Abstract
The osmotic release oral system (OROS) methylphenidate formulation is a prolonged-release medication for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children, adolescents, and adults. We conducted a seven-week open-label extension of a double-blind study to assess the safety and tolerability of OROS methylphenidate in a flexible dose regimen (18–90 mg daily) for the treatment of adults diagnosed with ADHD (N =370). Medication was adjusted to optimize efficacy and tolerability for each patient. Adverse events, vital signs, and laboratory parameters were assessed. Most patients (337; 91%) completed the seven-week treatment and the final dispensed dose was 18 mg (8%), 36 mg (29%), 54 mg (34%), 72 mg (20%), or 90 mg (9%). Adverse events were reported in 253 (68%) patients and most were mild or moderate in severity; most frequently reported included headache (17%), decreased appetite (13%), and insomnia (11%). Adverse events were rarely serious (<1%; 2/370). Small mean increases in systolic and diastolic blood pressure (both 2.4 mmHg) and pulse (3.2 bpm) were observed. Body weight decreased slightly (−1.5 kg). The results provide additional support for the safety and tolerability of prolonged-release OROS methylphenidate in a flexible dose regimen (18–90 mg/day) for the treatment of adults with ADHD.
Introduction
There is increasing recognition of the need for treatment of attention-deficit/hyperactivity disorder (ADHD) in adults, with mounting evidence that symptoms persist beyond childhood.Citation1 Adults show similar responsiveness to methylphenidate treatment for ADHD as seen in children.Citation2–Citation12 Recent literature reviews of the safety of methylphenidate in the treatment of ADHD in adults and other conditions concluded that the medication is effective and well tolerated.Citation13,Citation14 With growing support for methylphenidate treatment of ADHD in adults, the safety and tolerability of methylphenidate formulations and regimens in treatment of ADHD in adults merit further investigation.Citation15–Citation17
The osmotic release oral system (OROS) methylphenidate formulation is designed to deliver methylphenidate in a controlled manner for approximately 12 hours, thereby allowing extended coverage of symptoms during the day. This long-acting formulation, designed for once-a-day administration, was shown to be an effective and safe treatment of ADHD in children and adolescents,Citation18–Citation20 and more recently in adults.Citation21–Citation24 This prolonged-release medication (Concerta®; McNeil Pediatrics Division of Ortho-McNeil-Janssen Pharmaceuticals, Inc, Titusville, NJ, USA) is approved for the treatment of ADHD in children and adolescents in various countries, and for the treatment of ADHD in adults in the United States and Canada.
Recently, Medori and colleaguesCitation23 reported findings from a double-blind, placebo-controlled trial in which three fixed doses of OROS methylphenidate were administered once daily over five weeks for the treatment of ADHD in 401 adults. The three dosages of medication (18, 36, 72mg) were each an effective treatment of ADHD in adults, with a safety and tolerability profile consistent with methylphenidate use in children and adolescents. The trial included a subsequent open-label treatment phase to evaluate the safety of the medication under treatment conditions more consistent with actual clinical practice. In the open-label phase, patients received a flexible dose regimen of the medication (18–90mg daily) to optimize efficacy and tolerability for each patient based on the investigators’ judgment of clinical response. Flexible dosing more closely parallels clinical practice than fixed dosing and therefore may provide additional clinically relevant findings.
We report the results of the seven-week open-label extension of the Medori and colleagues trial.Citation23 Patients who completed the double-blind phase or discontinued study medication due to poor tolerability received a flexible dose regimen of the medication (18–90 mg daily) so as to optimize efficacy and tolerability for each patient based on the investigators’ judgment of clinical response. The primary purpose of the open-label phase was to evaluate the safety and tolerability of flexible dosages of OROS methylphenidate through assessments of adverse events, vital signs, clinical laboratory tests, and physical examination.
Methods
Background
Medori and colleagues reported a double-blind phase of a trial in which adults with ADHD (18–65 years) were randomly assigned to receive OROS methylphenidate (18, 36, or 72mg/day) or placebo for five weeks. Study procedures included administration of efficacy measures, monitoring of adverse events, clinical laboratory tests (hematology, biochemistry), vital signs (supine and standing blood pressure, pulse), and physical examination at baseline and at the end of weeks 1, 3, and 5, or at early withdrawal. Patients who completed the double-blind phase or discontinued the double blind phase of the study due to poor tolerability were eligible to participate in a seven-week open-label extension during which they received a flexible dose regimen of prolonged release (PR) OROS methylphenidate (18–90 mg/day) with adjustment to optimize efficacy and tolerability for each patient based on the investigators’ judgment of clinical response.
Of the original 401 patients enrolled in the double-blind trial, 370 (92%) continued to the open-label phase of the study. Of these 370 patients, 363 (98.1%) had previously completed the double-blind study and seven (1.9%) had not due to lack of efficacy (five patients) or adverse events (two patients). The discontinuations due to adverse events included one patient in the 36 mg/day treatment group described as having ongoing delusion of reference that resolved during the treatment interruption, and a second patient in the 72 mg/day group who reported anxiety, psychomotor agitation, disturbance in attention, and irritability.
The trial was conducted at 51 investigator sites in 13 European countries (listed in Acknowledgments) from April 2005 to June 2006. Ethics Committee approval was obtained and patients gave informed consent to participate.
Patients
The trial included adult men and women with a diagnosis of ADHD according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV),Citation25 and confirmed by the Conners’ Adult ADHD Diagnostic Interview for DSM-IV (CAADID).Citation26 Other requirements for inclusion were: age 18–65 years; chronic course of ADHD symptomatology from childhood to adulthood with some symptoms present before 7 years of age, as determined by investigators following the CAADID interview; and total score of ≥24 at screening on Conners’ Adult ADHD Rating Scale (CAARS; Investigator-rated).Citation26 The Structured Clinical Interview for DSM-IV AxisI Disorders (SCID-I) was used to evaluate the presence of other comorbidities.Citation27 The diagnosis of ADHD was not made if symptoms were better accounted for by another psychiatric disorder (eg, mood, anxiety, psychotic, personality disorder). Patients were excluded if: the investigator judged they (or their child) had a history of poor response or intolerance to methylphenidate; diagnosed with any current clinically unstable psychiatric condition (eg, acute mood disorder, bipolar disorder, acute obsessive-compulsive disorder), as determined by the investigator; or diagnosed with substance use disorder (abuse/dependence) according to DSM-IV criteria within the last six months. Other exclusion criteria included family history of schizophrenia or affective psychosis; serious illnesses (eg, hepatic or renal insufficiency, significant cardiac, gastrointestinal, psychiatric, or metabolic disturbances); hyperthyroidism, myocardial infarction, or stroke within six months of screening; and history of seizures, glaucoma, or uncontrolled hypertension.
Patients were eligible to continue in the seven-week open-label extension if they still met the initial inclusion and exclusion criteria (except for criteria concerning prior treatment with methylphenidate and CAARS score ≥24) and they completed the double-blind phase or discontinued treatment after a minimum of seven treatment days. Patients who had received a stable dose of an antidepressant (except MAO inhibitors) for at least three months prior to screening could continue this treatment at the same dose during the open-label phase. The continued use of antidepressants (eg, citalopram, escitalopram) by only a small number of patients (3.5%) was not expected to influence the assessment of OROS methylphenidate treatment. Patients treated at German or Spanish centers who discontinued the double-blind phase due to poor tolerability were not permitted to enter the open-label phase due to Ethics Committee requirements.
Procedure
Patients who entered the open-label extension received flexible dosages of OROS methylphenidate (18, 36, 54, 72 or 90mg/day) for seven weeks. Patients began with 36 mg/day and were titrated to the most appropriate OROS methylphenidate dose to preserve the blinding of the treatment received in the double-blind phase. The 36mg/day dosage was considered an appropriate starting dose to maintain some efficacy for patients who were receiving 72 mg/day OROS methylphenidate during the double-blind phase, while avoiding tolerability issues for those patients who received 18 mg/day or placebo during the double-blind phase. Patients at German centers began with 18mg/day due to Ethics Committee requirements. Titration of dosing was based on clinical observations of response and tolerability. The dosage could be increased by 18mg increments to improve efficacy, to a maximum of 90 mg/day, or decreased by 18 mg increments to improve tolerability. Investigators were encouraged to wait at least seven days to change the dosage.
The final visit of the double-blind phase was considered the baseline visit for the open-label phase, and included administration of efficacy measures, clinical laboratory tests (hematology, biochemistry), vital signs, physical examination, and confirmation that the patient met the required inclusion and exclusion criteria. Study visits during the open-label phase occurred at the end of weeks 1, 3, and 7 (or early termination); procedures included vital signs, concomitant medication review, adverse event monitoring, dispensing of study medication, and study drug accountability and dosing compliance, and the CAARS efficacy measure. The final visit also included clinical laboratory tests (hematology, biochemistry) and physical examination. Designated time intervals after first intake of medication was defined as week1 (1–13 days), week 3 (14–31 days), or week 7 (≥32 days) during the open label phase. A post-study visit occurred one week after the last dose of study medication.
Assessments
The safety and tolerability of OROS methylphenidate treatment were addressed via assessments of adverse events (using the Medical Dictionary for Regulatory Activities),Footnotea clinical laboratory tests, vital signs, and physical examination. A treatment-emergent adverse event was defined as any sign, symptom, syndrome, or new illness that appeared or worsened during the open-label phase, and included laboratory findings or results of other diagnostic procedures considered medically important. Adverse events were recorded throughout the open-label phase, along with action taken (none, dose reduced, drug stopped temporarily, drug stopped permanently). Based on standard definitions, the investigator judged the severity of an adverse event (mild, moderate, or serious) and relationship to the study drug (not related, doubtful, possible, probable, very likely). The evaluation of the safety data included all subjects who took at least one dose of study medication during the open-label phase. The incidence of adverse events was summarized overall and by daily dose over weeks 1, 3, and7. Descriptive statistics were computed for each laboratory analyte, vital sign, and weight measurements.
An efficacy analysis evaluated the change in CAARS total score from end of double-blind treatment (baseline) at each visit (last observation carried forward) in patients who received at least one dose of medication and had at least one post-baseline efficacy measurement during the open-label phase. Within-group statistical testing for change from double-blind end point was performed using a two-sided paired t-test. All statistical analyses were performed using SAS (Version 8.02; SAS Institute, Cary, NC, USA).
Results
Patient disposition
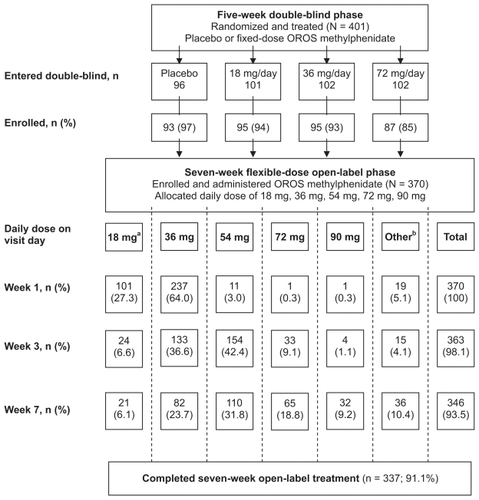
The flow of patients from the double-blind phase through the open-label phase of the trial is presented in . A total of 370 patients enrolled in the open-label phase and were administered study drug. Of these patients, 337 (91.1%) completed the seven-week treatment period. Eighteen (4.9%) patients permanently discontinued treatment due to one or more adverse events and 15 (4%) patients discontinued for other reasons (lost to follow-up [5], noncompliance [4], lack of efficacy [1], and other [5]).
Figure 1 Flow of patients from the double-blind phase through the open-label phase of the trial.
Abbreviation: OROS, osmotic release oral system.
Patients in the open-label phase did not differ in demographic and clinical characteristics from those randomized in the double-blind phase, which have been described elsewhere.Citation23 For the 370 patients in the open-label phase, median age was 35 years (M =34.3, SD =10.29), median weight was 77kg (M =78.0, SD =17.13), 54% were male, and 98% were Caucasian. Median age at diagnosis of ADHD was 32 years (range < 1–63), with the majority (74%) having documented ADHD combined subtype as a child (adult diagnosis). Currently active and stable psychiatric comorbidities in the study population included mood and anxiety disorders in 12% of patients and personality disorders in 1% of patients.
Dosage and exposure
Mean duration of exposure was 47.6 days (range 2–90), with a mean average daily dose (excluding no dose days) of 47.5 mg, mean minimum dose of 29.4 mg, and mean maximum daily dose of 57.6 mg (). The mean daily dose at weeks 1, 3, and 7 was 36.4 mg, 49.2 mg, and 52.4 mg, respectively. The most frequently used daily dose was 36 mg at week 1 (64%), 54mg at week 3 (42%), and 54 mg at week 7 (32%). The 54 mg daily dose was most frequently administered as a last dose (34%) and maximum dose (36%). The other daily dosages, 18 mg, 36 mg, 72 mg and 90 mg, were administered as a last dose to 8%, 29%, 20%, and 9% of the patients, respectively.
Table 1 Study medication administered during the open-label phase
Adverse events
Adverse events are summarized in for the 370patients. Overall, 253 (68%) patients experienced at least one treatment-emergent adverse event. The most frequently reported adverse events were headache, decreased appetite, and insomnia, and the highest incidence was for the category nervous system disorders (35%), psychiatric disorders (23%), and gastrointestinal disorders (21%). The most frequently occurring adverse events considered by the investigator as at least possibly related to study medication by the investigator were decreased appetite (12%), headache (9%), and insomnia (9%). The majority of the adverse events were mild or moderate in severity. Two patients experienced a serious adverse event during open-label treatment. No deaths occurred in this trial.
Table 2 Adverse events during the open-label phase
The incidence of adverse events by daily dose over weeks 1, 3, and 7 is provided in . The incidence (%) of adverse events decreased for all five dosages over the seven-week treatment period. The most frequently occurring adverse events recorded at the final treatment visit (week 7) according to the patient’s daily dose at this visit are presented in .
Table 3 Percent of patients who reported an adverse event by daily dose and treatment period
Table 4 Adverse events by daily dose at final treatment visit
Serious adverse events
Two patients reported a serious adverse event during open-label treatment. A 40-year-old man started at a daily dose of 18 mg that was increased to 36 mg after one week, and 22 days later, he was admitted to the hospital due to acute psychological stress. Treatment was stopped 19 days after admission, the patient was discharged two days later, and acute psychological stress resolved five days later. The investigator assessed the event as moderate in severity and not related to study medication. A second man (age 46) reported a foreign body in the urethra, and after it was removed, the patient was considered recovered and was discharged from hospital. The investigator did not consider the adverse event related to study drug.
Two patients reported serious adverse events during the post-study period. Five days after her last dose of medication (54mg daily), a 27-year-old woman experienced an increase in anxiety and was hospitalized in a psychiatric unit. Seven days later, the patient recovered from the event and was discharged from hospital. The investigator judged the event as severe and not related to trial medication. A 27-year-old man experienced temporal arteritis and severe headache six days after his last study medication (90 mg daily) and was hospitalized for less than 12 hours. The events lasted approximately two hours and were considered serious (ie, requiring hospitalization) and considered doubtfully drug related by the investigator.
Discontinuations due to adverse events
Seventeen (5%) patients permanently discontinued trial medication due to one or more adverse events. These adverse events included upper abdominal pain, decreased weight, decreased appetite, depressed mood and depression each reported by two patients. The onset of these adverse events occurred at different daily dosages, with three events at 18 mg, 13 at 36 mg, seven at 54 mg, and two at 72 mg. Patients recovered from these adverse events, except for a report of depressed mood in one patient and decreased weight in another patient. The daily dosage was 36 mg at the onset of these two adverse events. The investigators considered decreased weight as moderate and probably related to the trial medication, and depressed mood as mild and doubtfully related to the trial medication.
Laboratory values
Abnormal laboratory values were infrequently reported as adverse events. There were no discontinuations due to laboratory-related adverse events and none of these adverse events were reported as serious or severe, except for one patient with an abnormally increased creatine phosphokinase level. The event was considered not drug-related and resolved three weeks after completion of the study. There were no other clinically notable mean changes in laboratory values over time.
Cardiovascular-related effects
Small mean increases in systolic and diastolic blood pressure and pulse were observed during the open-label phase. The mean (SD) increase in blood pressure from the end of double-blind treatment to the end of open-label was 2.4 (14.9) mmHg for systolic and 2.4 mmHg (9.5) diastolic (measured while patient was standing). The increase in standing pulse was 3.2 (14.4) beats per minute (bpm). These modest changes were not considered clinically significant.
Changes in blood pressure and pulse during open label treatment were evaluated for patients who received placebo versus OROS methylphenidate during the double-blind phase. In placebo-treated patients, mean (SD) blood pressure changed from 125.6 (16.6) to 128.2 (16.6) mmHg for systolic and from 80.3 (11.0) to 84.2 (10.6) mmHg for diastolic from the end of double-blind treatment to the end of open-label. The corresponding changes were similar in OROS methylphenidate-treated patients; 124.4 (14.3) to 126.7 (15.5) mmHg and 80.5 (10.7) to 82.9 (10.6) mmHg. Pulse increased from 80.2 (13.3) to 87.4 (14.3)bpm in placebo-treated and 84.6 (14.3) to 87.4 (13.7)bpm in OROS methylphenidate-treated patients.
Clinically relevant criteria for elevated/increased systolic (≥ 140mmHg) or diastolic (≥90mmHg) blood pressure, or pulse (≥90 bpm) were predefined. The number (%) of patients who met these criteria at the last treatment visit (week 7) are presented by the patient’s daily dose at this visit in . For blood pressure and pulse, the percentage of patients who met these criteria appears similar across the lower three doses (18, 36, 54 mg), with a trend for a larger percentage at the higher two doses (72 and 90 mg). The percentage of patients who met these criteria before entering the open-label phase varied across their double-blind treatment group: systolic (13%–21%), diastolic (14%–25%), and pulse (22%–41%).
Table 5 Patients who met clinically relevant criteria for cardiovascular-related measurements by daily dose at final treatment visit
One patient discontinued trial medication due to raised blood pressure (systolic 125 mmHg and diastolic 90mmHg), which the investigator assessed as moderate in severity and probably drug-related. A second patient discontinued trial medication due to worsening of high blood pressure and paraesthesia. High blood pressure was reported at baseline of the double-blind phase (systolic 142mmHg and 94 diastolic mmHg) and increased at the time of discontinuation of the open-label phase (systolic 184mmHg and diastolic 103mmHg). The investigator assessed worsening of high blood pressure and paraesthesia feeling in right arm as moderate and mild in severity and very likely and possible related to study medication, respectively.
Tachycardia was reported as an adverse event in 10 patients (4%) and 12 patients (3%) experienced palpitations. One patient experienced tachycardia and discontinued trial medication (36 mg daily) because of this adverse event. The investigator considered the adverse event as mild in severity and probably drug-related.
Body weight
Decreased weight was reported as an adverse event for seven subjects. One patient discontinued study medication because of decreased weight; a second patient discontinued trial medication because of decreased weight and decreased appetite; and a third patient reported decreased appetite as reason for discontinuing trial medication, along with other adverse events. There was a small mean (SD) decrease in weight of −1.5kg (2.29) over the seven-week treatment period.
Efficacy assessment
The primary efficacy measure during the open-label phase was the mean change in CAARS total score from the end of double-blind treatment, which was assessed using within-group two-sided paired t-tests. Patients who received placebo in the double-blind phase showed improvement with a significant mean decrease (M = −3.5, SD = 8.49) in CAARS total score after one week of treatment in the open-label phase, and their total score further decreased at week 3 (M = −6.7, SD = 8.79) and week 7 (M = −8.5, SD = 9.65) (p values < 0.001). Patients treated with OROS methylphenidate during double-blind phase showed a small improvement at week 1 (M = −0.6, SD = 7.96) and a larger significant decrease in their total score at week 3 (M = −4.3, SD = 8.21) and week 7 (M = −6.5, 9.31) (p values < 0.001 at week 3 and 7).
Discussion
This seven-week open-label, flexible dosage extension of a five-week double-blind, fixed dose study provided further support for the safety and tolerability of OROS methylphenidate in a flexible dose regimen (18–90mg daily) for the treatment of adults diagnosed with ADHD. The flexible dosing more closely parallels clinical practice than fixed dosing and consequently provides additional clinically relevant findings. Adverse events seldom lead to discontinuation of study medication and overall there was a very low discontinuation rate. The adverse events most frequently reported by patients included headache, decreased appetite, and insomnia. Adverse events were rarely serious. There were small increases in systolic and diastolic blood pressure and pulse.
The safety findings are comparable to other open-label studies of OROS methylphenidate in the treatment of ADHD in adults, although previous studies treated a smaller number of patients. Biederman and colleagues reported a six-week study in 36 adults with late-onset ADHD, Fallu and colleagues treated 30 adults with ADHD for 38 days, and Ramos-Quiroga and colleagues treated 70 adults with ADHD for 90 days, and all studies observed that the medication was well tolerated.Citation28–Citation30 With a much larger sample of 370 patients, the open-label findings reported herein significantly enhances our confidence that prolonged-release OROS methylphenidate is a well tolerated therapy for the treatment of adults with ADHD.
The seven-week open-label safety findings were generally consistent with the five-week fixed-dose double-blind phase of the trial.Citation23 The small increases in systolic and diastolic blood pressure and pulse during the open-label phase were comparable to the double-blind phase. Medications used to treat ADHD including methylphenidate compounds are associated with small increases on heart rate and blood pressure, but the changes are usually modest with no clinically significant changes.Citation13,Citation14 As discussed in guidelines for the use of these medications for the treatment of ADHD in children and adults, the potential cardiovascular effects are generally considered acceptable and manageable and can be monitored as the physician feels necessary.Citation9,Citation31 As well, all drug products for the treatment of ADHD provide patient medication guides to inform about possible cardiovascular effects associated with of the medication.Citation15
As discussed by Godfrey and Rostain, when choosing a medication for treating adults with ADHD, important considerations are the control of symptoms throughout the working day with a regimen that may enhance adherence, and a formulation that can minimize the possible misuse of the medication.Citation13,Citation14 The osmotic controlled-release technology of the once-daily OROS methylphenidate formulation achieves a rapid onset of effect and delivers methylphenidate in a controlled manner for approximately 12 hours, thereby allowing extended coverage of symptoms during the day.Citation19,Citation32 In addition, the physical properties of the OROS mechanism of delivery reduce the risk of possible methylphenidate substance abuse.Citation33
In this open-label study, investigators adjusted the dose to optimize efficacy and tolerability for each patient based on their judgment of clinical response. The majority of patients (85%) received 36 mg, 54 mg, or 72 mg as their last daily dose; the 54 mg dose was most frequently administered as a last dose and maximum dose. For the three most frequently administered dosages, the incidence of adverse events decreased over time, suggesting that investigators were adjusting the dosage to optimize tolerability. However, this decrease may reflect, at least partially, longer exposure to the medication and increased tolerance in the patients. Along with the decrease in adverse events, patients continued to show improvements on the measure of efficacy (ie, decreases in CAARS total score) during the open-label phase. This result, along with the safety findings, suggests that investigators were able to optimize the daily dosage for a patient to achieve a balance between maintaining efficacy and minimizing adverse events.
There are a number of limitations associated with the reported study. The results covered a seven-week treatment period and did not provide information on the long-term safety of OROS methylphenidate treatment of ADHD in adults. Patients were not randomized which may limit the overall conclusions. Sample size was relatively large compared to other studies but not of the magnitude required to evaluate the possibility of rare adverse events. Patients with other Axis I disorder or patients with a high cardiovascular risk profile were excluded and there was limited ethnical/racial diversity. Concomitant psychotherapy or other therapeutic options were not administered to improve the treatment effect.
In conclusion, the seven-week open-label results provided additional support for the safety and tolerability of prolonged-release OROS methylphenidate in a flexible dose regimen for the treatment of adults diagnosed with ADHD. Moreover, the findings provided further guidance for usual medical care for the treatment of ADHD in adults in that most patients have the optimal trade-off between efficacy and absence of side effects with dosages of 36, 54, or 72 mg/day.
Acknowledgments
This study was supported by Janssen Pharmaceutica NV, Belgium. Susan Glasser, PhD, of Johnson and Johnson Pharmaceutical Research and Development, LLC, provided editorial review. The following investigators enrolled patients in the study: Czech Republic: Ceskova, Eva, MD; Raboch, Jiri, MD; Denmark: Arngrim, Torben, MD; Brødsgaard, Mogens Anders, MD; Erenbjerg, Ane-Marie, MD; Nicholson, Klavs, MD; Wernlund, Hans Henrik, MD; Finland: Henttonen, Antti Juhani, MD; Korkeila, Jyrki, MD; Niemelä, Asko Aukusti, MD; Sorvaniemi, Marko Petri, MD; France: Bouvard, Manuel, MD, PhD; Konofal, Eric, MD; Germany: Colla, Michael, MD; Gastpar, Markus, MD; Heinz, Andreas, MD; Imhof, Lothar, MD; Klein, Martin, MD; Krause, Johanna, MD; Lee, Sun-Hee, MD; Niemczyk, Wolfgang, MD; Nissen, Thomas, MD; Philipsen, Alexandra, MD; Rösler, Michael, MD; Sobanski, Esther, MD; Trott, Götz-Erik, MD; Greece: Christianopoulos, Kriton, MD; Koumoula, Anastasia, MD; Soldatos, Constantin, PhD, MD; Netherlands: Buitelaar, Jan, MD, PhD; Kooij, Sandra, MD, PhD; Norway: Auglænd, Odd, MD; Hustoft, Hilde, MD; Nyrerød, Hans Jørgen, MD; Portugal: Ferreira, Luis, MD; Filipe, Carlos, MD ; Spain: Casas, Miguel, MD, PhD; Sweden: Ginsberg, Ylva, MD; Guldberg-Kjär, Niels, MD; Lindström, Eva, Assistant PhD MD; Maahr, Eija, MD; Woxler, Per, MD; Switzerland: Eich, Dominique, MD, MD; Grossenbacher, Jürg, MD; Hofecker Fallahpour, Maria, MD; United Kingdom: Adamou, Marios, MD; Kumar, Vinod, MD; Rogers, Danny, MD.
Disclosures
Bradford Challis, PhD, is a full-time employee of Johnson & Johnson Pharmaceutical Research and Development, LLC, and Dr Medori was a full-time employee of Janssen-Cilag. Joachim Dejonckheere, PhD, is a consultant of SGS Life Sciences and provided statistical services under a contract with Janssen-Cilag. Drs Buitelaar, Casas, Kooij, and Ramos-Quiroga have served as consultants for Janssen-Cilag.
Notes
a MedDRA®: international medical terminology developed under the auspices of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).
References
- FeifelDCommentary: why diagnose and treat ADHD in adults?Postgrad Med20081203131518824821
- AshersonPClinical assessment and treatment of attention deficit hyperactivity disorder in adultsExpert Rev Neurother20055452553916026236
- AshersonPChenWCraddockBTaylorEAdult attention-deficit hyperactivity disorder: recognition and treatment in general adult psychiatryBr J Psychiatry20071904517197649
- FaraoneSVBiedermanJMickEThe age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studiesPsychol Med20053517
- FayyadJDe GraafRKesslerRCross-national prevalence and correlates of adult attention-deficit hyperactivity disorderBr J Psychiatry200719040240917470954
- KesslerRCAdlerLBarkleyRThe prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey ReplicationAm J Psychiatry2006163471672316585449
- KesslerRCAdlerLABarkleyRPatterns and predictors of attention-deficit/hyperactivity disorder persistence into adulthood: results from the national comorbidity survey replicationBiol Psychiatry200557111442145115950019
- GreenhillLLPliszkaSDulcanMKSummary of the practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adultsJ Am Acad Child Adolesc Psychiatry200140111352135511699811
- NuttDJFoneKAshersonPEvidence-based guidelines for management of attention-deficit/hyperactivity disorder in adolescents in transition to adult services and in adults: recommendations from the British Association for PsychopharmacologyJ Psychopharmacol2007211104117092962
- KooijJJBuitelaarJKvan den OordEJFurerJWRijndersCAHodiamontPPInternal and external validity of attention-deficit hyperactivity disorder in a population-based sample of adultsPsychol Med200535681782715997602
- KooijJJBurgerHBoonstraAMVan der LindenPDKalmaLEBuitelaarJKEfficacy and safety of methylphenidate in 45 adults with attention-deficit/hyperactivity disorder. A randomized placebo-controlled double-blind cross-over trialPsychol Med200434697398215554568
- BanaschewskiTCoghillDSantoshPLong-acting medications for the hyperkinetic disorders. A systematic review and European treatment guidelineEur Child Adolesc Psychiatry200615847649516680409
- GodfreyJSafety of therapeutic methylphenidate in adults: a systematic review of the evidenceJ Psychopharmacol200923219420518515459
- RostainALAttention-deficit/hyperactivity disorder in adults: evidence-based recommendations for managementPostgrad Med20081203273818824823
- US Food and Drug AdministrationFDA Directs ADHD Drug Manufacturers to Notify Patients about Cardiovascular Adverse Events and Psychiatric Adverse EventsRockville, MDNational Press Office2212007 Press0726
- NissenSEADHD drugs and cardiovascular riskN Engl J Med2006354141445144816549404
- OkieSADHD in adultsN Engl J Med2006354252637264116790695
- PelhamWEGnagyEMBurrows-MacleanLOnce-a-day Concerta methylphenidate versus three-times-daily methylphenidate in laboratory and natural settingsPediatrics20011076E10511389303
- SwansonJGuptaSLamADevelopment of a new once-a-day formulation of methylphenidate for the treatment of attention-deficit/hyperactivity disorder: proof-of-concept and proof-of-product studiesArch Gen Psychiatry200360220421112578439
- WolraichMLGreenhillLLPelhamWRandomized, controlled trial of oros methylphenidate once a day in children with attention-deficit/hyperactivity disorderPediatrics2001108488389211581440
- AdlerLAZimmermanBStarrHLEfficacy and safety of OROS methylphenidate in adults with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, double-blind, parallel group, dose-escalation studyJ Clin Psychopharmacol200929323924719440077
- BiedermanJMickESurmanCA randomized, placebo-controlled trial of OROS methylphenidate in adults with attention-deficit/hyperactivity disorderBiol Psychiatry200659982983516373066
- MedoriRRamos-QuirogaJACasasMA randomized, placebo-controlled trial of three fixed dosages of prolonged-release OROS methylphenidate in adults with attention-deficit/hyperactivity disorderBiol Psychiatry2008631098198918206857
- ReimherrFWWilliamsEDStrongREMestasRSoniPMarchantBKA double-blind, placebo-controlled, crossover study of osmotic release oral system methylphenidate in adults with ADHD with assessment of oppositional and emotional dimensions of the disorderJ Clin Psychiatry20076819310117284136
- American Psychiatric AssociationDiagnostic and Statistical Manual of Mental DisordersFourth Edition, Text RevisionWashington, DCThe American Psychiatric Association2000
- ConnersCErhardtDSparrowEConners’ Adult ADHD Rating Scales (CAARS): Technical ManualNorth Tonawanda, NYMulti-Health Systems1999
- FirstMSpitzerRGibbonMWilliamsGStructured Clinical Interview for DSM-IV Axis I Disorders, Patient Edition (SCID-P), Version 2New York, NYNew York State Psychiatric Institute, Biometrics Research1994
- BiedermanJMickESpencerTAn open-label trial of OROS methylphenidate in adults with late-onset ADHDCNS Spectr200611539039616641844
- FalluARichardCPrinzoRBinderCDoes OROS-methylphenidate improve core symptoms and deficits in executive function? Results of an open-label trial in adults with attention deficit hyperactivity disorderCurr Med Res Opin200622122557256617166338
- Ramos-QuirogaJABoschRCastellsXEffect of switching drug formulations from immediate-release to extended-release OROS methylphenidate : a chart review of Spanish adults with attention-deficit hyperactivity disorderCNS Drugs200822760361118547128
- VetterVLEliaJEricksonCCardiovascular monitoring of children and adolescents with heart disease receiving stimulant drugs: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young Congenital Cardiac Defects Committee and the Council on Cardiovascular NursingCirculation2008117182407242318427125
- ModiNBLindemulderBGuptaSKSingle- and multiple-dose pharmacokinetics of an oral once-a-day osmotic controlled-release OROS (methylphenidate HCl) formulationJ Clin Pharmacol200040437938810761165
- BuksteinOSubstance abuse in patients with attention-deficit/hyperactivity disorderMedscape J Med20081012418324334