Abstract
Heat shock proteins (HSP) are molecular chaperones implicated in facilitation of protein folding and translocation between distinct compartments, and hence in preventing protein from aggregation. In terms of proteolysis, HSP act as a double-edged sword, stimulating proteasome-dependent proteolysis while preventing the degradation of the same proteins, even though in both cases association of unfolded proteins with HSP is the initial step. The proteasomal degradation products are utilised as ligands of major histocompatibility complex (MHC) class I molecules to be recognised by CD8+ T cells, leading to activation of cytotoxic T cell immunity indispensable in fighting virus infections and cancers. In this context, HSP-mediated antigen traffic towards proteasomal degradation is coupled with acquired T cell immunity. In addition, exogenous antigens internalised by dendritic cells (DC) are also forwarded to the proteasome, possibly through the ER-associated degradation (ERAD) system, based on the fusion of the ER-membrane to the endosome containing the antigens. Thus, antigens within endosomes might be translocated to the cytosol, possibly through the Sec61 complex recruited from ER and degraded by the proteasome, rendering their peptides presentable by MHC class I molecules, a process known as cross-presentation. Since binding protein (Bip) facilitates degradation of most ER luminal soluble proteins in yeast, it is possible that endosomal HSP in DC, mimicking the action of Bip, facilitate the degradation of internalised soluble antigens. This may explain why the HSP-peptide/protein complex is extremely efficient in terms of cross-presentation ability. In this review, we discuss how HSP are linked to the ubiquitin-dependent proteasome system to generate peptides presentable by MHC molecules.
Introduction
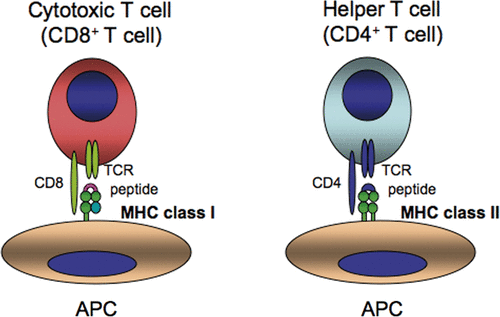
CD8+ and CD4+ T lymphocytes recognise peptides in the context of MHC class I and class II molecules, respectively (). The peptides associated with MHC class I molecules consist of 8–11 amino acids that are mostly derived from degradation products of endogenous cellular proteins. The peptide binding portion formed by the α1/α2 domain of MHC class I creates a β-sheet floor surrounded by two long α-helices, yielding pockets to which amino acids bind, and typically a peptide binds in the groove in an extended conformation Citation[1],Citation[2]. Peptides associated with the MHC class II peptide-binding groove consist of 10–34 amino acids and are mostly derived from exogenous proteins that enter the cell via phagocytosis/endocytosis. Based on the open ends of the peptide-binding groove of MHC class II, the bound peptides are not limited to a precise length.
Figure 1. CD8+ T cells and CD4+ T cells recognise peptides in the context of MHC class I and MHC class II molecules, respectively. TCR, T cell antigen receptor; APC, antigen presenting cell.
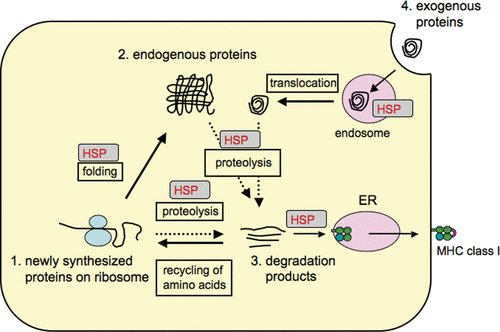
In the strict rules of the antigen processing/presentation system, there is ample evidence to suggest that heat shock proteins (HSP) play critical roles in supporting protein-traffic towards protein degradation to produce antigenic peptides presentable by MHC molecules. HSP are known as molecular chaperones and many members of this family are induced by heat shock and other conditions that cause protein denaturation and unfolding. The main functions of HSP are to (1) facilitate protein folding/refolding and prevent aggregation of newly synthesised proteins on the ribosome; (2) maintain the normal function of mature proteins; (3) target misfolded and/or damaged mature proteins for degradation by the proteasome; (4) transport proteins between distinct microorganisms (cellular compartments, see (). All proteins denoted here finally undergo degradation by the proteasome in order to recycle amino acids and their bi-products (degradation products, peptides) are simultaneously utilised as antigen peptides mainly presented by MHC class I molecules. Thus, the use of proteasomal degradation products as ligands of MHC molecules makes perfect sense in the immune system to alert against any antigenic alterations to the T cell immune system as well as to the life cycle of all proteins with no waste. Each step towards proteolysis by the proteasome is, at least in part, regulated by HSP and will be discussed in some detail.
Figure 2. HSP mediate degradation of a few percent of newly synthesised proteins as well as mature proteins. In dendritic cells (DC), HSP seem to facilitate the degradation of even internalised exogenous proteins following their translocation to the cytoplasm. The proteolysis step depends on the ubiquitin-proteasome system to produce peptides presentable by MHC class I molecules.
Role of HSP in processing endogenous antigen
General features of endogenous antigen processing
CD8+ T cells recognise naturally processed peptides in the context of MHC class I molecules Citation[3]. Peptides presented by MHC class I molecules are mainly products of cellular proteins degraded by the ubiquitin-dependent proteasome system Citation[4]. During ribosomal de novo protein synthesis, less than 3% of the proteins seem to undergo misfolding, which occurs mainly due to mutational changes in the encoding gene, transcriptional and translational mistakes, and unsuccessful protein folding. These misfolded proteins are called defective ribosomal products (DRiPs) and are implicated as the main source of antigen peptides presented by MHC class I molecules Citation[5],Citation[6]. The peptides produced by the proteasome are translocated through transporter associated with antigen processing (TAP) molecules into the endoplasmic reticulum (ER), where they associate with MHC class I-β2m hetero complex to form stable tri-molecular complexes Citation[7]. These endogenous peptides are displayed on the cell surface in the context of MHC class I molecules to activate CD8+ T cells. HSP participate in the selection of cellular proteins that are poised for degradation by the proteasome and in transportation of proteasomal degradation products to the ER for loading peptides onto MHC class I-β2 m hetero complex.
Role of HSP in trafficking of endogenous peptide towards processing into MHC class I ligands
Unfolded proteins are recognised by molecular chaperones, Hsc70/Hsp40 and Hsp90, and can be polyubiquitinated by various E3 ubiquitin ligases, such as carboxyl terminus of Hsc70 interacting protein (CHIP), which is eventually followed by degradation by the proteasome Citation[8],Citation[9]. Kunisawa and Shastri Citation[10] found that proteins to be degraded into MHC class I ligands by the proteasome are associated with cytosolic Hsp90α (but not Hsp90β), and group II chaperonin TRiC Citation[11], and that the quantity of proteins (and/or large proteolytic intermediates) chaperoned by Hsp90 were increased when cells were treated with proteasome inhibitors. They also found that geldanamycin (GA), a Hsp90 inhibitor, reduced the associated proteins and down-regulated endogenous antigen processing. The relationship between DRiPs and Hsp90-associated proteins is not clear, but it is possible that DRiPs are first captured by Hsp90 (and/or Hsp70) after their formation and degraded by the ubiquitin-proteasome system.
CHIP was originally identified as a co-chaperone of Hsp70 that inhibits Hsp40-stimulating ATPase activity of Hsp70 Citation[12], and is also known to play important roles in protein degradation Citation[13]. CHIP contains two functional domains: a C-terminal U-box-like domain that can act as E3-ligase for ubiquitinylation of substrates and an N-terminal tetratricopeptide repeat (TPR) domain that associates with Hsp70 or Hsp90 through their C-terminal-EEVD motif Citation[13]. Therefore, CHIP ubiquitinylates misfolded proteins chaperoned by Hsp70/Hsp40 or Hsp90, and thus serves as a bridge between HSP-associated substrates and their delivery to the 26S proteasome Citation[14]. Inhibitors of Hsp90 such as GA and radicicol that bind ATP binding pockets, abrogate the ATP-dependent chaperone cycle of Hsp90 Citation[15]. Treatment of cells with such specific inhibitors suppresses the association between Hsp90 and proteins, which could result in failure of protein ubiquitinylation by CHIP, leading to unsuccessful processing by the proteasome. Therefore, Hsp90 inhibitors spoil proteins that would, otherwise, be properly processed by the proteasome to be MHC class I ligands and/or their N-terminal extended precursors, which in turn explains the down-regulation of MHC class I molecules on the cell surface Citation[10]. This is a reasonable illustration to understand how peptides are produced from misfolded proteins and processed in the cytosol.
This concept, on the other hand, seems to be in conflict with the general observation that GA stimulates Hsp90 to release client proteins that eventually undergo proteolysis by the proteasomes Citation[16–18]. In such case, degradation of proteins associated with Hsp90 is abrogated. Therefore, GA treatment should stimulate degradation of many client proteins, resulting in production of many T cell epitopes. However, several studies including ours indicate that Hsp90 inhibitors down-regulate MHC class I antigen processing Citation[10],Citation[19],Citation[20]. In this context, endogenous antigen processing might be transiently up-regulated during the early phase of incubation (i.e.<1 hour after GA treatment), while longer incubation (>8–12 hours) decreases this process due to consumption of the endogenous peptides and proteins that are pooled on Hsp90, an issue that needs careful evaluation. Nonetheless, it remains a mystery why Hsp90 actively participates in ubiquitin-dependent proteolysis of some proteins; while in contrast, it prevents degradation of others, although the association of proteins with Hsp90 is an initial step in both cases. Indeed, analogous observation was reported, in which cystic fibrosis transmembrane conductance regulator (CFTR) is either folded or degraded by GA: in one case, GA enhanced degradation of CFTR in mammalian cells Citation[21], while it blocked the degradation in the rabbit reticulocyte lysate Citation[22]. It is possible that Hsp90 is not a unidirectional chaperone but plays a diverse number of functions, probably depending on the conformation of the associated substrates and on its co-chaperones.
Recently, Callahan et al. Citation[20] showed that only the N-terminally extended proteolytic intermediates of MHC class I ligands are found associated with Hsp90 in cells, indicating that the Hsp90-associated peptides are post-proteasomal degradation products. The N-terminally elongated peptides are likely to be ferried to TAP molecules to enter the ER lumen by Hsp90. The same group found that radicicol treatment empties Hsp90 by releasing the peptides, resulting in unloading of such peptides onto MHC class I molecules in the ER, thereby, down-regulating the cell surface MHC class I molecules Citation[20]. The peptides isolated in that study were only N-terminally extended versions, whereas the findings of Kunisawa and Shastri Citation[11] indicated the association of both N- and C-extensions with Hsp90, although both experiments used the same KOVAK system (K-OVA-K; both N- and C-terminal flanking residues of OVA 257-264 are replaced with lysine (K) by which the epitope becomes cleavable by trypsine and carboxypeptidase B, respectively. Also K within the epitopic sequence is replaced with histidine (H) to make this epitope refractory to trypsin) to analyse the structures of the peptides associated with HSP. The reason for this discrepant observation remains to be elucidated. However, different isolation procedures for Hsp90 could reconcile the two observations. Isolation of Hsp90 by affinity chromatography with a specific antibody, as reported by Kunisawa and Shastri Citation[11], might purify mainly the Hsp90-protein complex. On the other hand, the use of an antibody-independent purification method for HSP-peptide complex, as reported by Callahan et al. Citation[20] might exclude the Hsp90 protein complex. Apart from the purification method, it is likely that Hsp90 directs antigen trafficking in both pre- and post-proteasomal events towards peptide loading onto MHC class I molecules in the ER.
Another issue to be kept in mind is that Hsp90 inhibitors transiently (within three hours after treatment) disrupt the structures of the 26S proteasome in mammalian cells Citation[23] and delay the re-assembly after ATP-depletion or heat shock Citation[24] in yeast. The results indicate that Hsp90 plays a role in the maintenance of the structure of the 26S proteasome, although other unknown molecules can redundantly overcome the assembly after Hsp90 inhibitor treatment. It is controversial whether the 26S proteasome releases the lid and base of PA700 regulatory particle during ATP-dependent proteolysis of ubiquitinylated substrates Citation[25],Citation[26]. The regulatory mechanisms for disassembly and re-assembly of the 26S proteasome need to be further clarified, and the dynamics of the 26S proteasome structure could influence the processing of endogenous antigens.
Processing of exogenous antigens and cross-presentation
General features of exogenous antigen processing
Macrophages can take up soluble- or cell-associated antigens in the endosome and/or phagosome and digest them with lysosomal enzymes. Hence, the proteolytic fragments (peptides) are loaded onto MHC class II molecules that have been sorted from the ER, and MHC class II-peptide complexes are moved up on the cell surface to display those peptides to CD4+ T cells Citation[27], by which antibody production from B cells is stimulated. Interestingly, Hsp90 might be involved in this antigen presentation to activate CD4+ T cells because one group argued that presentation of exogenous antigen in the context of MHC class II molecules was completely blocked by GA Citation[28], although the mechanism was not fully understood. Regardless of the involvement of Hsp90, internalised exogenous antigens undergo proteolysis in the endosome/lysosome and the resulting peptides are presented to CD4+ T cells. Should this be the only pathway for presentation of exogenous antigen by APC, CD8+ T cells specific to viral or tumour antigens would not be activated. However, one would not have to worry about this because exceptionally specialised machinery seems to be utilised in DC to secure the priming of CD8+ T cells after internalisation of exogenous antigens Citation[27],Citation[29], as described below.
Cross-presentation is essential for activation of CD8+ T cells by exogenous antigens
DC are the most powerful antigen presenting cells (APC) and able to prime naïve T cells to confer protective cytotoxic T cell (CD8+ T cells) immunity against cancers and infectious agents Citation[30]. During viral infection or malignant transformation, intracellular changes in non-APC must be reported to CD8+ T cells, which is indispensable to fighting virus-infected cells and cancer cells Citation[31]. However, the latter cells cannot act by themselves as APC to prime T cells, although they express tumour antigens on the cell surface. In addition, DC do not become infected by certain viruses, and thus, cannot present such viral antigens directly to CD8+ T cells. Therefore, DC must internalise those adjacent tumour or infected cells, digesting them, and then present antigen peptides to CD8+ T cells in the context of MHC class I molecules (). This pathway is called cross-presentation Citation[32], and is believed to be specific to DC, not other APC types such as macrophages or B cells. In DC, significant proportions of exogenous antigens within the endosome are translocated to the cytosol Citation[33], ubiquitinylated and degraded by the 26S proteasome to render the peptides presentable by MHC class I molecules. The produced peptides enter the ER through TAP molecules to associate with MHC class I molecules Citation[7], and move up on the cell surface to activate CD8+ T cells.
Mechanistic insight into cross-presentation
In cross-presentation, the pathway through which exogenous antigens can access MHC class I molecules remains obscure. The delivery of exogenous proteins within the endosome to the cytosol might require the same protein complex machinery, responsible for dislocation of misfolded proteins in the ER. ER-misfolded proteins dislocated to the cytosol are polyubiquitinylated for degradation by the 26S proteasome, a process known as the ER-associated degradation (ERAD) pathway Citation[34],Citation[35]. A mechanistic explanation of the cross-presentation can be found in the indirect observation that the ER membranes possibly fuse with the nascent phagosome, referred to as ER-phagosome fusion Citation[36]. ER-phagosome fusion allows internalised exogenous antigen to dislocate to the cytosol, followed by degradation by the proteasome, resulting in production of antigen peptides presentable by MHC class I molecules Citation[37], Citation[38]. If this is true, the pore structure for translocation of the antigen to the cytosol is the Sec61 complex, which is utilised for dislocation of ER-misfolded proteins to the cytosol by the ERAD pathway. Pseudomonas aeruginosa Exotoxin A, an inhibitor for retro-translocation of ER-misfolded proteins, substantially blocked cross-presentation Citation[33]. However, there is still controversy on whether or not the Sec61 complex structure is required for cross-presentation, because the toxin was reported to bind AAA ATPase Cdc48 (in yeast)/p97 (in mammals) but not Sec61 complex Citation[33].
Exogenous proteins in the endosome must be relatively unfolded before being translocated to the cytosol, since the size of the membrane pore is not large enough for escape while keeping its native form Citation[39],Citation[40]. Such unfolded proteins are reminiscent of the DRiPs; the main source of peptides presented by MHC class I molecules as described above. If the unfolded antigen is similar to DRiPs, it must first be recognised by HSP, both in the endosome and cytosol, for its smooth translocation and then re-folded or degraded by the proteasome Citation[41]. In this context, HSP might play critical roles that will be discussed below.
Role of HSP in cross-presentation
HSP facilitate ERAD, and then stimulate cross-presentation?
Phagosome fusion with ER-membranes to form ER-phagosome might enable translocation of internalised exogenous antigen to the cytosol, as described in Sections 2–3, which is suggested to be an important mechanism for cross-presentation.
To consider the roles of molecular chaperones in ERAD, we need to discriminate between misfolded proteins in the ER lumen and integral membrane proteins of the ER Citation[42]. In yeast, the ER luminal proteins are dislocated to the cytosol and degraded by the ubiquitin-proteasome system. This process requires ER luminal chaperones such as Bip and its J-domain containing co-chaperones, Jem1p and Scj1p and E3 ubiquitin ligase, Hrd1p, together with E2 enzyme, Ubc7p. Bip is not only required for maintaining the solubility of misfolded proteins but also for stimulating the translocation of the substrates to the cytosol through Sec61p complex. Indeed, Bip seems to accelerate the degradation of almost all known soluble ER substrates Citation[43]. In this context, substrates with high affinity to Bip would be selected as good candidates for retro-translocation.
On the other hand, integral membrane proteins require cytosolic chaperone Hsp70 (Ssa1p) and J-domain containing Hlj1p and Ydj1p, in addition, E3 ubiquitin ligase Doa10p in conjunction with the E2 enzymes, Ubc6p and Ubc7p, for their dislocation Citation[44],Citation[45]. Polyubiquitinylation is a prerequisite for both ER luminal and membrane spanning proteins to be translocated to the cytosol in a manner dependent on Cdc-48 and its cofactors, Npl4p and Ufd1p. Cdc48p might pull these substrates and segregate them from the ER membranes and forward them to the 26S proteasome. Nakatsukasa et al. Citation[46] recently showed that cytosolic Hsp70/Hsp40 facilitates the association of Ste6p and/or CFTR, both of which are membrane spanning proteins, with Doa10p, an E3 ubiquitin ligase known to stimulate the ubiquitinylation of the substrates in conjunction with Ubc6p and Ubc7p. Ubiquitinylated Ste6p is recognised by Cdc48p complex and released into the cytosol, and its ubiquitin chain is further extended by E4 ubiquitin ligase, Ufd2p, then, degraded by the 26 S proteasome.
Overall, the aforementioned results indicate that cytosolic and ER luminal Hsp70/Hsp40 chaperones enhance ERAD of membrane spanning and soluble proteins, respectively. Taking these findings into account, and assuming that ER-phagosome fusion is required for cross-presentation, it is probable that internalised soluble antigens require Bip to stimulate their retro-translocation to the cytosol. Whether this is the case or not remains to be elucidated, but internalised ovalbumin (OVA) was previously found to associate with Bip in DC Citation[47]. Cross-presentation of cellular antigens might require cytosolic Hsp70/Hsp40, an issue that remains to be clarified. It is well known that the cross-presentation efficiency of cellular antigens is considerably higher than soluble forms, which might indicate that Bip recruited from ER to phagosome is not abundant enough to fully activate ERAD-like mechanism for internalised soluble antigen. In contrast, cytosolic Hsp70/Hsp40, which is already abundant in the cytoplasm, might play a prominent role in ERAD-like mechanism of internalised cellular antigens. These aspects of cross-presentation are very important and must be investigated carefully. Based on these ideas, we should move next to consider why HSP-peptide (and/or protein) complex, but not free peptide (and/or protein), is efficient in cross-presenting the associated peptides to CD8+ T cells.
Role of HSP-peptide complex in antigen donor cells for cross-presentation
Vaccination with heat shock proteins (HSP) protects mice against challenge with tumour from which the HSP were isolated Citation[48],Citation[49]. The antigenicity of HSP vaccination is based on HSP-associated endogenous MHC class I peptides or their precursors Citation[48],Citation[50]. The vaccination effect is mediated by CD8+ T cells, indicating that HSP can cross-prime T cells in vivo, acting as a carrier for antigens Citation[51]. Srivastava and colleagues Citation[52] pioneered these studies and CD91 (a receptor for α2M macroglobulin, α2M) was identified as a receptor for Hsp90, Hsp70, Gp96 and calreticulin for cross-presentation Citation[53],Citation[54]. Rabbit polyclonal antibodies to CD91 and α2M protein were reported to inhibit the Gp96-mediated cross-presentation Citation[53]. Intriguingly, CD91 is also a receptor for P. aeruginosa Exotoxin A Citation[55] that blocks ERAD and cross-presentation. The results indicate that the route from internalisation of HSP-peptide complex to its retro-translocation to the cytosol overlaps with the one for Exotoxin A.
Vaccination using cellular antigens (antigen-bearing parenchymal cells) is extremely efficient compared to the use of soluble antigens. In this context, HSP in antigen donor cells seems to play an important role. HSF-1 is a major transcription factor for induction of many HSP and is indispensable for triggering heat shock response to various stimuli. Immunisation using HSF-1 deficient cells (whose expression of Hsp90 and Hsp70 is less than those of normal cells) confers less efficient generation of cytotoxic T lymphocyte (CTLs) to multiple antigens Citation[56], providing a genetic evidence for the role of HSF-1 in regulating cross-presentation of MHC class I-associated antigens. Even vaccination using cell lysates containing antigens can cross-prime CD8+ T cells and generate CTLs specific to β-galactosidase and OVA. Intriguingly, depletion of Hsp90, Hsp70, Gp96, and calreticulin by specific antibody-absorption from the total lysates abrogated the cross-priming ability Citation[57]. Although these results clearly indicate that antigen peptides (and/or protein fragments) chaperoned by HSP are critical for cross-priming, there are reports in disagreement with these observations Citation[58]. Intact proteins to be degraded by the proteasome are reported to be sufficient to cross-prime T cells because depletion of normal steady-state OVA from the cell fractions eliminated its cross-priming activity Citation[58]. Although this issue is still debatable, at least, it is very clear that intact proteins by themselves remain to be poor vaccine candidates unless they are mixed with stimulators for innate immunity such as Toll-like receptor (TLR)-ligands.
In considering why the HSP peptide complex is so efficient in terms of cross-presentation, one possibility is that internalised HSP into the endosome can mimic the function of Bip, which is normally recruited from the ER to the endosome, leading to the enhancement of ERAD-like mechanism. The peptides chaperoned by HSP might be efficiently translocated to the cytosol and processed by the 26S proteasome. Since HSP-associated peptides seem to be short steretched peptides, ubiquitinylation of such peptides might not be necessary for their translocation and degradation by the 26S proteasome. A similar mechanism would operate when proteins chaperoned by HSP are internalised by DC, although they require polyubiquitinylation for the degradation. Indeed, proteins captured by HSP110 were reported to cross-prime T cells efficiently Citation[59],Citation[60]. A complex between HSP110 and the intracellular domain (ICD) of human epidermal growth factor receptor 2 protein (HER-2)/neu was formed by heat shock Citation[59],Citation[60]. This HSP110-ICD vaccine elicited both CD8+ and CD4+ T-cell responses against ICD. Proteins internalised into the endosome encounter a low pH that might induce alteration of their structure, resulting in misfolding. The association of HSP with such proteins might keep their solubility and efficient retro-translocation to the cytoplasm would be promised. The association of substrates with HSP in these cases is non-covalent and sensitive to ATP hydrolysis. It is not clear at this stage whether only peptides or both peptides and HSP are retro-translocated to the cytosol.
On the other hand, genetically engineered fusion proteins comprising HSP and short peptides such as T cell epitopes and even proteins, whose association is covalent, if the structure of HSP is not altered to maintain their solubility, seem to work as a vehicle for cross-presentation, similar to the HSP-peptide complex Citation[59],Citation[60]. We generated a recombinant fusion protein consisting of full-length Hsc70 and Kb-restricted dominant T-cell epitope, OVA257-264 and four other different peptides to investigate the mechanism of HSP-mediated cross-presentation Citation[61],Citation[62]. Injection of Hsc70-OVA257-264 stimulated proliferation of adoptively transferred OT-I (transgenic TCR specific for OVA257-264 plus H-2Kb) CD8+ T cells in the spleen and generated specific CTLs Citation[61],Citation[62]. Thus, OT-I CD8+ T cells proliferate, differentiate into active CTLs and produce IFNγ. The Hsp70-mediated cross-priming was abrogated in TAP1KO mice and DC-depleted CD11c/DTR mice (unpublished results). By using this system and various TLRs KO mice, we are currently investigating the cross-priming mechanism by Hsp70 in the context of MyD88 signalling.
Recent evidence suggests that TLR-MyD88 signalling is essential for cross-presentation of soluble antigens Citation[63]. One reason for the requirement of MyD88 is that the recruitment of TAP molecules to early endosomes is dependent on TLR4-MyD88 signalling because the OVA protein generally used in antigen presentation assay contains few but biologically active endotoxins such as LPS Citation[64]. Peptides generated from exogenous protein in the cytoplasm enter, through TAP molecules, the lumen of early endosomes from which it dislocates, and associate with MHC class I molecules. The recruitment of TAP to early endosomes might result from facilitation of ER-phagosome fusion. Another possible mechanism–acceleration of ERAD for exogenous proteins–might operate in a TLR4-MyD88-dependent manner, which remains to be investigated.
Is cross-presentation governed by receptors through which exogenous antigens are internalised?
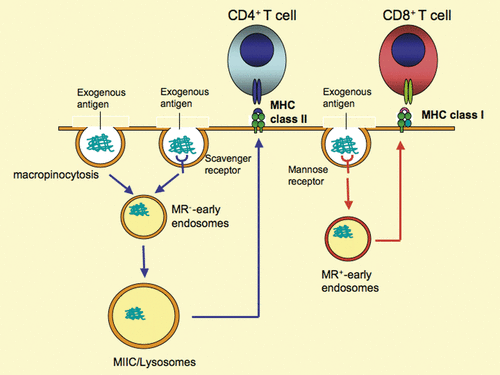
OVA can be internalised by DC in a fluid phase-, scavenger receptor- and mannose receptor-mediated manner. Of these three distinct internalisation routes, mannose receptor is the only one required for cross-presentation Citation[27] (). OVA endocytosed by other receptors seems to be presented to CD4+ T cells in the context of MHC class II molecules Citation[27] (). This evidence suggests that there are some specialised receptors by which only internalised exogenous antigens are cross-presented in the context of MHC class I molecules. Thus, receptors utilised for internalisation of exogenous antigens might control the pathway through which they are presented to either CD4+ or CD8+ T cells. The mechanism underlying the segregation of the pathways is linked with whether the internalised antigen is retro-translocated to the cytosol or not. Leakage of internalised OVA into the cytoplasm was found in normal DC, but not in mannose receptor-deficient DC Citation[27]. Thus, the mannose receptor-mediated internalisation is linked to the ERAD-like pathway, whereas other receptors are not. In this context, CD91-mediated internalisation is associated with the ERAD-like pathway because this pathway is blocked by Exotoxin A, as described. The reason why mannose receptor- or CD91-dependent internalisation can utilise the ERAD-like pathway is unknown. One cannot exclude the possibility of a slow rate of maturation of the fusion of the endosome with the lysosome following mannose receptor- and CD91-mediated internalisation compared to those by other routes. The slow maturation of the endosome might give enough time during which internalised antigens are dislocated by the ERAD-like pathway, otherwise they will be rapidly degraded by lysosomal enzymes. Several minutes would be sufficient for dislocation of internalised proteins and their appearance in the cytosol. Thus, there might be only a few minutes delay of mannose receptor-mediated endosomal maturation even if it is slower than that of scavenger receptor-mediated internalisation. Alternatively, it is possible that the mannose receptor-mediated endosome somehow can fuse with ER-membranes, purchasing ERAD-like pathway, while scavenger receptor-mediated endosome cannot. The exact mechanism needs to be elucidated in the future.
Figure 3. Different fates of internalised antigens by distinct cell surface receptors. The antigen internalised by macropinocytosis and/or scavenger receptor-mediated endocytosis is presented by MHC class II molecules to CD4+ T cells. In contrast, the same antigen internalised by mannose receptor-dependent endocytosis is presented by MHC class I molecules to CD8+ T cells. The molecular mechanisms responsible for the two distinct processes remain to be elucidated. However, the difference between the two pathways could be linked to the diverse mechanisms responsible for antigen trafficking associated with HSP, which remain elusive. MIIC, MHC class II compartment; MR, mannose receptor.
Conclusions
We focused mainly on the role of HSP in protein degradation and peptide production by the proteasome towards MHC class I antigen presentation. Proteolysis and protein/peptide trafficking is certainly not a random process but is elaborately regulated by HSP, leading to the control of production of peptides presentable by MHC molecules. In this context, acquired (T cell) immunity is tightly coupled with HSP-proteasome interplay. T cell recognition of antigenic peptides causes self and non-self discrimination, however, we could alternatively say that to be or not to be degraded by the proteasome system regulates the consequence, which all might boil down to decisions by HSP that can sense subtle conformational changes of proteins. It would be wonderful if we can manipulate the functions and expression levels of individual chaperones to regulate the immune system associated with host defence against infectious diseases and cancers, and even autoimmune diseases.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Townsend A, Bodmer H. Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol 1989; 7: 601–624
- Rammensee HG, Falk K, Rotzschke O. Peptides naturally presented by MHC class I molecules. Annu Rev Immunol 1993; 11: 213–244
- Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol 1999; 17: 739–779
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000; 404: 770–774
- Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): A major source of antigenic peptides for MHC class I molecules?. J Immunol 1996; 157: 1823–1826
- Qian SB, Reits E, Neefjes J, Deslich JM, Bennink JR, Yewdell JW. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J Immunol 2006; 177: 227–233
- Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev 2005; 207: 145–157
- Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2001; 2: 1133–1138
- Murata S, Chiba T, Tanaka K. CHIP: A quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol 2003; 35: 572–578
- Kunisawa J, Shastri N. Hsp90α chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity 2006; 24: 523–534
- Kunisawa J, Shastri N. The group II chaperonin TRiC protects proteolytic intermediates from degradation in the MHC class I antigen processing pathway. Mol Cell 2003; 12: 565–576
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 1999; 19: 4535–4545
- McDonough H, Patterson C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 2003; 8: 303–308
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 2001; 3: 100–105
- Workman P. Overview: Translating Hsp90 biology into Hsp90 drugs. Curr Cancer Drug Targets 2003; 3: 297–300
- Lu C, Chen D, Zhang Z, Fang F, Wu Y, Luo L, Yin Z. Heat Shock Protein 90 regulates the stability of c-Jun in HEK293 Cells. Mol Cells 2007; 24: 210–214
- Zhang L, Nephew KP, Gallagher PJ. Regulation of death-associated protein kinase. Stabilization by HSP90 heterocomplexes. J Biol Chem 2007; 282: 11795–11804
- Matei D, Satpathy M, Cao L, Lai YC, Nakshatri H, Donner DB. The platelet-derived growth factor receptor α is destabilized by geldanamycins in cancer cells. J Biol Chem 2007; 282: 445–453
- Yamano T, Murata S, Shimbara N, Tanaka N, Chiba T, Tanaka K, Yui K, Udono H. Two distinct pathways mediated by PA28 and hsp90 in major histocompatibility complex class I antigen processing. J Exp Med 2002; 196: 185–196
- Callahan MK, Garg MM, Srivastava PK. Heat-shock protein 90 associates with N-terminal extended peptides and is required for direct and indirect antigen presentation. Proc Natl Acad Sci USA 2008; 105: 1662–1667
- Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J 1998; 17: 6879–6887
- Fuller W, Cuthbert AW. Post-translational disruption of the delta F508 cystic fibrosis transmembrane conductance regulator (CFTR)-molecular chaperone complex with geldanamycin stabilizes delta F508 CFTR in the rabbit reticulocyte lysate. J Biol Chem 2000; 275: 37462–37468
- Yamano T, Mizukami S, Murata S, Chiba T, Tanaka K, Udono H. Hsp90-mediated assembly of the 26 S proteasome is involved in major histocompatibility complex class I antigen processing. J Biol Chem 2008; 283: 28060–28065
- Imai J, Maruya M, Yashiroda H, Yahara I, Tanaka K. The molecular chaperone Hsp90 plays a role in the assembly and maintenance of the 26S proteasome. EMBO J 2003; 22: 3557–3567
- Babbitt SE, Kiss A, Deffenbaugh AE, Chang YH, Bailly E, Erdjument-Bromage H, Tempst P, Buranda T, Sklar LA, Baumler J, et al. ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell 2005; 121: 553–565
- Kriegenburg F, Seeger M, Saeki Y, Tanaka K, Lauridsen AM, Hartmann-Petersen R, Hendil KB. Mammalian 26S proteasomes remain intact during protein degradation. Cell 2008; 135: 355–365
- Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007; 316: 612–616
- Rajagopal D, Bal V, Mayor S, George A, Rath S. A role for the Hsp90 molecular chaperone family in antigen presentation to T lymphocytes via major histocompatibility complex class II molecules. Eur J Immunol 2006; 36: 828–841
- Rock KL. Exiting the outside world for cross-presentation. Immunity 2006; 25: 523–525
- Steinman RM. Dendritic cells in vivo: A key target for a new vaccine science. Immunity 2008; 29: 319–324
- Huang AY, Bruce AT, Pardoll DM, Levitsky HI. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity 1996; 4: 349–355
- Bevan MJ. Cross-priming. Nat Immunol 2006; 7: 363–365
- Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006; 25: 607–617
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 2002; 4: 134–139
- Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol 2002; 3: 246–255
- Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 2002; 110: 119–131
- Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003; 425: 397–402
- Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M. Phagosomes are competent organelles for antigen cross-presentation. Nature 2003; 425: 402–406
- Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC. Rapoport TA. 2004. X-ray structure of a protein-conducting channel. Nature 2004; 427: 36–44
- Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol 2008; 86: 353–362
- Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem J 2007; 404: 353–363
- Nishikawa S, Brodsky JL, Nakatsukasa K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem 2005; 137: 551–555
- Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annu Rev Genet 2001; 35: 149–191
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem 2004; 279: 38369–38378
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 2004; 165: 41–52
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 2008; 132: 101–112
- Imai J, Hasegawa H, Maruya M, Koyasu S, Yahara I. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int Immunol 2005; 17: 45–53
- Udono H, Srivastava PK. Heat shock protein 70-associated peptides elicit specific cancer immunity. J Exp Med 1993; 178: 1391–1396
- Tamura Y, Peng P, Liu K, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 1997; 278: 117–120
- Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, Ono T, Hizuta A, Tanaka N, Srivastava PK, Nakayama E. Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 1999; 162: 1303–1309
- Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995; 269: 1585–1588
- Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 2002; 20: 395–425
- Binder RJ, Han DK, Srivastava PK. CD91: A receptor for heat shock protein gp96. Nat Immunol 2000; 1: 151–155
- Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001; 14: 303–313
- FitzGerald DJ, Fryling CM, Zdanovsky A, Saelinger CB, Kounnas M, Winkles JA, Strickl D, Leppla S. Pseudomonas exotoxin-mediated selection yields cells with altered expression of low-density lipoprotein receptor-related protein. J Cell Biol 1995; 129: 1533–1541
- Zheng H, Li Z. Cutting edge: Cross-presentation of cell-associated antigens to MHC class I molecule is regulated by a major transcription factor for heat shock proteins. J Immunol 2004; 173: 5929–5933
- Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol 2005; 6: 593–599
- Shen L, Rock KL. Cellular protein is the source of cross-priming antigen in vivo. Proc Natl Acad Sci USA 2004; 101: 3035–3040
- Manjili MH, Henderson R, Wang XY, Chen X, Li Y, Repasky E, Kazim L, Subjeck JR. Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res 2002; 62: 1737–1742
- Facciponte JG, Wang XY, Subjeck JR. Hsp110 and Grp170, members of the Hsp70 superfamily, bind to scavenger receptor-A and scavenger receptor expressed by endothelial cells-I. Eur J Immunol 2007; 37: 2268–2279
- Udono H, Yamano T, Kawabata Y, Ueda M, Yui K. Generation of cytotoxic T lymphocytes by MHC class I ligands fused to heat shock cognate protein 70. Int Immunol 2001; 13: 1233–1242
- Mizukami S, Kajiwara C, Ishikawa H, Katayama I, Yui K, Udono H. Both CD4+ and CD8+ T cell epitopes fused to heat shock cognate protein 70 (hsc70) can function to eradicate tumors. Cancer Sci 2008; 99: 1008–1015
- Palliser D, Ploegh H, Boes M. Myeloid differentiation factor 88 is required for cross-priming in vivo. J Immunol 2004; 172: 3415–3421
- Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol 2008; 9: 558–566