution is indicated in patients with a prior history of deep vein thrombosis or patients on warfarin therapy. Low platelets are seen at the highest concentrations but are reversible when the drug is held.
Although EKG changes including ST-T wave changes and QT prolongation were observed, they were not clinically significant. Before starting HDAC-I therapy it is advised to monitor and if necessary correct the K+ and Mg+ ion concentrations. Although there may be a class effect with respect to cardiac toxicity, there may be differences among HDAC-inhibitors.
Vorinostat is indicated for the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma (CTCL) who have progressive, persistent or recurrent disease on or following two systemic therapies.
Future direction of vorinostat
As of 7 March 2007, according to the National Institutes of Health Clinical Trials.gov website, there are 41 other clinical trials recruiting patients for vorinostat alone or in combination for a wide range of cancer types. Of these trials, 36 are recruiting patients and 5 are approved but not yet recruiting patients. Vorinostat has activity in multiple other solid and hematologic malignancies and is first in its class of HDAC-inhibitor therapy for cancer. HDAC inhibitors may prove even more effective when they are combined with other agents such as retinoids, ie, bexarotene, phototherapy and extracorporeal photophoresis with not-overlapping toxicities (). Vorinostat in combination studies with other agents such as radiation therapy, epirubicin, and fluorouracil have shown synergistic or additive effects in a variety of cancers (), so the future is very exciting and there is unlimited potential for novel approaches to be developed (CitationAlmenara et al 2002; CitationNimmanapalli et al 2003; CitationMarchion et al 2004; CitationOcker et al 2005; CitationBolden et al 2006; CitationLakshmikanthan et al 2006).
Table 9 Proposed HDAC inhibitor combination strategies in CTCL
Table 10 Vorinostat in combination studies
Disclosures
MD has received clinical and basic research support from Aton/Merck for conducting the clinical trial. MD was the recipient of salary support from NIH K24CA86815 and participated on a Scientific advisory board for Merck. JV declares no competing financial interest.
References
- AlmenaraJRosatoRGrantS2002Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA)Leukemia1613314312094258
- ApisarnthanaraxNTalpurRWardS2004Tazarotene 0.1% gel for refractory mycosis fungoides lesions: an open-label pilot studyJ Am Acad Dermatol50600715034511
- AssafCBagotMDrummerR2006Minimizing adverse side-effects of oral bexarotene in cutaneous T-cell lymphoma: an expert opinionBr J Dermatol155261616882161
- AxelrodPILorberBVonderheidEC1992Infections complicating mycosis fungoides and Sezary syndromeJAMA267135481740857
- BergerCLHanlonDKanadaD2002The growth of cutaneous T-cell lymphoma is stimulated by immature dendritic cellsBlood9929293911929784
- BlanderGGuarenteL2004The Sir2 family of protein deacetylasesAnnu Rev Biochem734173515189148
- BoldenJEPeartMJJohnstoneRW2006Anticancer activities of histone deacetylase inhibitorsNat Rev Drug Discov57698416955068
- BrenemanDDuvicMKuzelT2002Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphomaArch Dermatol1383253211902983
- ButlerLMAgusDBMarksPA2000Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivoCancer Res6051657011016644
- ButlerLMAgusDBScherHI2000Suberoylanilide hydoxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivoCancer Res6051657011016644
- ButlerLMLiapisVBouralexisS2006The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAILInt J Cancer1199445416550602
- ButlerLMZhouXXuWS2002The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxinProc Natl Acad Sci USA9911700512189205
- CohenLAAminSMarksPA1999Chemoprevention of carcinogen-induced mammary tumorigenesis by the hybrid polar cytodifferentiation agent, suberanilohydroxamic acid (SAHA)Anticancer Res194999500510697502
- CohenLAMarksPARifkindRA2002Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen-induced mammary tumorsAnticancer Res22149750412168829
- de RuijterAJvan GennipAHCaronHN2003Histone deacetylases (HDACs): characterization of the classical HDAC familyBiochem J3707374912429021
- DeethsMJChapmanJTDellavalleRP2005Treatment of patch and plaque stage mycosis fungoides with imiquimod 5% creamJ Am Acad Dermatol522758015692473
- DesaiDDasACohenL2003Chemopreventive efficacy of suberoylanilide hydroxamic acid (SAHA) against 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in female A/J miceAnticancer Res231A49950312680257
- DiwanAHPrietoVGHerlingM2005Primary Sezary syndrome commonly shows low grade cytologic appearance and absence of epidermotropismAm J Clin Pathology12351015
- DuvicMMartinAGKimY2001Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphomaArch Dermatol1375819311346336
- DuvicMZhangC2006Clinical and laboratory experience of vorinostat (suberoylanilide hydroxamic acid) in the treatment of cutaneous T-cell lymphomaBr J Cancer95Suppl 1S1319
- DuvicMTalpurRNiX2007Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL)Blood10931916960145
- FossFDemierreMFDivenutiG2005A phase-1 trial of bexarotene and denileukin diftitox in patients with relapsed or refractory cutaneous T-cell lymphomaBlood106454715811959
- FurumaiRMatsuyamaAKobashiN2002FK228 (Depsipeptide) as a natural prodrug that inhibits class I histone deacetylasesCancer Res6249162112208741
- Garcia-ManeroGYangHSanchez-GonzalezB2005Final results of a phase I study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with leukemia and myelodysplastic syndrome [abstract]Blood106785a
- GiardiMHealdPWWilsonLD2004The pathogenesis of mycosis fungoidesN Engl J Med35019788815128898
- GoreSDCarducciMA2000Modifying histones to tame cancer: clinical development of sodium phenylbutyrate and other histone deacetylase inhibitorsExp Opin Invest Drugs9292334
- GregorettiIVLeeYMGoodsonHV2004Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysisJ Mol Biol338173115050820
- GuiCYNgoLXuWS2004Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1Proc Natl Acad Sci USA1011241614734806
- GuoFSiguaCTaoJ2004Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cellsCancer Res642580915059915
- HeLZTolentinoTGraysonP2001Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemiaJ Clin Invest10813213011696577
- HeiderUKaiserMSterzJ2006Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphomaEur J Haematol76425016343270
- JackowCMCatherJCHearneV1997Association of erythrodermic cutaneous T-cell lymphoma, superantigen-positive Staphylococcus aureus, and oligoclonal T-cell receptor V beta gene expansionBlood8932408978274
- JackowCMMcHamJBFrissA1996HLA-DR5 and DQB1*03 class II alleles are associated with cutaneous T-cell lymphomaJ Invest Dermatol10737368751973
- KazakovDVBurgGKempfW2004Clinicopathological spectrum of mycosis fungoidesJ Eur Acad Dermatol Venereol1839741515196152
- KellyWKMarksPA2005Drug insight: Histone deacetylase inhibitors-development of the new targeted anticancer agent suberoylanilide development of the new targeted anticancer agent suberoylanilide hydroxamic acidNat Clin Paract Oncol21507
- KellyWKO’ConnorOAKrugLM2005Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancerJ Clin Oncol2339233115897550
- KellyWKRichonVMO’ConnorO2003Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenouslyClin Cancer Res910 Pt 135788814506144
- KimEJHessSRichardsonSK2005Immunopathogenesis and therapy of cutaneous T cell lymphomaJ Clin Invest11579881215841167
- KrakenWAWardSRDuvicM2003Bexarotene is a new treatment option for lymphomatoid papulosisDermatology206142712592082
- LainTTalpurRDuvicM2003Long-term control of mycosis fungoides of the hands with topical bexaroteneInt J Dermatol422384112653924
- LakshmikanthanVKaddour-DjebbarILewisRW2006SAHA-sensitized prostate cancer cells to TNFalpha-related apoptosis-inducing ligand(TRAIL): mechanisms leading to synergistic apoptosisInt J Cancer119221816450389
- LedentVVervoortM2006Comparative genomics of class 4 histone deacetylases indicates a complex evolutionary historyBMC Biol42416884538
- LimHWEdelsonRL1995Photopheresis for the treatment of cutaneous T-cell lymphomaHematol Oncol Clin North Am91117268522488
- LindemannRKGabrielliBJohnstoneRW2004Histone-deacetylase inhibitors for the treatment of cancerCell Cycle37798815153801
- MarchionDCBicakuEDaudAI2004Sequence-specific potentiation oftopoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilidehydroxamic acidJ Cell Biochem922233715108350
- MarchionDOBicakuEDaudAI2004Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamine acidJ Cell Biochem922233715108350
- MarksPRifkindRARichonVM2001Histone deacetylases and cancer: causes and therapiesNat Rev Cancer119420211902574
- MarksPABreslowR2007Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anti-cancer drugNature Biotechnology258490
- MarksPADokmanovicM2005Histone deacetylase inhibitors: discovery and development as anticancer agentsExpert Opin Investig Drugs141497511
- MarksPAJiangX2005Histone deacetylase inhibitors in programmed cell death and cancer therapyCell Cycle45495115738652
- McGinnisKSUbrianiRNewtonS2005The addition of interferon gamma to oral bexarotene therapy with photopheresis for Sézary syndromeArch Dermatol1411176816172331
- MeiSHoADMahlknechtU2004Role of histone deacetylase inhibitors in the treatment of cancer (Review)Int J Oncol25150915547685
- MillerABHoogstratenBStaquetM1981Reporting results of cancer treatmentCancer47207147459811
- MoradeiOMarounCRPaquinI2005Histone deacetylase inhibitors: latest developments, trends, and prospectsCurr Med Chem Anticancer Agents55296016178777
- NiXZhangCTalpurR2005Resistance to activation-induced cell death and bystander cytotoxicity via the Fas/Fas ligand pathway are implicated in the pathogenesis of cutaneous T cell lymphomasJ Invest Dermatol1247415015816832
- NimmanapalliRFuinoLStobaughC2003Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cellsBlood1013236912446442
- OckerMAlajatiAGanslmayerM2005The histone-deacetylase inhibitor SAHA potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cellsJ Cancer Res Clin Oncol1313859415754201
- O’ConnorOAHeaneyMLSchwartzL2006Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignanciesJ Clin Oncol241667316330674
- O’ConnorOA2005Developing new drugs for the treatment of lymphomaEur J HaematolSuppl 66150816004608
- OlsenEDuvicMFrankelA2001Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphomaJ Clin Oncol193768811208829
- OlsenEKimYHKuzelT2007Phase IIB. Multicenter trial of Vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphomaJ Clin Oncol618 Epub
- PicardoDAQuerfeldCGuitartJ2004Cutaneous T-cell lymphoma: a paradigm for biological therapiesLeukemia and Lymphoma4517556515223633
- PiekarzRBatesS2004A review of depsipeptide and other histone deacetylase inhibitors in clinical trialsCurr Pharm Des1022899815279609
- PiekarzRLRobeyRSandorV2001Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case reportBlood982865811675364
- PiekarzRLRobeyRWZhanZ2004T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistanceBlood10346364314996704
- PiekarzRLRobeyRWZhanZ2004T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide onmolecular markers, therapeutic targets, and mechanisms of resistanceBlood10346364314996704
- PrinceHMGeorgeDJJohnstoneR2006LBH589 a novel deacetylase inhibitor (DACi), treatment of patients with cutaneous T-cell lymphoma (CTCL): skin gene expression profiles in the first 24 hours related to clinical response following therapyPresented at 2006 ASH Annual MeetingOrlando, FL
- QuerfeldCRosenSTGuitartJ2005The spectrum of cutaneous T-cell lymphomas: new insights into biology and therapyCurrent Opinion in Hematology12273815928483
- ReddyPMaedaYHotaryK2004Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effectProc Natl Acad Sci USA1013921615001702
- RichonVMEmilianiSVerdinE1998A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylasesProc Natl Acad Sci USA95300379501205
- RichonVMSandhoffTWRifkindRA2000Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylationProc Natl Acad Sci USA97100141910954755
- RookAHGottliebSLWolfeJT1997Pathogenesis of cutaneous T-cell lymphoma: implications for the use of recombinant cytokines and photopheresisClin Exp Immunol107Suppl 116209020930
- RosatoRRAlmenaraJADaiY2003Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cellsMol Cancer Ther212738414707268
- SakajiriSKumagaiTKawamataN2005Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell linesExp Hematol33536115661398
- SandorVSenderowiczAMertinsS2000P21-dependent G1 arrest withdownregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylaseinhibitor FR901228British Journal of Cancer838172510952788
- ShaoYGaoZMarksPA2004Apoptotic and autophagic cell death induced by histone deacetylase inhibitorsProc Natl Acad Sci USA10118030515596714
- ShimakageMSasagawaTKawaharaK2001Expression of Epstein-Barr virus in cutaneous T-cell lymphoma including mycosis fungoidesInt J Cancer922263111291050
- SomechRIzraeliSSimonJ2004Histone deacetylase inhibitors – a new tool to treat cancerCancer Treat Rev304617215245778
- SommerVHClemmensenOJNielsenO2004In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3Leukemia1812889515141228
- SteeleNVidalL2005A phase 1 pharmacokinetic (PK) andpharmacodynamic (PD) study of the histone deacetylase (HDAC) inhibitor PXD101in patients (pts) with advanced solid tumoursJ Clin Oncol2316 Suppl3035
- TanRSButterworthCMMcLaughlinH1974Mycosis fungoides-a disease of antigen persistenceBr J Dermatol91607164281316
- TokuraYYagiHOhshimaA1995Cutaneous colonization with staphylococci influences the disease activity of Sezary syndrome: a potential role for bacterial superantigensBr J Dermatol1336127669641
- TurksonJ2004STAT proteins as novel targets for cancer drug discoveryExpert Opin Ther Targets84092215469392
- UngerstedtJSSowaYXuWS2005Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitorsProc Natl Acad Sci USA102673815637150
- VegaFLuthraRMedeirosLJ2002Clonal heterogeneity in mycosis fungoides and its relationship to clinical courseBlood10033697312384439
- VranaJADeckerRHJohnsonCR1999Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53Oncogene1870162510597302
- ZhangCRichonVNiX2005Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic actionJ Invest Dermatol12510455216297208