Abstract
This paper is an overview of the diagnosis, differential diagnosis and cellular and molecular mechanisms of glucocorticoid resistant asthma. It addresses the clinical definition and rationale for the diagnosis of therapy resistant asthma. It purports that, since glucocorticoid resistant asthmatics are not globally physiologically glucocorticoid resistant, then the phenomenon is most likely acquired, probably in immune cells (and most probably in T cells and monocyte/macrophages), as a result of local inflammatory and environmental influences. The molecular mechanisms which have been uncovered to date which could account for glucocorticoid resistance are discussed, in particular the roles of AP-1 and p38 MAP kinase signaling, the role of the β-isoform of the glucocorticoid receptor and the role of histone proteins and DNA folding. Finally, there are suggestions for clinical management of these patients based on accumulated evidence.
Introduction
Glucocorticoids are very effective therapy for asthma and numerous studies have shown that they reduce asthma symptoms, exacerbations and bronchial hyperresponsiveness. The benefit/risk ratio of glucocorticoid therapy was enhanced further in the 1960’s when topical delivery devices were introduced. Inhaled glucocorticoids are now the first line therapy in patients with all but the mildest disease.
The vast majority of asthmatics are controlled satisfactorily with regular inhaled glucocorticoids with or without the addition of short- or long-acting bronchodilators. In such patients, particularly those on low to moderate dosages of inhaled glucocorticoids, it is hard to conceive of a safer or more effective therapy.
Unfortunately, however, a proportion of patients develop severe disease which is relatively or totally refractory to glucocorticoid therapy. The possible etiology of this condition will be discussed in subsequent paragraphs, but the existence of the phenomenon implies that asthma is heterogeneous in terms of its susceptibility to inhibition by glucocorticoids, and possibly therefore also in terms of its etiology.
While the percentages of patients with glucocorticoid resistant asthma are small, these patients consume a significant proportion of medical resources in terms of both time and money (CitationBuist 1995). Regardless of costs, there is an urgent need to provide alternative therapies for these patients, who often have severely impaired quality of life not only from the severity of their symptoms but from the effects of excessive glucocorticoid exposure.
Difficult asthma
Definition
Although any clinician managing asthmatic patients will have a clear picture of “difficult to treat” disease, it is in fact extraordinarily difficult to define this condition accurately, particularly for the purposes of research.
Definition of “difficult asthma” could include the following:
Confirmation of the diagnosis of asthma
An unusually poor response to therapy
Chronicity of the problem.
Problems arise with all of these concepts. Obviously it is essential to be as certain as possible of a primary diagnosis of asthma. This diagnosis of course rests on a clinical history of typical symptoms and physiological evidence of variable and reversible airways obstruction. Often, however, on presentation to a specialist, it may not be possible to obtain contemporary evidence of these features if the patient is already established on therapy. Clearly it is essential to search for and eliminate other possible alternative or associated diagnoses (). Allergic bronchopulmonary aspergillosis and pulmonary eosinophilic syndromes (for example, pulmonary eosinophilia or Churg-Strauss syndrome) may be considered unique diseases which encompass some of the clinical features of asthma and which are often difficult to treat: these are probably best considered outside the definition of “difficult” asthma. Vocal cord dysfunction characterized by paradoxical adduction of the vocal cords may masquerade as and co-exist with asthma (CitationNewman et al 1995). Other respiratory conditions such as chronic bronchitis or bronchiectasis may also co-exist with asthma, but there are very few studies assessing the effect of these in the long term on asthma severity or control.
Table 1 Diagnoses that may masquerade as difficult/therapy-resistant asthma
“Resistance to therapy” is usually identified as failure of asthma “control” despite therapy. This does, however, imply that all patients who are resistant to glucocorticoid therapy will necessarily have severe disease, which may not be the case. It also begs the question of what is used to define asthma “control”. This is usually assessed in terms of symptoms and requirement for short-acting β2-agonist medication. Patients vary in their perception of airflow limitation and poor perceivers may be particularly prone to severe attacks (CitationKikuchi et al 1994). Other techniques commonly used to define asthma “control” include objective monitoring of airways obstruction and the numbers of exacerbations experienced by patients defined according to various criteria (such as, for example, those requiring systemic glucocorticoid therapy). The international guidelines (Global Initiative for Asthma 1995) define asthma severity in terms of airways obstruction as measured by PEF or FEV1. Nevertheless, the presence of persistent airways obstruction does not necessarily reflect loss of asthma “control”. The value of using asthma biomarkers to assess the amount of airways inflammation and the relationship of these measurements to asthma control is also currently under close scrutiny. There is little evidence to date, however, that short-term measurements of biomarkers can predict the severity and chronicity of symptoms. Consequently, the concept of “difficult asthma” at present requires our third dimension of chronicity of observation. Increasingly, the amount of inhaled glucocorticoid and other therapy needed for the control of asthma, rather than quantification of symptoms or of lung function is being used to define asthma severity (CitationCockcroft and Swystun 1996).
A number of factors may contribute to poor asthma “control”, and must be taken into account before labeling the patient as glucocorticoid refractory or resistant (). Since response to therapy is part of the definition of “difficult” asthma, the question of patient adherence to therapy must be considered. Compliance with inhaled glucocorticoid therapy in asthma has been reported to be very poor (CitationKelloway et al 1994; CitationRobinson et al 2003) and it might be expected, although there is actually little hard evidence (CitationCochrane 1992; CitationMilgrom et al 1996) that this contributes to poor asthma control. Insufficient therapy is probably a much bigger contributor to poor asthma control worldwide (CitationRabe et al 2004). Psychosocial factors, which may be linked with or compounded by poor patient compliance and lack of appropriate medical care (CitationMiller and Strunk 1989; CitationWareham et al 1993; Chung et al 1999; CitationAmerican Thoracic Society 2000) have also been implicated in poor asthma control and asthma deaths. Incorrect diagnosis of respiratory symptoms as asthma is a cause of apparent poor responsiveness to asthma therapy, and this is not as uncommon as might be expected, even in specialist asthma centres (Robinson et al 2000). Although a precise mechanistic link between gastro-esophageal reflux and a decline in asthma control is not established, varying degrees of improvement in asthma have been observed when concomitant gastro-esophageal reflux has been treated (CitationSpaulding et al 1982). Targeted treatment of rhinosinusitis, often present concurrently with asthma, can lead to specific improvement in asthma control (Corren et al 1982). Although attention to these comorbid factors seems pertinent, there is little evidence that they exert a global impact on the response of asthmatics to therapy: indeed one study (CitationHeaney et al 2003), although showing a prevalence of comorbid conditions in patients with poorly controlled asthma, failed to show that such conditions were more prevalent in asthmatics who responded poorly to glucocorticoids. Moreover, targeted treatment of identified comorbidities barely altered asthma-related quality of life in those patients with glucocorticoid-insensitive disease.
Table 2 Factors that may be responsible for poor asthma control Incorrect diagnosis
Clinical and pathophysiological phenotypes of “difficult” asthma
Studies attempting to delineate clinical phenotypes of “difficult” asthma are ongoing in the United States and Europe (CitationENFUMOSA 2003; CitationDolan et al 2004; CitationMiller et al 2005). The ENFUMOSA study (CitationENFUMOSA 2003) showed that severe asthmatics were more likely to be female, more likely to be aspirin sensitive and less likely to be atopic. Although some clinical subgroups such as “brittle” asthmatics and glucocorticoid resistant asthmatics are recognized as being difficult to treat, these studies have not yet provided a viable framework upon which to predict responsiveness to therapy on clinical grounds.
Attempts have also been made to delineate pathophysiological features of asthma which predict a poor response to therapy. The problem with such studies in severe asthmatics is that it is difficult to discriminate a priori abnormalities from the effects of the high dosages of glucocorticoid and other therapies that these patients are inevitably receiving. Much circumstantial evidence implicates eosinophils in asthma pathogenesis, although as with all inflammatory leukocytes the precise mechanisms by which these cells cause the clinical features of asthma, and why these mechanisms might be therapy resistant, remain unclear. One study described a group of apparently therapy resistant severe asthmatics with high numbers of airways eosinophils (CitationSilkoff et al 2005), but there is evidence (Citationten Brinke et al 2004) that such patients may respond if treated with sufficiently high dosages of glucocorticoid. On the other hand, Wenzel and colleagues (CitationWenzel et al 1997, Citation1999) have shown that it is possible to delineate groups of severe, glucocorticoid dependent asthmatics with numbers of airways eosinophils within the range observed in normal controls. One of these studies suggested that these patients also had increased airways neutrophils, while the other did not. The airways changes associated with asthma which are collectively termed “remodeling” (lay down of new proteins, mucous hyperplasia, neovascularisation, smooth muscle hypertrophy/hyperplasia) could conceivably be resistant to therapy and cause irreversible airways blockage, although this has never been formally demonstrated. Clinically, patients with severe asthma and irreversible airways obstruction show tomographic abnormalities suggestive of remodeling (CitationBumbacea et al 2004). Whether remodeling is caused by inflammatory cells or cytokines, or both, is still not clear. In animals, over expression of the cytokine IL-13 alone in the airways reproduced all the features of airways remodeling (CitationKibe et al 2003); glucocorticoid administration abolished the associated cellular infiltrate but not the remodeling changes. In human studies, anti-IL-5 therapy was shown to reduce lay down of extracellular matrix proteins in the lungs while partly but not completely abolishing the infiltration of eosinophils (CitationFlood-Page et al 2003), leaving the question open whether or not leukocytes such as eosinophils play an indispensable role in this process. It has been suggested that one etiological factor in intrinsic asthma is autoimmune attack of the airways, for example by autoantibodies (Lasalle et al 1993; CitationNahm et al 2002).
In summary, despite this wealth of possible aetiological factors which may contribute to loss of asthma control, it is still not clear which of these can be clearly implicated in the genesis of glucocorticoid refractory or resistant asthma. It is furthermore not clear whether these aetiological factors lead to specific features of asthma pathology or contribute specifically to any feature of the natural history of the disease.
Environmental factors and “difficult” asthma
Oxidative stress and anti-oxidants
Markers of oxidative stress such as 8-isoprostane are elevated in severe asthma (CitationKatsoulis et al 2003; CitationKharitonov and Barnes 2003). Cigarette smoking and poor dietary anti-oxidant intake are potential sources of oxidative stress, and it has been shown that asthmatics who smoke are clinically more resistant to glucocorticoid therapy (CitationChaudhuri et al 2003). Oxidative stress can affect several aspects of glucocorticoid activity, for example through activation of the transcriptional regulatory protein AP-1 or by reducing nuclear translocation of the glucocorticoid receptor (see below).
Viral and other infections
Recurrent respiratory tract infections, most of these viral, increase the risk of asthma exacerbation (Citationten Brinke et al 2005), although it is not clear whether they alter responsiveness to glucocorticoid therapy. Very little is known about the mechanisms by which viral infections exacerbate asthma, and this field deserves much more study. There is also some evidence for the involvement of latent chlamydial (CitationHahn et al 1991) and, at least in animal models, adenoviral infections (CitationYamada et al 2000).
Allergen exposure
There is abundant evidence (summarized by CitationLeung and Bloom 2003; CitationBusse et al 2005) that exposure of sensitized asthmatics to clinically relevant allergens requires increased glucocorticoid therapy for disease control. It has been shown (CitationNimmagadda et al 1997) that seasonal allergen exposure of atopic asthmatics increases refractoriness of their T cells to glucocorticoid in an allergen-specific and IL-2 and IL-4 dependent fashion (see also below).
Microbial superantigens
There is evidence in other atopic diseases, particularly atopic dermatitis that colonization of the target organ (which is common) or infection with Staphylococcus aureus confers resistance to glucocorticoid therapy. Preferential expansion of T cells expressing particular antigen receptor Vβ chains has been observed in poorly controlled asthmatics (CitationHauk et al 1999), suggesting activation by microbial superantigen. In addition to T cell activation, superantigen has been shown to confer T cell resistance to glucocorticoid by induction of the β-isoform of the glucocorticoid receptor and by phosphorylation of the glucocorticoid receptor (CitationHauk et al 2000; CitationLi et al 2004).
The concept of glucocorticoid resistant asthma
The diagnosis of glucocorticoid resistant asthma is essentially one of exclusion. Before it can be diagnosed, it must be ensured that the diagnosis of asthma is correct and that factors contributing to poor asthma control have been eliminated as far as is possible. These stages have been formalized (CitationWoolcock 1993) as follows:
Establish/confirm the diagnosis of asthma (consider differential diagnosis – see );
Ensure that adequate dosages of glucocorticoids are reaching the airways (compliance, inhaler technique, metabolic factors which may increase glucocorticoid clearance);
Exclude ongoing exposure to provoking agents (smoke, irritants, allergens, etc.);
Rule out and eliminate as far as possible other potential aggravating conditions (chronic sinusitis, esophageal reflux, drugs which may exacerbate asthma, etc.);
Rationalize inhaled β2-agonist therapy;
Introduce a strict management plan to assess response to therapy which will typically last for weeks or sometimes months.
Even when all factors which may abrogate the effects of glucocorticoid therapy are eliminated or minimized as far as possible, there remains a group of patients who show little or no response of airways obstruction to glucocorticoid therapy. This concept was first proposed over 30 years ago (CitationSchwartz et al 1968). In this study, the authors described asthmatic patients in whom disease was poorly controlled, and the typical peripheral blood eosinopaenic response diminished, on large oral dosages of glucocorticoids. Later, it was recognised that there were some asthmatics in whom the diurnal pattern of airways obstruction is little altered by glucocorticoid therapy (CitationClark and Hetzel 1977).
The first attempt to define glucocorticoid resistant asthma in objective terms (CitationCarmichael et al 1981) was based on changes in base line FEV1 following a 14 day course of oral prednisolone (40 mg/day). Patients showing improvements of <15% of baseline were classified as resistant, whereas those showing improvements of 30% or more were considered glucocorticoid sensitive. All patients, in contrast, showed marked improvement in FEV1 in response to inhaled β2-agonists, indicating that, at least in this group of patients, the glucocorticoid response did not reflect “irreversible” airways obstruction (although, as discussed, this may be a factor in other patients). Clearly, these FEV1 responses represent opposite ends of a continuum of clinical response. Most subsequent studies have employed definitions of glucocorticoid sensitive and resistant asthma similar or identical to the above, in both adults and children (CitationKamada et al 1992; CitationAlvarez et al 1992). The possibility that glucocorticoid “resistant” subjects who show no clinical response following two weeks of oral glucocorticoid therapy might nevertheless respond following more protracted therapy has never been formally addressed, although it was shown (CitationKamada et al 1993) that 90% of severe asthmatic children showing an improvement in FEV1 >15% of baseline on high-dosage oral glucocorticoid therapy did so within 10 days.
On the basis of these studies, a workshop of experts on glucocorticoid resistant asthma proposed that this should be defined by the failure to improve baseline morning pre-bronchodilator FEV1 by more than 15% of the baseline value following at least 14 days of therapy with prednisolone 40 mg daily or its equivalent (CitationLee et al 1996). One weakness of this definition is that patients with refractory asthma are typically taking large dosages of inhaled glucocorticoid, the therapeutic effect of which may have reached a “ceiling” limiting further clinical response. Nevertheless, patients with complete glucocorticoid resistance show not only a failure of response in terms of FEV1, but also, in general, an ability to tolerate reduction of glucocorticoid dosages without significant change in disease activity. In addition, they typically show little increase in FEV1 even with more protracted, much higher dosages of systemic glucocorticoids. This is in distinction to glucocorticoid “dependent” asthmatics, who may not show a response in FEV1 of 15% or more during a 14 day trial of systemic glucocorticoid therapy, but rapidly deteriorate when this therapy is withdrawn. Although it is assumed that these patients fall at the end and near end of a spectrum of glucocorticoid responsiveness in asthma, it is not certain whether these two groups of patients have fundamentally different abnormalities leading to glucocorticoid refractoriness. Although glucocorticoid resistant patients may show a degree of fixed airways obstruction, many show marked diurnal variability in PEF and a brisk bronchodilator response (CitationAlvarez et al 1992; CitationKamada et al 1992).
It is inherent in the diagnosis of glucocorticoid resistant asthma that the diagnosis of asthma has been made correctly and that factors confounding therapy have been eliminated or reduced as far as possible. Most clinical descriptions of glucocorticoid resistant asthma emphasize the following common clinical features (CitationSpahn et al 1997):
Persistent symptoms despite optimal therapy;
Chronic airflow limitation with FEV1<60% predicted in adults and <80% predicted in children;
Failure to achieve an increase in morning pre-bronchodilator FEV1 of >60% predicted despite systemic glucocorticoid therapy (at least 40 mg/day of prednisolone or its equivalent given for at least 14 days);
Frequent nocturnal symptoms with significant “morning dipping” of PEF;
Poor clinical and spirometric response to oral glucocorticoid therapy, with <15% improvement in pre-bronchodilator FEV1 following a trial of oral glucocorticoid therapy as specified.
It should be noted finally that, although glucocorticoid resistant patients are identified by definition on the basis of having severe disease which does not respond to therapy, it cannot be excluded that patients with milder disease may also nevertheless be glucocorticoid resistant. It would require a large and protracted study to investigate this hypothesis, but the point has a bearing on whether or not the severity of disease itself actually regulates, at least in part, glucocorticoid responsiveness (see below).
Mechanisms of glucocorticoid resistant asthma
Despite several decades of usage of glucocorticoids as anti-inflammatory agents, in general very little is known about the precise mechanisms by which they ameliorate inflammatory diseases. A good review is provided by CitationIto et al (2006). Glucocorticoids exert a number of generalized anti-inflammatory activities, such as capillary vasoconstriction and reduction of vascular permeability, which may be relevant to suppression of inflammation however caused. In the case of asthma, it is now generally accepted that bronchial mucosal inflammation is a fundamental feature of disease pathogenesis. Despite the doubts about the precise roles (if any) of effector inflammatory leukocytes discussed above, a working scenario is that eosinophils are regarded as pro-inflammatory effector cells the products of which damage the bronchial mucosa, causing variable airways obstruction and bronchial hyperresponsiveness. Selective eosinophil accumulation and activation in asthma is in turn brought about by the release of eosinophil-active cytokines, particularly IL-3, IL-5 and GM-CSF, principally from activated T cells but also partly from other inflammatory cells, including mast cells and eosinophils themselves (CitationCorrigan and Kay 1992).
If a mechanistic role for leukocytes such as eosinophils in asthma has become less clear recently, there remains much better evidence that glucocorticoid therapy which results in amelioration of asthma is associated with reduced activation of, and synthesis of asthma-relevant cytokines by activated T cells. For example, elevated percentages of peripheral blood CD4, but not CD8 T cells from patients with exacerbation of asthma expressed mRNA encoding IL-3, IL-5 and GM-CSF but not IL-2 and IFN-γ as compared with controls (CitationCorrigan et al 1995). Spontaneous secretion of the corresponding cytokines was also demonstrable in these patients using an eosinophil survival-prolonging assay. The percentages of CD4 T cells expressing mRNA encoding asthma-relevant cytokines, as well as spontaneous secretion of these cytokines was reduced in association with glucocorticoid therapy and clinical improvement. In a double-blind, parallel group study (CitationRobinson et al 1993), therapy of mild atopic asthmatics with oral prednisolone, but not placebo, resulted in clinical improvement associated with a reduction in the percentages of BAL fluid cells expressing IL-5 and IL-4 and an increase in those expressing γ. These and other studies have provided overwhelming evidence in support of the general hypothesis that, in asthma, activated CD4 T cells secrete cytokines that are relevant to asthma pathogenesis, through direct actions on the airways mucosa or activation of inflammatory leukocytes such as eosinophils, and that glucocorticoids exert their anti-asthma effect at least partly by reducing the synthesis of cytokines by these cells.
Functional T cell abnormalities in glucocorticoid resistant asthma
A reasonable hypothesis is, then, that glucocorticoid resistant asthma may reflect refractoriness of T cells to glucocorticoid inhibition. A pioneering study in this field (CitationPoznansky et al 1984) showed that when peripheral blood mononuclear cells from glucocorticoid sensitive and resistant asthmatics were cultured with the T cell mitogen phytohaemagglutinin (PHA) in soft agar in vitro, methylprednisolone (10−8 M) produced a lesser degree of inhibition of colony formation by the cells from the resistant asthmatics, suggesting impaired T cell inhibition by glucocorticoids in these patients. This observation was followed up with two reports from the author’s laboratory (CitationCorrigan et al 1991a, Citation1991b) characterizing peripheral blood T cells of glucocorticoid sensitive and resistant asthmatics. In summary, it was demonstrated in these reports that PHA-induced proliferation of peripheral blood T cells was inhibited by dexamethasone at therapeutic concentrations in glucocorticoid sensitive, but not resistant asthmatics. This resistance was not absolute but relative, reflecting a shift in the concentration-response curve for inhibition. In other words, T cells from glucocorticoid resistant asthmatics can be inhibited by glucocorticoids, but only at concentrations requiring glucocorticoid dosages that most physicians would not contemplate using for protracted periods in clinical practice. Consistent with this, it was demonstrated (CitationCorrigan et al 1991b) that elevated percentages of peripheral blood T cells expressed the activation markers in CD25 and HLA-DR in glucocorticoid resistant, as compared with sensitive asthmatics, with no differences in total cell numbers, suggesting persistent T cell activation in the resistant patients despite glucocorticoid therapy.
In glucocorticoid resistant asthmatics, clinical resistance to therapy could not be accounted for by differences in absorption and clearance of plasma prednisolone derived from orally administered prednisone. We have subsequently shown (CitationHaczku et al 1994) that the inhibition of PHA-induced proliferation of peripheral blood T cells from asthmatics by glucocorticoids in vitro is reproducible both in the short term and when patients are re-tested after intervals of several months. This suggests that the degree of glucocorticoid sensitivity of peripheral blood T cells from asthmatics, and by inference their clinical sensitivity to glucocorticoid therapy, remains relatively constant, although there is evidence (see below) that sensitivity in individual patients may vary to some degree according to the ongoing severity of their disease.
Glucocorticoid resistant asthmatics are not Addisonian and do not have elevated plasma cortisol concentrations (CitationCorrigan et al 1991a), suggesting that the impaired glucocorticoid responsiveness observed in their peripheral blood T cells is not a generalised, systemic phenomenon. One possibility is that impaired T cell glucocorticoid responsiveness in asthma may be induced by the action of pro-inflammatory cytokines within the local environment of the inflammatory process. In this regard, there exists evidence that the glucocorticoid receptor ligand binding affinity of peripheral blood T cells in asthmatics may be altered in the short term according to disease severity in vivo and by exposure to cytokines in vitro. The glucocorticoid receptor ligand binding affinities of peripheral blood T cells from a group of poorly controlled asthmatics were reduced as compared with normal controls (median Kd 29.0 vs 8.0 nM). Glucocorticoid therapy of these asthmatics, which was accompanied by clinical improvement, was associated with a significant increase in affinity of the T cell glucocorticoid receptors (CitationSpahn et al 1995). In a detailed study of glucocorticoid receptor binding in peripheral blood mononuclear cells from glucocorticoid sensitive and resistant asthmatics, two distinct abnormalities were observed (CitationSher et al 1994). The majority of the asthmatics (15 out of 17) demonstrated a significantly reduced receptor binding affinity (mean Kd 42.1 nM) as compared with sensitive patients (mean Kd 21.6 nM) and normal controls (mean Kd 7.9 nM). This abnormality was confined to T cells, reverted to normal after culture of the T cells in vitro for 48 hours, but could be sustained by culture in the simultaneous presence of high concentrations of IL-2 and IL-4. The remaining two glucocorticoid resistant asthmatics had abnormally low numbers of nuclear glucocorticoid receptors with normal binding affinity. This abnormality was not confined to T cells and was not influenced by the presence of exogenous cytokines. It was further shown (CitationKam et al 1993) that this abnormality could be induced by culture of peripheral blood T cells from normal donors with IL-2 and IL-4 in vitro, and that this induction was associated with a reduced inhibitory effect of methylprednisolone on the proliferation of the T cells induced by phorbol ester and ionomycin. A similar effect of exogenous IL-13 alone was subsequently demonstrated in monocytes (CitationSpahn et al 1996). The intrinsic physiological significance of these relatively small changes in ligand binding affinity of the glucocorticoid receptor is difficult to assess, but clearly they may contribute to glucocorticoid refractoriness. More likely, they may represent an epiphenomenon reflecting more fundamental changes in the properties of the glucocorticoid receptor in T cells following exposure to cytokines (see below).
There exists similar evidence for a differential effect of glucocorticoids on T cells from sensitive and resistant asthmatics in vivo (CitationLeung et al 1995). In this study, bronchoalveolar lavage was performed in glucocorticoid sensitive and resistant asthmatics before and after a course of oral prednisone, and expression by BAL cells of mRNA encoding cytokines was measured by in situ hybridization. Whereas prednisone therapy of the glucocorticoid sensitive asthmatics was associated with reductions in the percentages of BAL cells expressing mRNA encoding IL-4 and IL-5 and elevation of the percentages of cells expressing mRNA encoding IFN-γ, only a decrease in the percentages of cells expressing mRNA encoding γ was observed in association with prednisone therapy of the glucocorticoid resistant asthmatics. In addition, compared with the sensitive asthmatics, the resistant patients had elevated percentages of BAL cells expressing mRNA encoding both IL-2 and IL-4 at baseline. These data are compatible with the hypothesis that glucocorticoids exert differential effects on cytokine mRNA expression in T cells from glucocorticoid sensitive and resistant asthmatics in vivo. The authors also speculated that the local elevated expression of IL-2 and IL-4, at least at the level of mRNA, in these patients might have been responsible for some of the glucocorticoid refractoriness of the cells described in the in vitro experiments above.
Although glucocorticoids generally reduce inflammation by inhibiting the production of pro-inflammatory cytokines, one interesting facet of glucocorticoid action, which is receiving increasing attention, is their ability to increase the production of the anti-inflammatory cytokine interleukin-10 (IL-10). Studies in patients, initially in transplantation (CitationWan et al 1996), and more recently in other conditions including asthma (CitationJohn et al 1998), have demonstrated that administration of glucocorticoids, intravenously or by inhalation, results in a significant increase in systemic or local IL-10 synthesis respectively. Parallel studies from our own laboratories have shown a concentration-dependent induction by glucocorticoids of T cell IL-10 expression in vitro (CitationRichards et al 2000). Interest in the possible therapeutic benefit of IL-10 in asthma already exists, based on its proposed role in regulating immune homeostasis in the lung (CitationAkbari et al 2001). IL-10 inhibits pro-inflammatory cytokine production, antigen presentation, T cell activation and mast cell and eosinophil function (reviewed by CitationMoore et al 2001). Furthermore, synthesis of IL-10 is deficient in the airways of asthmatics as compared with controls (CitationBorish et al 1996), and polymorphisms of the IL-10 gene leading to impaired expression of IL-10 are associated with a more severe disease phenotype (CitationLim et al 1998). We have recently described a marked deficiency in the capacity of CD4+ T cells from glucocorticoid resistant asthmatics to synthesize IL-10 following in vitro stimulation in the presence of dexamethasone, as compared with sensitive patients of equivalent disease severity (CitationHawrylowicz et al 2002). We have furthermore shown that vitamin D3 in combination with glucocorticoids can restore the ability of blood CD4 T cells from patients with glucocorticoid resistant asthma to manufacture IL-10 in quantities comparable with those of sensitive patients, and that oral administration of vitamin D3 to glucocorticoid resistant asthmatics for seven days enhanced ex vivo responses of their T cells to glucocorticoid (CitationXystrakis et al 2006).
Molecular basis of glucocorticoid action and resistance
Observations made in the past few years have greatly increased our knowledge of how the GR regulates transcription, and how this process may be modified both in vitro and in vivo. The GR comprises three domains, the N-terminal or immunogenicity domain, the central DNA-binding domain and the C-terminal ligand-binding domain (CitationKino and Chrousos 2001; CitationIto et al 2006). The GR in its ligand-unbound state is located primarily in the cytoplasm as part of hetero-oligomeric complexes containing the heat shock proteins 90, 70 and 50. Upon binding to ligand, the GR undergoes conformational changes, dissociates from the heat shock proteins, homodimerizes and translocates to the nucleus. There, the ligand-activated GR may interact with DNA sequences (glucocorticoid response elements) or with other transcriptional regulators through protein/protein interactions, directly influencing the activity of the latter on their target genes ().
Figure 1 Mechanism of glucocorticoid action. After diffusion across the cytoplasmic membrane, glucocorticoid (GC) binds to the glucocorticoid receptor (GR) which dissociates from heat shock protein 90 (HSP90) then dimerises. In the cell cytoplasm it may interact with transcriptional regulatory proteins such as AP-1; such interactions are mutually inhibitory. Nuclear translocation of the ligand bound GR is regulated by chemical modifications, such as phosphorylation, and possibly other mechanisms. Once inside the nucleus, the ligand bound receptor may interact directly with binding regions (glucocorticoid regulatory elements, GRE) adjacent to genes, either increasing (A) or decreasing (B, negative GRE) transcription of these genes. More usually, however, the ligand-bound GR interacts with DNA as part of a highly dynamic complex of proteins including transcriptional regulatory proteins such as AP-1, NFκB and cyclic AMP response element binding protein (CREB) (see text). The complexity of these interactions in individual cells accounts for the remarkable variability of glucocorticoid actions on cells according to their function and phenotype.
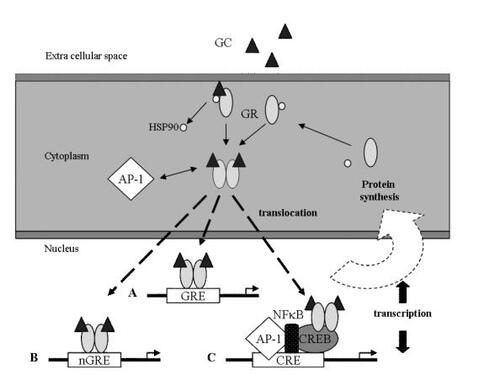
Proteins which may bind directly to the GR and modulate its activity in this way include AP-1, NFκB, signal transduction and activators of transcription (STATs) and certain of the CCAAT-enhancer binding proteins (C/EBP). These interactions appear to be particularly important in glucocorticoid-mediated suppression of inflammation, and may allow the activated GR to transrepress expression of cytokine genes without binding to DNA at all (CitationReichhardt et al 2001).
Interaction of GR with AP-1
The pro-inflammatory transcriptional element activator protein-1 (AP-1) is an important contributor to the expression of the asthma-relevant Th2 cytokines IL-4, IL-5 and IL-13. AP-1 comprises of variable heterodimers of Jun (c-Jun, JunB and JunD) and Fos (c-Fos, FosB, Fra1 and Fra2) family members. AP-1 is inducible by a variety of cytokines and growth factors (CitationLee et al 1987), and also by oxidative stress (see above). It is activated through the phosphorylation of c-Jun and the transcriptional regulation of c-Fos (). Phosphorylation of c-Jun is the end result of the action of a trilayer of kinases (CitationEnglish and Cobb 2002). c-Jun itself is phosphorylated by Jun-N-terminal kinase (JNK), a member of the extracellular signal-related kinases/mitogen-activated protein kinases (ERK/MAPK) family of serine/threonine kinases. JNK is in turn activated by phosphorylation by JNK kinase, a member of the MAPK/ERK kinase (MEK) family of kinases that phosphorylate on both a tyrosine and a threonine or serine residue. Of the seven members of the MEK family, MEK4 or Jun-N-terminal kinase kinase is principally responsible for the phosphorylation of JNK. At the top end of the trilayer, the most upstream kinases in the cascade, are the MEK kinases (MEKK), serine/threonine kinases that are diverse in sequence and structure ().
Figure 2 Regulation of activation of c-fos and c-jun, the components of AP-1. Transcription of c-fos is induced by serum response factor (SRF) which binds to the serum response element (SRE) adjacent to the c-fos gene. SRF is activated by phosphorylation by ELK-1, which is in turn activated by phosphorylation by a range of MAP kinases including ERK-1/2 and JNK (see text). Phosphorylated JNK also phosphorylates and activates c-jun, which together with c-fos forms the heterodimer complex AP-1. AP-1 increases the transcription of a number of asthma-relevant cytokine genes such as interleukin (IL)-4 and IL-5. This activity is inhibited by interaction with the ligand bound glucocorticoid receptor (GR) (see text and above). IL-4 and IL-5 may in turn further stimulate MAP kinase activity, forming a positive, feed-forward loop. Many other stimuli, including oxidative stress may also activate MAP kinases.
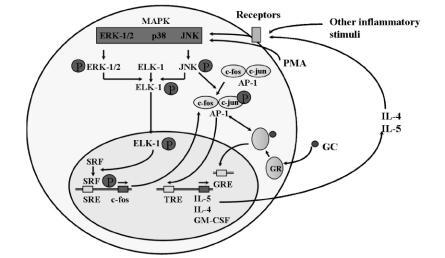
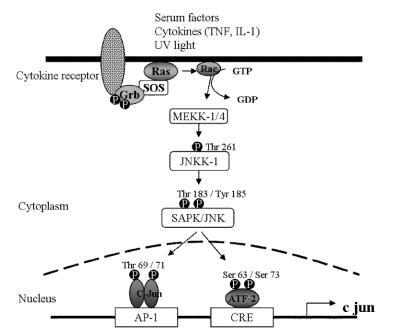
Figure 3 The tri-layer of kinases which activate c-jun. Environmental cytokines, ultraviolet light, serum factors, oxidative stress and many other stimuli phosphorylate and activate MEKK-1/4 through the Ras/Rho family of small GTP-binding proteins as shown. MEKK1 in turn phosphorylates JNKK1, which phosphorylates JNK, which binds to and phosphorylates c-jun, the active component of AP-1 (see text and above), and the regulatory protein activating transcription factor-2 (ATF-2). Phosphorylation may occur on threonine or tyrosine residues.
Abbreviations: GT(D)P, guanosine tri(di)phosphate; JNK, c-jun N-terminal kinase; JNKK1, c-jun N-terminal kinase kinase-1; MEKK-1/4, MAPK/ERK kinase kinase-1/4; SAPK, serum activated protein kinase; Ser, serine; SOS, son of sevenless; Thr, threonine; Tyr, tyrosine.
In collaboration with Professor Ian Adcock at Imperial College, London, we have implicated abnormal regulation of AP-1 in the mechanism of glucocorticoid resistance. We showed that glucocorticoid-exposed peripheral blood mononuclear cells from glucocorticoid-resistant, as compared with glucocorticoid-sensitive asthmatics have fewer activated GR available for DNA binding (Adcock et al 1995a), but elevated DNA binding of AP-1 following phorbol ester stimulation (Adcock et al 1995b). These cells also demonstrated significantly elevated basal, as well as phorbol ester-stimulated, transcription and translation of c-Fos. Furthermore, phorbol ester stimulation of cells from glucocorticoid-sensitive patients induced a glucocorticoid-resistant phenotype, which was associated with direct interaction between the activated GR and c-Fos, detected by co-immunoprecipitation (CitationLane et al 1998). Binding of GR to other pro-inflammatory transcriptional activators (such as CREB and NFκB) was unaffected. We interpret these data to suggest that mononuclear cells from glucocorticoid-resistant asthmatics inappropriately over-express AP-1, which sequesters and neutralises activated GR, thus causing refractoriness to glucocorticoid-induced inhibitory responses. More recently we have shown that (CitationLoke et al 2006) that systemic glucocorticoid therapy of glucocorticoid sensitive, but not resistant, asthmatics is associated with down-regulation of phosphorylation of both c-Jun and JNK within inflammatory cells within the bronchial mucosa, further underlining a critical role for AP-1 components in regulating glucocorticoid response in asthma. The reason for this failure of glucocorticoid to down-regulate the c-Jun/JNK/MEK cascade in clinically resistant patients is presently unclear, but may reflect influences of the local cytokine milieu in these patients (such as elevated IL-2/IL-4 expression, as described above, which may enhance AP-1 production (CitationWang et al 1994)) and/or the influences of environmental stimuli such as oxidative stress or microbial superantigens as described above.
The role of p38 MAP kinase
p38 MAP kinase is another member of the family of MAPK/ERK molecules. Its activity is regulated by phosphorylation, principally by the p38 MAP kinases MEK3 and MEK6. A recent study (CitationIrusen et al 2002) has raised the intriguing possibility that changes in the binding affinity of nuclear GR induced by exposure to IL-2/IL-4 may be caused by direct phosphorylation of the GR at serine 226 secondary to the resulting activation of p38 MAP kinase. Although this study did not directly demonstrate that p38 MAP kinase phosphorylation of the GR is responsible for its reduced binding affinity for glucocorticoids or its defective induction of an inhibitory signal, the hypothesis is certainly plausible. Similarly TNF-α has been shown to induce glucocorticoid resistance in normal human monocytes, possibly through activation of p38 MAP kinase in addition to NF-κB (CitationFranchimont et al 1999). Several other protein kinases, such as JNK, may also modify activity of the GR in this way, either directly or through phosphorylation of co-factor molecules (CitationKrstic et al 1997; CitationRogatsky et al 1998).
The findings with p38 MAP kinase raise the possibility that the small alteration in ligand binding affinity of the nuclear GR induced by IL-2/IL-4 exposure is an epiphenomenon reflecting more fundamental alterations in the function of the GR brought about by phosphorylation. This is in line with the fact that the observed changes in ligand-binding affinity of the GR are relatively small and of doubtful physiological significance per se. Such a fundamental alteration in GR function is also suggested by the fact that serine 226 is located in the N-terminal domain of the GR, remote from the ligand-binding pocket which resides in the C-terminal portion of the molecule. There is some precedent for the possibility that remote regions of the GR may mutually interact. For example, there are two trans-activation domains, activation functions (AF) 1 and 2, situated respectively in the immunogenic and DNA-binding domains of the GR, which co-contribute to the full activity of the GR molecule on its responsive promoters. Conceivably, phosphorylation at a remote site might effect such interactions. Furthermore, both AF1 and AF2 interact with several other nuclear proteins and protein complexes, such as members of the p160 family and the co-activators p300/cyclic AMP-responsive element-binding protein (CREB)-binding protein (CRP) (CitationHittelman et al 1999) which can affect the glucocorticoid-titration response of GR transactivation of its responsive promoters (CitationSzapary et al 1999).
The role of mitogen-activated extracellular signal-related kinases (MEK)
In one further recent study (CitationLi et al 2004), stimulation of T cells with enterotoxin superantigen produced by Staphylococcus aureus rendered them resistant to GC inhibition in vitro, and this was accompanied by phosphorylation of both the mitogen-activated extracellular signal-related kinase (MEK/ERK) signaling protein and the GC receptor itself, as well as its impaired nuclear translocation. Treatment of cells with MEK/ERK inhibitors abrogated all of these effects. The authors inferred that activation of the MEK/ERK signaling pathway renders T cells resistant to GC inhibition by direct, ERK-mediated phosphorylation of the GC receptor which impairs its nuclear translocation. The effects of external environmental factors such as staphylococcal colonization on GC responsiveness emphasize the possibility that environmental factors may influence this responsiveness in individual patients.
The role of the GRβ-isoform
The β-isoform of the GR is a splice variant of the “normal” GR, or GRα. The splicing varies in the choice of a splice acceptor site in exon 9. The cDNA sequence up until this point encodes a common region of 727 amino acids. Thereafter, the GRα splice adds 50 amino acids whereas the β isoform has only a further 15 residues. The consequence of this alternative splice was shown to be an inability of the β isoform to bind ligand (CitationOakley et al 1996). The reason for this has been clarified by the recent crystallization of the GR ligand-binding domain complexed with dexamethasone and a peptide that was homologous to the GR interaction domain of the co-activator TIF2 (CitationBledsoe et al 2002). This study demonstrated that the amino acids of the receptor bind to ligand through either hydrogen bonds or hydrophobic interactions. One residue forming a hydrogen bond (T739) and seven residues forming hydrophobic interactions (L732, Y735, C736, T739, I747, F749 and L753) are present within the a, but are absent from the β isoform.
Artificial transfection of cells with GRβ can inhibit GRα-mediated stimulation of gene expression, [CitationOakley et al 1996; CitationWebster et al 2001). The popular theory to explain this is that it acts as a dominant negative inhibitor of GRα activity (but see reservations below). Physiological expression of GRβ in neutrophils has been suggested as a possible cause of their relative refractoriness to glucocorticoid inhibition (CitationStrickland et al 2001). Several studies suggest that GRβ may be induced in cell lines to an extent sufficient to induce glucocorticoid resistance by pro-inflammatory cytokines such as TNF-α (CitationWebster et al 2001), possibly reflecting the location of a consensus NF-κB binding sequence in the 5-flanking sequences of the GR gene. Furthermore, increased GRβ immunoreactivity has been reported in peripheral blood mononuclear cells and bronchoalveolar lavage cells from patients with glucocorticoid-resistant asthma (CitationHamid et al 1999), although possible modulation of expression in association with glucocorticoid therapy was not explored. In studies from our own laboratory using a model of tuberculin-induced cutaneous inflammation, we reported elevated expression of GRβ immunoreactivity in inflammatory cells infiltrating the skin lesions in glucocorticoid resistant, as compared with sensitive asthmatics (CitationSousa et al 2000). Treatment of the patients with systemic glucocorticoids was associated with down regulation of GRα expression in the glucocorticoid sensitive, but not the resistant patients.
Notwithstanding these observations, it seems likely that there is much yet to be learned about the possible functional role of GRβ. In this context it is possible to speculate on how GRβ may influence transcriptional activation mediated by GRα and its binding to the glucocorticoid response element (GRE), transcriptional repression mediated by GRα binding to negative GREs and inhibition of transcriptional activation mediated by sequestration of transcription factors:
Effects of GRβ on transcriptional activation
In terms of transcriptional activation, the classical role of GRα is to bind ligand, to dimerize and then to bind to GRE. This binding permits the recruitment of co-activator complexes, because one of the consequences of ligand binding for nuclear hormone receptors is a reduction in their affinity for co-repressor complexes, and replacement of these complexes with co-activators. The crystallization data referred to above (CitationBledsoe et al 2002) indicate that GRβ lacks residues forming the charge clamp required for docking of the amino terminus TIF2 LXXLL motif, suggesting that GRβ may be unable to recruit co-factors required for transcriptional activation. Thus GRβ, by displacing GRα, could conceivably act as a dominant negative inhibitor of transcriptional activation. There are, however, uncertainties regarding this conclusion. In the first place, GRβ seems to be expressed in much lower quantities (typically 10- to 100-fold less, at least at the level of mRNA expression) than GRα. In the second place, whereas in the case of GRα ligand binding appears to be a prerequisite for nuclear translocation, it is not known what governs the cytoplasmic/nuclear partitioning of GRβ or if it is present in sufficient quantities in the cellular nucleus. Finally, it is unlikely that GRβ could dimerise efficiently, so that its ability to compete for binding with high affinity to GREs is questionable.
Effects of GRβ on transcriptional repression
Repression mediated by ligand-bound GR is thought to occur through binding to atypical sites on DNA, for example non-consensus NFAT/AP-1 sites, rather than binding to GREs. At these sites, it is thought that the conformation of the DNA binding site, and the influence of locally bound factors might cause the GR to adopt a structure that is not permissive for recruitment of co-activators, but rather permissive for recruitment of co-repressors. There is at present no information as to whether or not GRβ can bind to such sites and, more importantly, recruit co-repressor complexes.
Inhibition of transcriptional activation mediated by sequestration of transcription factors
So far, there has been no clear demonstration for a role for the extreme carboxy-terminus of the GR in the sequestration (and inactivation) of transcriptional activating proteins such as AP-1. It is conceivable, therefore, that both GRα and GRβ could exert repressive effects on gene expression by sequestration of such transcriptional activators.
The possible role of histone proteins
DNA is packaged into chromatin, a highly organized and dynamic protein-DNA complex. The N-terminal tails of the core histone proteins contain highly conserved lysine residues that are sites for post-translational acetylation. Acetylation of histone residues results in unwinding of the DNA coiled around the histone core. This process opens up the chromatin structure, allowing transcriptional factors and RNA polymerase II to bind more readily to DNA, thereby increasing gene transcription (CitationImhof and Wolffe 1998). The large co-activator molecule CREB binding protein (CBP) that binds to the basal transcriptional apparatus has intrinsic histone acetyl transferase (HAT) activity. Additionally, associated co-activator proteins, including steroid receptor co-activator-1 (SRC-1), transcription factor intermediary factor–2 (TIF2), p300/CBP co-integrator-associated protein (p/CIP) and glucocorticosteroid receptor enhancing protein-1 (GRIP-1) may enhance local HAT activity. In genes which are induced by glucocorticoids, high concentrations of glucocorticoids cause binding of the activated GR to CBP and/or associated co-activators, resulting in histone acetylation on lysines 5 and 16 of histone H4 and increased gene transcription. HAT activity may be further enhanced by binding of transcriptional regulatory proteins such as AP-1 and NF-κB (CitationJanknecht and Hunter 1996; CitationImhof and Wolffe 1998). Repression of genes is conversely associated with a reversal of this process by histone deacetylation, mediated by histone deacetylases (HDACs) (CitationPazin and Kadonaga 1997). HDACs comprise of a growing family of enzymes of which at least 10 mammalian members have been described (CitationBertos et al 2001). Some, such as HDAC4 and HDAC8 are able to shuttle between the cellular nucleus and cytoplasm.
Histone acetylation is a dynamic process in which small changes in acetylase and deacetylase activity can considerably alter the net HAT activity at any particular gene promoter site. Suppression of HAT activity, as well as recruitment of HDAC activity to active transcriptional complexes may play a role in glucocorticoid regulation of gene transcription (CitationIto et al 2000). This may occur in a variety of ways. Glucocorticoid repression may reflect competition between the activated GR and the binding sites on CBP for other transcriptional activating proteins, such as AP-1 (CitationKamei et al 1996), NF-κB, Sp1, Ets, NF-AT and STATs, which may alter local HAT activity. Alternatively, and not exclusively, the GR may bind to one of several co-repressor molecules such as RIP140, NCoR1 and GRIP1which in turn associate with proteins having differing HAT activity (CitationDing et al 1998). These complex interactions probably play a major role in the genesis of the infinitely variable and subtle effects of glucocorticoids on individual target cells.
Clearly, then, intrinsic abnormalities of, or external influences on the regulation of HAT activity in individual cells may influence their glucocorticoid responsiveness. Already there are suggestions that external influences such as exposure to cigarette smoke, an oxidative stress, may inhibit the anti-inflammatory actions of glucocorticoids on cells in the lungs of smokers by reducing HDAC expression and activity (CitationIto et al 2005). Similar phenomena may contribute to glucocorticoid resistance in asthma.
Genetic basis of glucocorticoid resistance
The glucocorticoid receptor gene itself is a plausible candidate for a genetic basis to glucocorticoid resistance. A missense mutation in this gene had previously been found to be responsible for the phenomenon of familial glucocorticoid resistance (CitationHurley et al 1991). These patients have a markedly reduced affinity of the glucocorticoid receptor for ligand and clinically have features of Addison’s disease, which is not the case with glucocorticoid resistant asthmatics. Analysis of the sequence of the glucocorticoid receptor protein in six resistant and sensitive patients showed no mutations in any of the subjects (CitationLane et al 1994). Although this work is ongoing, there is no obvious mutation that might account for glucocorticoid resistance. Obviously, relevant mutations do not necessarily have to be within the coding region of the receptor gene itself, but could be in genes encoding products in downstream signaling pathways.
In addition to this, glucocorticoid responsiveness may be governed at least partly by genetic factors. For example, one study suggested that inherited anomalies might render approximately 7% of the normal population relatively hypersensitive to glucocorticoids (CitationLamberts et al 1996). Indeed, there is marked variability in the sensitivity of T cells even from normal individuals to glucocorticoid inhibition in vitro, suggesting that the phenomenon may not be induced entirely by inflammation but may be partly heritable.
The technique of positional cloning provides a way to identify genes or gene complexes which predispose to disease in an unbiased fashion. Using this technique, five genes or gene complexes (ADAM33, PHF11, DPP10, GPRA and SPINK5) have been linked with predisposition to asthma (reviewed by CitationCookson and Moffatt 2004), and studies are ongoing to investigate the functions of these genes, some of which, along with others to be discovered, may play a role in therapy resistant asthma. These studies involve large numbers of patients, but larger studies still will be needed to discern the relative contribution of genetics and environment in regulating asthma severity and responsiveness to therapy in individual patients. A further complication is that different genes may contribute variably to asthma in different racial groups.
The alternative approach to genetic analysis is to look for variability in the expression of “candidate genes” (that is, those hypothesized to be an important part of the disease process or in governing responsiveness to therapy), for example between asthmatics and controls or between glucocorticoid sensitive and resistant asthmatics. The list of such “candidate genes” is potentially very large, and the problem with this approach is that it is impossible to know the relative importance, and possible physiological significance of any differences uncovered. This process is being facilitated by microarray gene fingerprinting, which allows analysis of the differential expression of potentially many thousands of genes on a single chip. Using this technique in an Icelandic population it was possible to define a gene expression fingerprint which predicted glucocorticoid resistant, as compared with sensitive asthma not only in the population of origin but also in an independent cohort (CitationHakonarson et al 2005). Again such analysis might be confounded by ethnicity. Genes may also regulate drug metabolism variably in different individuals (CitationHall 1998).
Management of glucocorticoid resistant asthma
In view of the above, a proposed list of techniques for the investigation of “difficult” asthma, including glucocorticoid resistant asthma, is proposed in . Such investigation would also include a formal trial of glucocorticoid therapy to investigate possible glucocorticoid resistance defined by the criteria discussed above. Key features of the management of severe asthma have much in common with the management of any patient with asthma. It is critical to make an accurate diagnosis, minimize factors which may induce loss of asthma control and ensure good compliance with therapy.
Table 3 Investigation of difficult/therapy resistant asthma
By definition, the “difficult” asthmatic will be receiving high dosages of inhaled glucocorticoid therapy, often together with oral glucocorticoids. Generally all patients with severe asthma, particularly those admitted as an emergency will be treated with systemic glucocorticoids at high dosage for at least two weeks. It is only in the fullness of time that a pattern of glucocorticoid responsiveness is established. In patients thought to be truly glucocorticoid resistant (that is, patients showing the proscribed failure in FEV1 response when the diagnosis is confirmed and aggravating conditions excluded), further long-term therapy with systemic glucocorticoids is probably inadvisable since there is little evidence that these drugs will influence asthmatic symptoms and disease activity, but may on the other hand cause considerable unwanted effects. In such cases it may be sensible to reduce or withdraw glucocorticoids and treat instead with adequate dosages of alternative anti-asthma drugs such as bronchodilators, leukotriene receptor antagonists and immunosuppressive drugs (see below). In clinical practice, however, because there is little hard evidence to justify withdrawal of systemic glucocorticoids from any severe asthmatic patient, this process is often ignored or delayed. Furthermore, it is not clear on clinical grounds whether glucocorticoid responsiveness is induced or enhanced by the use of glucocorticoids themselves (although reduction of asthmatic inflammation would appear to be associated with an increased affinity of the glucocorticoid receptor for binding to its ligand, as discussed above). In view of the possibility that glucocorticoid sensitivity may change with time, it would seem prudent to re-assess glucocorticoid sensitivity periodically (CitationDemoly et al 1998) although, as discussed above, within individuals T cell glucocorticoid sensitivity appears to be relatively stable at least over a period of months.
The observation that glucocorticoid responsiveness of asthmatics correlates with glucocorticoid inhibition of their T cells suggests that other drugs that inhibit T cells might be useful for asthma therapy, in particular drugs that inhibit T cells by mechanisms distinct from those of glucocorticoids. We have shown (Corrigan et al 1991; CitationHaczku et al 1994) that the immunosuppressive drugs cyclosporin A, rapamycin and mycophenolate mofetil inhibit proliferation of T cells from glucocorticoid sensitive and resistant asthmatics to an equivalent extent. It has been shown (CitationAlexander et al 1992; CitationLock et al 1996) that cyclosporin A, when administered to patients with poorly controlled asthma despite continuous systemic glucocorticoid therapy, improved lung function while allowing reduction of oral glucocorticoid dosages in a proportion of the patients. Similarly concomitant therapy of glucocorticoid dependent asthmatics with methotrexate (CitationShiner et al 1990) or gold salts (CitationKlaustermeyer et al 1987) has been shown in some trials to spare glucocorticoid therapy, although no trials have suggested that these agents improve lung function. In general, none of these agents is particularly satisfactory in the sense that many patients fail to respond and it is impossible to predict responsiveness a priori. Furthermore, chronic immunosuppression raises the risk of development of serious infection or malignancy, and there is in addition a list of not insignificant unwanted effects associated, in some patients, with the use of each particular drug. An urgent appraisal of other immunosuppressive drugs or cytokine inhibitory strategies is needed in glucocorticoid dependent and glucocorticoid resistant asthma. It is a priority to produce a global definition of which patients are suitable for treatment, and what constitutes an adequate trial of therapy. It will also be essential to develop cheap and reliable biomarkers of airways inflammation and remodeling so that glucocorticoid responsiveness can be assessed accurately and objectively in the short and long term.
References
- AdcockIMLaneSJBrownCRDefective binding of glucocorticoid receptor to DNA in steroid-resistant asthmaJ Immunol1995154350057897230
- AdcockIMLaneSJBrownCRAbnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthmaJ Exp Med1995182195187500041
- AkbariODeKruyffRHUmetsuDTPulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigenNature Immunol200127253111477409
- AlexanderAGBarnesNCKayABTrial of cyclosporine in corticosteroid-dependent chronic severe asthmaLancet199233932481346410
- AlvarezJSursWLeungDYMSteroid resistant asthma: immunologic and pharmacologic featuresJ Allergy Clin Immunol199289714211545093
- American Thoracic SocietyProceedings of the ATS workshop on refractory asthma: current understanding, recommendations and unanswered questionsAm J Respir Crit Care Med200016223415111112161
- BertosNRWangAHYangXJClass II histone deacetylases: structure, function and regulationBiochem Cell Biol2001792435211467738
- BledsoeRKMontanaVGStanleyTBCrystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerisation and coactivator recognitionCell20021109310512151000
- BorishLAaronsARumbyrtJInterleukin-10 regulation in normal subjects and patients with asthmaJ Allergy Clin Immunol1996971288968648025
- BuistSACost effectiveness of asthma management strategiesEur Respir Rev199552924
- BumbaceaDCampbellDNguyenLParameters associated with persistent airflow obstruction in chronic severe asthmaEur Respir J200424122815293614
- BussePJWangJJHalmEAAllergen sensitization evaluation and allergen avoidance education in inner-city adult cohort with persistent asthmaJ Allergy Clin Immunol20051161465215990788
- CarmichaelJPatersonICDiazPCorticosteroid resistance in chronic asthmaBr Med J19812821419226784848
- ChaudhuriRLivingstonEMcMahonADCigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthmaAm J Respir Crit Care Med200316813081112893649
- ClarkTJHHetzelMRDiurnal variation of asthmaBr J Dis Chest1977718792861159
- CochraneGMTherapeutic compliance in asthma: its magnitude and implicationsEur Respir J1992512241577134
- CockcroftDWSwystunVAAsthma control versus asthma severityJ Allergy Clin Immunol1996981016188977499
- CooksonWMoffattMMaking sense of asthma genesN Engl J Med20043511794615496632
- CorrenJAdinoffADBuchmeierADNasal beclomethasone prevents the seasonal increase in bronchial responsiveness in patients with allergic rhinitis and asthmaJ Allergy Clin Immunol19929025061500629
- CorriganCJBrownPHBarnesNCGlucocorticoid resistance in chronic asthma: glucocorticoid pharmacokinetics, glucocorticoid receptor characteristics and inhibition of peripheral blood T-cell proliferation by glucocorticoids in vitroA Rev Respir Dis1991a144101625
- CorriganCJBrownPHBarnesNCGlucocorticoid resistance in chronic asthma: peripheral blood T-lymphocyte activation and a comparison of the T-lymphocyte inhibitory effects of glucocorticoids and cyclosporin AAm Rev Respir Dis1991b1441026321952427
- CorriganCJKayABThe role of T lymphocytes and eosinophils in the pathogenesis of asthmaImmunol Today19921350171361126
- CorriganCJHamidQNorthJPeripheral blood CD4 but not CD8 T-lymphocytes in patients with exacerbation of asthma transcribe and translate messenger RNA encoding cytokines which prolong eosinophil survival in the context of a TH2-type pattern: effect of glucocorticoid therapyAm J Respir Cell Mol Biol199512567787742019
- EnglishJMCobbMHPharmacological inhibitors of MAPK pathwaysTrends in Pharmacological Sciences20022340511804650
- DemolyPJaffuelDMathieuMGlucocorticoid insensitive asthma: a one-year clinical follow up pilot studyThorax199853106310195080
- DingXFAndersonCMMaHNuclear receptor-binding sites of coactivators glucocorticosteroid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificitiesMol Endocrinol199812302139482670
- DolanCMFraherKEBleeckerERDesign and baseline characteristics of the epidemiology and natural history of asthma: Outcomes and treatment regimens (TENOR) study: a large cohort of patients with severe or difficult-to-treat asthmaAnn Allergy Asthma Immunol20049232914756462
- ENFUMOSAThe ENFUMOSA cross-sectional European multicentre study of the clinical phnotype of chronic severe asthmaEur Respir J200322470714516137
- Flood-PagePMenzies-GowAPhippsSAnti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmaticsJ Clin Invest200311210293614523040
- FranchimontDMartensMHagelsteinMTTumour necrosis factor alpha decreases, and interleukin-10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptorJ Clin Endocrin Metab19998428349
- HaczkuAAlexanderABrownPThe effect of dexamethasone, cyclosporine and rapamycin on T-lymphocyte proliferation in vitro: comparison of cells from patients with glucocorticoid-sensitive and glucocorticoid-resistant chronic asthmaJ Allergy Clin Immunol199493510198120277
- HahnDLDodgeRWGolubjatnikovRAssociation of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onsetJAMA1991266225302056624
- HakonarsonHBjornsdottirUSHalapiEProfiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patientsProc Natl Acad Sci USA2005102147899416203992
- HallIPGenetic factors in asthma severityClin Exp Allergy1998285 Suppl16209988442
- HamidQAWenzelSEHankPJIncreased glucocorticoid receptor β in airway cells of glucocorticoid-insensitive asthmaJ Respir Crit Care Med19995916004
- HaukPJWenzelSETrumbleAEIncreased T-cell receptor vbeta8+ T cells in bronchoalveolar lavage fluid of subjects with poorly controlled asthma: a potential role for microbial superantigensJ Allergy Clin Immunol1999104374510400837
- HaukPJHamidQAChrousosGPInduction of corticosteroid insensitivity in human PBMCs by microbial superantigensJ Allergy Clin Immunol2000105782710756230
- HawrylowiczCMRichardsDFLokeT-KA defect in corticosteroid-induced IL-10 production in T lymphocytes from corticosteroid resistant asthmatic patientsJ Allergy Clin Immunol20021093697011842311
- HeaneyLGConwayEKellyCPredictors of therapy resistant asthma: outcome of a systematic evaluation protocolThorax200358561612832665
- HittelmanABBurakovDIniguez-LlukiJADifferential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteinsEMBO J1999185380810508170
- HurleyDMAcciliDStratakisCAPoint mutation causing a single amino acid substitution in the hormone-binding domain of the glucocorticoid receptor in familial glucocorticoid resistanceJ Clin Invest19918768061704018
- ImhofAWolffeAPTranscription: gene control by targeted histone acetylationCurr Biol19988R42249637914
- IrusenEMatthewsJGTakatiashiAMAP kinase-induced glucocorticoid-receptor phosphorylation reduces its activity: role in steroid-insensitive asthmaJ. Allergy Clin Immunol20021096495711941315
- ItoKBarnesPJAdcockIMGlucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1β-induced histone H4 acetylation on lysines 8 and 12Mol Cell Biol200020689190310958685
- ItoKItoMElliottWMDecreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Eng J Med2005352196776
- ItoKChungKFAdcockIMUpdate on glucocorticoid action and resistanceJ Allergy Clin Immunol20061175224316522450
- JanknechtRHunterTVersatile molecular glue: transcriptional controlCurr Biol1996695148805328
- JohnMLimSSeyboldJInhaled corticosteroids increase interleukin-10 but reduce macrophage inflammatory protein-1alpha, granulocyte-macrophage colony-stimulating factor and interferon-gamma release from alveolar macrophages in asthmaAm J Respir Crit Care Med1998157256629445307
- KamJCSzeflerSJSursWCombination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoidsJ Immunol1993151346068376786
- KamadaAKLeungDYMSzeflerSJSteroid resistance in asthma: our current understandingPediatr Pulmonol19921418061480444
- KamadaAKSzeflerSJLeungDYMThe growing problem of steroid resistant asthmaDrug Ther1993235568
- KameiYXuLHeinzelTA CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptorsCell199685403148616895
- KatsoulisKKontakiotisTLeonardopoulosISerum total antioxidant status in severe exacerbation of asthma: correlations with the severity of the diseaseJ Asthma2003408475414736084
- KellowayJSWyattRAAdlisSAComparison of patients’ compliance with prescribed oral and inhaled asthma medicationsArch Intern Med19941541349528002686
- KharitonovSABarnesPJNitric oxide, nitrotyrosine, and nitric oxide modulators in asthma and chronic obstructive pulmonary diseaseCurr Allergy Asthma Rep20033121912562551
- KibeAInoueHFukuyamaSDifferential regulation by glucocorticoid of interleukin-13-induced eosinophilia, hyperresponsiveness, and goblet cell hyperplasia in mouse airwaysAm J Respir Crit Care Med200316750612502476
- KikuchiYOkabeSTamuraGChemosensitivity and perception of dyspnea in patients with a history of near-fatal asthmaN Engl J Med19943301329348152444
- KinoTChrousosGPGlucocorticoid and mineralocorticoid resistance/hypersensitivity syndromesJ Endocrinol20011694374511375113
- KlaustermeyerWBNoritakeDTKwongFKChrysotherapy in the treatment of corticosteroid-dependent asthmaJ Allergy Clin Immunol19877972053106448
- KrsticMDRogatskyIYamamotoKPMitogen-activated and cyclin-dependent kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptorMol Cell Biol1997173947549199329
- LambertsSWJHuizengaATMde LandePClinical aspects of glucocorticoid sensitivitySteroids19961476906
- LaneSJArmJPStaynovDZChemical mutational analysis of the human glucocorticoid receptor cDNA in glucocorticoid-resistant bronchial asthmaAm J Respir Cell Mol Biol1994114288018337
- LaneSJAdcockIMRichardsDCorticosteroid-resistant bronchial asthma is associated with increased c-fos expression in monocytes and T lymphocytesJ Clin Invest19981022156649854051
- LassallePDelnesteYGossetPT and B-cell immune response to a 55 kDa endothelial cell-derived antigen in severe asthmaEur J Immunol1993237968038458370
- LeeTHBrattsandRLeungDCorticosteroid action and resistance in asthmaAm J Respir Crit Care Med1996154S51
- LeeWMitchellPTjianRPurified transcription factor AP-1 interacts with TPA-inducible enhancer elementsCell198749741523034433
- LeungDYMartinRJSzeflerSJDysregulation of interleukin 4, interleukin-5 and interferon gamma gene expression in steroid-resistant asthmaJ Exp Med199518133407807013
- LeungDYBloomJWUpdate on glucocorticoid action and resistanceJ Allergy Clin Immunol200311132212532089
- LiLBGolevaEHallCFSuperantigen-induced corticosteroid resistance of human T cells occurs through activation of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK-ERK) pathwayJ Allergy Clin Immunol200411410596915536411
- LimSCrawleyEWooPHaplotype associated with low interleukin-10 production in patients with severe asthmaLancet19983521139672280
- LockSHKayABBarnesNCDouble-blind, placebo-controlled study of cyclosporine A as a corticosteroid-sparing agent in corticosteroid-dependent asthmaJ Allergy Clin Immunol1996153509
- LokeTKMallettKHRatoffJSystemic glucocorticoid reduces bronchial mucosal activation of activator protein 1 components in glucocorticoid-sensitive but not glucocorticoid-resistant asthmatic patientsJ Allergy Clin Immunol20061183687516890760
- MilgromHBenderBAckersonLNoncompliance and treatment failure in children with asthmaJ Allergy Clin Immunol199698105178977504
- MillerBDStrunkRCCircumstances surrounding the deaths of children due to asthma. A case-control studyAm J Dis Child1989143129492816856
- MillerMKJohnsonCMillerDPSeverity assessment in asthma: an evolving conceptJ Allergy Clin Immunol2005116990516275365
- MooreKWde Waal MalefytRCoffmanRLIL-10 and the IL-10 receptorAnnu Rev Immunol20011968376511244051
- NahmDHLeeYEYimEJIdentification of cytokeratin 18 as a bronchial epithelial autoantigen associated with nonallergic asthmaAm J Respir Crit Care Med20021651536912045129
- National Institutes of HealthNHLB/WHO Workshop Report. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention: No. 95199536591176
- NewmanKBMasonUGSchmalingKBClinical features of vocal cord dysfunctionAm J Respir Crit Care Med1995152138267551399
- NimmagaddaSRSzeflerSJSpahnJDAllergen exposure decreases glucocorticoid receptor binding affinity and steroid responsiveness in atopic asthmaticsAm J Respir Crit Care Med199715587939001294
- OakleyRHSarMCidlowskiJAThe human glucocorticoid receptor β isoform: expression, biochemical properties, and putative functionJ Biol Chem1996271955098621628
- OakleyRHJewellCMYudtMRThe dominant negative activity of the human glucocorticoid receptor β isoformJ Biol Chem1999274278576610488132
- PazinMJKadonagaJTWhat’s up and down with histone deacetylation and transcription?Cell19978932589150131
- PoznanskyMCGordonACHDouglasJGResistance to methylprednisolone in cultures of blood mononuclear cells from glucocorticoid-resistant asthmatic patientsClin Sci198467639456488705
- RabeKFAdachiMLaiCKWorldwide severity and control of asthma in children and adults: the global asthma insights and reality surveysJ Allergy Clin Immunol200411440715241342
- ReichhardtHMTuckermannJPGottlicherMRepression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptorEMBO J20012071687311742993
- RichardsDFFernandezMCaulfieldJGlucocorticoids drive human CD8+ T cell differentiation towards a phenotype with high IL-10 and reduced IL-4, IL-5 and IL-13 productionEur J Immunol20003023445410940925
- RobinsonDSHamidQYingSPrednisolone treatment in asthma is associated with modulation of bronchoalveolar lavage cell interleukin-4, interleukin-5 and interferon-gamma cytokine gene expressionAm Rev Respir Dis19931484026
- RobinsonDSCampbellDADurhamSRSystematic assessment of difficult-to-treat asthmaEur Respir J2003224788314516138
- RogatskyILoganSKGarabedianMJAntagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinaseProc Natl Acad Sci USA199895205059482836
- SchwartzHLLowellFCMelbyJCSteroid resistance in bronchial asthmaAnn Intern Med19686949395673169
- SherERLeungDMSursWSteroid-resistant asthma cellular mechanisms contributing to inadequate response to glucocorticoid therapyJ Clin Invest1994933398282803
- ShinerRJNunnAJChungKFRandomised, double-blind, placebo-controlled trial of methotrexate in steroid-dependent asthmaLancet1990336137401973472
- SilkoffPELentAMBusackerAAExhaled nitric oxide identifies the persistent eosinophilic phenotype in severe refractory asthmaJ Allergy Clin Immunol200511612495516337453
- SousaARLaneSJCidlowskiJAGlucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor beta-isoformJ Allergy Clin Immunol20001059435010808175
- SpahnJDLeungDYSursWReduced glucocorticoid binding affinity in asthma is related to ongoing allergic inflammationJ Respir Crit Care Med1995151170914
- SpahnJDSzeflerSJSursWA novel action of IL-13: induction of reduced monocyte glucocorticoid receptor-binding affinityJ Immunol1996157265498805670
- SpahnJDLeungDYSzeflerSJNew insights into the pathogenesis and management of steroid-resistant asthmaJ Asthma199734177949168845
- SpauldingHSJrMansfieldLESteinMRFurther investigation of the association between gastro-oesophageal reflux and bronchoconstrictionJ Allergy Clin Immunol198269516217076992
- StricklandIKisichKHaukPJHigh constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroidsJ Exp Med20011935859411238589
- SzaparyDHuangYSimonsSSOpposing effects of corepressors and coactivators in determining the dose-response curve of agonists, and residual agonist-activity of antagonists, for glucocorticoid receptor-regulated gene expressionMol Endocrinol19991321082110598585
- ten BrinkeAZwindermanAHSterkPJ“Refractory” eosinophilic airway inflammation in severe asthma: effect of parenteral corticosteroidsAm J Respir Crit Care Med2004170601515215154
- ten BrinkeASterkPJMascleeAARisk factors of frequent exacerbations in diffiult-to-treat asthmaEur Respir J2005268121816264041
- WanSDeSmetJMAntoineMSteroid administration in heart and heart-lung transplantation: is the timing adequate?Ann Thoracic Surg1996616748
- WangCYBassukAGBoiseLHActivation of the granulocyte-macrophage colony-stimulating factor promoter in T cells requires cooperative binding of Elf-I and AP-1 transcription factorsMol Cell Biol199414115398289796
- WarehamNJHarrisonBDJenkinsPFA district confidential enquiry into deaths due to asthmaThorax1993481117208296254
- WebsterJCOakleyRHJewellCMProinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative β-isoform: a mechanism for the generation of glucocorticoid resistanceProc Natl Acad Sci USA20019868657011381138
- WenzelSESzeflerSJLeungDYBronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoidsAm J Respir Crit Care Med1997156737439309987
- WenzelSESchwartzLBLangmackELEvidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristicsAm J Respir Crit Care Med19991601001810471631
- WoolcockAJSteroid-resistant asthma: what is the clinical definition?Eur Respir J1993674378519386
- XystrakisEKusumakarSBoswellSReversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patientsJ Clin Invest20061161465516341266
- YamadaKElliottWMHayashiSLatent adenoviral infection modifies the steroid response in allergic lung inflammationJ Allergy Clin Immunol20001068445111080705