Abstract
Purpose: Molecular mechanisms underlying hyperthermia-induced cellular injury are not fully understood. The aim of this study was to identify the components of mitochondrial oxidative phosphorylation affected by mild hyperthermia and to quantify the contribution of each component to changes in system behaviour.
Methods: Temperature effects on the oxidative phosphorylation in isolated rat-heart mitochondria were assessed using modular kinetic analysis. Mitochondrial H2O2 production and lipid peroxidation were measured for estimation of temperature-induced oxidative damage.
Results: The increase of temperature in the febrile range (40°C) slightly activated mitochondrial function through stimulation of the respiratory module, without affecting the kinetics of the proton leak and phosphorylation modules. At 42°C, state 3 respiration rate remained unchanged, the proton leak across the inner mitochondrial membrane was substantially increased, the respiratory module slightly inhibited, leading to decreased membrane potential (Δψ) and diminished ATP synthesis (16% lower phosphorylation flux). Increase of temperature above 42°C caused dissipation of Δψ and abolishment of ATP synthesis indicating complete uncoupling of oxidative phosphorylation. The changes in mitochondrial functions induced by incubation at 42°C were completely reversible in contrast to only partial recovery after incubation at higher temperature (45°C). Furthermore, hyperthermia stimulated the production of H2O2 and membrane lipid peroxidation with maximal rates observed at 40°C.
Conclusions: We demonstrated for the first time that febrile temperature (40°C) activates mitochondrial energy supplying functions, whereas further temperature increase by only a few degrees leads to severe impairment of mitochondrial ability to maintain ΔΨ and synthesise ATP.
Introduction
The events following the exposure of cells to moderate heating involve interplay of multiple factors operating at different regulatory levels and inducing changes in the metabolic activities, signal transduction and gene expression. The death or survival of different cells upon hyperthermia is determined by molecular mechanisms that are not yet well established. Despite physiological importance, the information about the molecular events induced by fever in various types of cells is scarce Citation1. Supra-physiological hyperthermia (usually in the range of 42°–45°C) is clinically applied for cancer treatment Citation2, Citation3. However, it is still not clear why cells of different types of tumours display different temperature sensitivity Citation4, Citation5, as well as why some normal types of cells are more sensitive (e.g. neurons, cardiomyocytes) than other normal cells Citation6–9. Both for normal and tumour cells quite little is known about differences in response to mild heating compared to more severe hyperthermic conditions.
It was suggested that mitochondria are important players in the development of the heat-stress induced apoptosis in certain tumour cell lines Citation10, Citation11 as well as in cardiomyocytes Citation9 and hematopoietic cell lines Citation12. These organelles perform multiple functions in cell life, including energy supply, maintenance of ion balance and transduction of apoptotic signals. However, the response of mitochondria from different tissues to heating was investigated only by a few research groups Citation9, Citation13–15. These authors report that hyperthermia induces increase in state 3 (i.e. state with maximal ADP phosphorylation) respiration together with uncoupling of oxidative phosphorylation (increase in state 2 respiration), and decrease in the efficiency of ADP phosphorylation (drop in the ADP/O ratio).
The estimation of functional parameters (state 2 and state 3 respiration or the ADP/O ratio) is a classical approach for assessing temperature effects on mitochondrial metabolism. However, it provides very limited information about the sites of action of possible multi-site effectors (such as hyperthermia) on a complex system of oxidative phosphorylation. Modular kinetic analysis (or top-down elasticity analysis) has been shown to be a suitable tool to identify the sites of action of multi-site effectors within complex systems (reviewed in Citation16), including the effects of low temperatures on the system of oxidative phosphorylation Citation17. Using this approach, a complex system (process) is simplified by division into a small number of modules involved in the production or consumption of one intermediate (a so called connecting, or common intermediate). In this study we divided oxidative phosphorylation into three modules (the proton leak, respiratory and phosphorylation modules) that interact through the membrane potential (Δψ) as the connecting intermediate Citation18, Citation19. In order to detect the components of the system that are influenced by an effector, the effector-induced shift in the kinetics of each module (i.e. the dependence of the flux through the module on the level of the connecting intermediate Δψ) is determined.
The aim of our study was to perform modular kinetic analysis in the hyperthermic temperature range and to compare the effects on physiologically relevant fluxes of respiration, phosphorylation and proton leak induced by the febrile temperature (40°C) and higher temperatures commonly used for hyperthermic treatment (42° and 45°C). The results indicate that in these two temperature ranges the response of heart mitochondria is essentially different–febrile temperature slightly activates mitochondrial energy producing functions, whereas only a few degrees above the fever temperature causes severe impairment of mitochondrial ability to maintain Δψ and synthesise ATP.
Materials and methods
Isolation of mitochondria
Mitochondria were isolated from the hearts of male Wistar rats weighing 275–300 g as described in Citation19. The animals were killed according to the rules defined by the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Purposes (Licence no. 0006). The heart was quickly removed and placed into ice-cold 0.9% KCl solution. The tissue was cut into small pieces and homogenised in a glass-teflon homogeniser. Homogenisation medium contained 160 mM KCl, 20 mM Tris, 10 mM NaCl, 5 mM ethylene glycol tetra-acetic acid (EGTA), 1 mg/mL bovine serum albumin (BSA), pH 7.7. The homogenate was centrifuged at 750 × g for 5 min, and the supernatant was centrifuged at 6800 × g for 10 min. The mitochondrial pellet was resuspended in a suspension buffer containing 180 mM KCl, 20 mM Tris, 3 mM EGTA (pH 7.3) and stored on ice. The protein concentration was determined by the burette method Citation20 using BSA as a standard.
Measurement of mitochondrial respiration and Δψ
Mitochondrial respiration and Δψ were measured at different temperatures (37° ± 0.1°C, 40° ± 0.1°C, 42° ± 0.1°C and 45° ± 0.1°C) in a closed, stirred and thermostatted 1.5 mL glass vessel equipped with Clark-type oxygen electrode and TPP+ tetraphenylphosphonium (TPP+)-sensitive electrode (A. Zimkus, Vilnius University, Lithuania) allowing simultaneous monitoring of Δψ and mitochondrial respiration. For each incubation the TPP+-sensitive electrode was calibrated by small additions of tetraphenylphosphonium chloride (TTPCl) solution to a final concentration of 267 nM. Δψ was calculated from the distribution of TPP+ using Nernst equation and TPP+ binding correction factor of 0.16 µL/mg protein Citation18.
The assay medium contained 30 mM Tris, 5 mM KH2PO4, 110 mM KCl, 10 mM NaCl, 1 mM EGTA, 5 mM nitrilotriacetic acid, 1 mM dithiothreitol, 50 mM creatine, 5.17 mM MgCl2 (1 mM free Mg2+), and 0.875 mM CaCl2 (1 µM free Ca2+), pH 7.2. Excess of creatine kinase (0.1 mg/mL) was added to maintain steady state respiration Citation21. The experiments were performed using 1 mM pyruvate plus 1 mM malate as the respiratory substrate. Mitochondria (0.3 mg protein/mL) were incubated in the assay medium with the respiratory substrate (state 2) for 3 min at 37°, 40°, 42° or 45°C. State 3 respiration was initiated by adding 1 mM ATP.
The rate of mitochondrial respiration in state 2 (V2), state 3 (V3) and the respiratory control index (RCI = V3/V2) were defined according to the conventional terminology Citation22. Mitochondrial swelling at different temperatures was determined spectrophotometrically by the changes in absorbance at 540 nm in the assay medium used in the respiration and Δψ measurements.
Determination of dissolved molecular oxygen concentration
The concentration of molecular oxygen dissolved in the assay medium at different temperatures (37°–47° ± 0.1°C) was determined polarographically using glucose oxidase reaction Citation23. The pH of the medium was maintained at pH 7.2 at each temperature. The reaction was initiated by adding 4.7 U/mL of glucose oxidase followed by 100 nmol of glucose each minute until complete oxygen consumption. The concentration of dissolved oxygen was calculated from the obtained polarographic curves. The molar ratio coefficient of the reaction between D-glucose and O2 was defined in the medium with known concentration of dissolved oxygen (0.9% NaCl solution in the deionised water, oxygen solubility 420.5 µmol O/L at 37°C, 760 mmHg atmospheric pressure) Citation24. Dissolved oxygen concentrations determined in the medium were 394.63, 371.78, 356.54 and 333.69 µmol O/L at 37°, 40°, 42° and 45°C, respectively.
Modular kinetic analysis
Mitochondrial oxidative phosphorylation system was conceptually divided into three functional modules, interacting through a linking intermediate Δψ () Citation18. Although the true linking intermediate is the protonmotive force (Δp), it has been shown previously that measurement of Δψ instead of Δp does not introduce signifficant error in the determination of module kinetics, since ΔpH (second component of Δp) is small and does not change under our experimental conditions Citation25.
Figure 1. Division of the oxidative phosphorylation system to three subsystems connected by the membrane potential Δψ. JO, flux through the respiratory subsystem; JL, flux through the membrane leak subsystem; JP, flux through the phosphorylation subsystem.
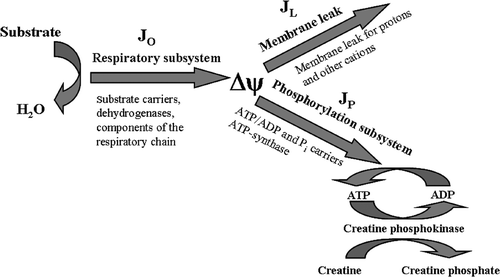
Δψ is generated by the respiratory module (comprised of the substrate carriers, matrix dehydrogenases and respiratory chain complexes), and consumed by the phosphorylation module (comprised of ATP synthase, adenine nucleotide translocator and phosphate carrier) and the membrane leak module (comprised of the passive permeability of the mitochondrial inner membrane to protons, and any cation cycling reactions).
The dependence of the flux through the respiratory module (JO) on Δψ was determined by gradually inhibiting the flux through the phosphorylation module with the inhibitor of adenine nucleotide translocator carboxyatractyloside (0–1.75 nmol/mg protein) and concomitantly measuring the respiration flux JO and Δψ corresponding to each new steady state. The dependence of the flux through the proton leak module (JL) on Δψ was determined by titrating the flux through the respiratory module with rotenone (0–0.08 nmol/mg protein) when the flux through the phosphorylation module was fully blocked with oligomycin (2 µg/mg mitochondrial protein). The dependence of the flux through the phosphorylation module (JP) on Δψ was determined by titrating the flux through the respiratory module with the respiratory chain inhibitor rotenone (0–0.06 nmol/mg protein) and concomitantly measuring the respiration flux (JO) and Δψ corresponding to each new steady state. Subsequently, JP was calculated by subtracting JL from JO at the same value of Δψ (i.e. JP = JO − JL). The experiments of modular kinetic analysis were paired: the measurements were performed at 37° and 40°C, or at 37° and 42°C using the same mitochondrial preparation.
Assessment of intramitochondrial H2O2 formation
Mitochondria were loaded with H2DCFDA (final concentration 5 µM) and incubated for 30 min on ice in the dark. After incubation, mitochondrial suspension was diluted 1 : 50 with suspension buffer, centrifuged 10 min at 6800 × g, 4°C, and resuspended in fresh suspension buffer. H2DCF oxidation was measured in mitochondria (0.5 mg/1.5 mL) supplemented with 1 mM pyruvate plus malate (state 2) in a standard assay medium at 37°, 40°, 42° and 45°C using thermostatted fluorometer Tecan GENios Pro™ (Tecan Group, Menedorf, Switzerland) (excitation and emission wavelengths 485 and 535 nm, respectively). 100 µM of H2O2 was used as a positive control. To correct for non-specific mitochondria-derived background fluorescence the same measurements were performed using mitochondria which were not loaded with dye. Data were acquired and analysed using XFluor™ software (Tecan). H2DCF oxidation rate was expressed as relative fluorescence units per min per mg of protein.
Measurements of MDA formation
Mitochondria (5 mg of mitochondrial protein) were incubated in 1 mL standard assay medium supplemented with 1 mM pyruvate plus malate (state 2) in a thermostatted chamber for 3 min. After incubation, medium with mitochondria was transferred to a glass tube containing 1 mL ice-cold 0.5% 2-tiobarbituric acid solution in 20% trichloracetic acid, and vortexed. The tube was sealed and incubated in a boiling water bath for 30 min. The solution was cooled and centrifuged for 10 min at 10000 × g, 4°C. The absorbance of the supernatant was measured at 532 nm and 600 nm. MDA concentration was calculated using molar extinction coefficient of 1.56 × 105 M−1· cm−1.
Data presentation and statistical analysis
Data are presented as means ± SEM (n = 3). Statistical significance of the temperature effects was evaluated using Student's t-test (paired). The differences were assumed to be statistically significant when p < 0.05.
Results
Modular kinetic analysis of temperature effects on oxidative phosphorylation
In this study we assessed the effect of increase in temperature in the range 37°–45°C on the functional activity of isolated heart mitochondria. We aimed to apply modular kinetic analysis for estimation of hyperthermia-induced changes in the kinetics of the respiratory, phosphorylation and proton leak modules of the oxidative phosphorylation system.
We have reported earlier Citation26 that the effect of hyperthermia did not depend on free Ca2+ concentration in the medium when it is maintained in the submicromolar range 5 nM−1 µM. However, mutual negative synergy of the effects of hyperthermia and Ca2+ overload (30 µM) on respiration of heart mitochondria in state 2 and state 3 was demonstrated. Moderate heating (40°–45°C) significantly and progressively reduced uncoupling and inhibitory Ca2+ effect on state 3 respiration due to the reduced Ca2+ uptake at lower Δψ. The effects of hyperthermia (uncoupling and inhibition of the respiratory chain) are more pronounced when mitochondria respire at optimal Ca2+ concentration and are significantly smaller in Ca2+ overload-inhibited heart mitochondria Citation27. Therefore in this study we used concentration of extramitochondrial Ca2+ that is optimal for the oxidation of the most physiological substrates (1 µM).
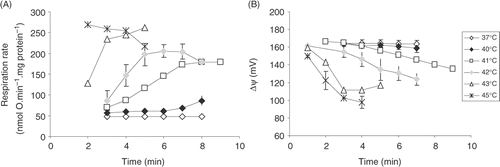
Modular kinetic analysis is applicable only to the systems that are able to attain steady state Citation16. Therefore, we evaluated the ability of isolated heart mitochondria to respire at a constant rate and to maintain Δψ in metabolic state 2 at elevated temperature ().
Mitochondrial respiration rate in metabolic state 2 Citation22 is registered when no external ADP is present in the medium (the phosphorylation subsystem does not operate, oxygen consumption rate is low and the membrane potential is high). State 3 respiration is measured after addition of saturating ADP concentration, that leads to the use of Δψ for ATP synthesis (lower Δψ), and higher respiration rate. State 2 respiration rate is mostly determined by the permeability of the inner mitochondrial membrane to protons Citation18, Citation19, and therefore increase in state 2 (or similar state 4) respiration rate is explained as an increase in the membrane permeability to ions.
We found that at 37°C the rate of mitochondrial respiration in state 2 (V2) and membrane potential ΔΨ remained constant (48 ± 2 nmol O · min−1 · mg protein−1 and 164 ± 0 mV, respectively) during 7 min of incubation.
These values were not significantly different at 40°C (59 ± 5 nmol O · min−1 · mg protein−1 and 164 ± 0 mV, respectively). At 41°C (the upper limit of fever temperature) the respiration rate and Δψ after 3 min were similar to the ones at 37°C up to 4 min of incubation. However, further incubation at 41°C led to a gradual increase in V2 and decrease in Δψ: at minute 7 V2 increased to 174 ± 2 nmol O · min−1 · mg protein−1, and Δψ decreased to 146 ± 2 mV. This possibly indicates a progressive uncoupling due to increasing permeability of the inner mitochondrial membrane. The uncoupling effect became much more pronounced when incubation temperature was increased to 42°C: a very fast increase in V2 and decrease in Δψ was observed until the values of 206 ± 16 nmol O · min−1 · mg protein−1 and 130 ± 8 mV were reached after 5 min incubation. At 45°C the uncoupling effect was almost immediate: after 2 min incubation V2 reached maximal value of 269 ± 6 nmol O · min−1 · mg protein−1. However, further incubation led to inhibition of state 2 respiration (after 5 min V2 decreased to 218 ± 4 nmol O · min−1 mg protein−1). Meanwhile Δψ decreased to 98 ± 7 mV after 3 min incubation. Thus, at 45°C oxidative phosphorylation was completely uncoupled and there is evidence for a time-dependent inhibition of the respiratory chain.
Our data () show that increases of temperature in the febrile range have no effect on state 2 respiration and membrane potential. That means that membrane barrier properties are well preserved if temperature does not exceed 40°C. State 2 respiration increases only at 41°C at longer incubation (more than 4 min). Increase in state 2 respiration rate at higher temperatures indicates substantial increase in the membrane permeability. Despite the rate of oxygen consumption is increased, this respiration is ‘free’, not coupled to phosphorylation.
Figure 2. The dependence of state 2 respiration rate (A) and Δψ (B) in heart mitochondria on different incubation temperature and time. Averages from n = 3 independent experiments ± SEM.
From the obtained results we concluded that the maximal temperature for applying modular kinetic analysis is 42°C, since the further increase in temperature is related to strong and time-dependent uncoupling. However, the analysis at 42°C is possible only with some reservations, i.e. if during inhibitor titrations the respiration rates and Δψ obtained with different concentrations of inhibitors are compared at the same time points (e.g. standardised at minutes 4 and 5) but different runs.
The aim of the further experiments was to evaluate how temperature elevation from 37°C to 40° and 42°C affects the kinetics of the proton leak, the respiratory and the phosphorylation modules on Δψ in heart mitochondria oxidising pyruvate plus malate. The results show that the increase in temperature from 37° to 40°C (, upper panels and ) had no effect on the kinetics of phosphorylation () and proton leak (), but led to a slight activation of the reactions of the respiratory module (). The respiration (JO) and phosphorylation (JP) fluxes through the modules in state 3 did not change significantly. Furthermore, Δψ increased by 4 mV (). That is consistent with the activation of the Δψ producing respiratory module. Our data indicate that temperature increase in the febrile range does not lead to uncoupling but rather causes slight activation of oxidative phosphorylation.
Figure 3. Comparison of the kinetics of the modules of oxidative phosphorylation at 37°C versus 40°C and 37°C versus 42°C. Upper panels (A), (B), (C) represent comparison of inhibitor titration results at 37°C versus 40°C; lower panels (D), (C), (E) at 37°C versus at 42°C. (A), (D), kinetics of the phosphorylation module obtained by titration with rotenone. Phosphorylation flux Jp was calculated as Jp = Jo − JL at the same value of Δψ. (B), (E), kinetics of the respiratory module obtained by titration with carboxyatractyloside. (C), (F), kinetics of the proton leak module obtained by titration with rotenone when ADP phosphorylation is blocked with oligomycin. Averages from n = 3 independent experiments ± SEM. Open symbols, experiments were carried out at 37°C; closed symbols, experiments were carried out either at 40°C (A, B, C) or at 42°C (D, E, F).
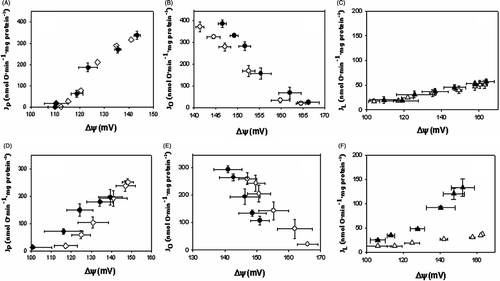
Table I. Effect of increasing the temperature from 37°C to 40°C on the fluxes through the modules of oxidative phosphorylation and the membrane potential. Jo, Jp, JL, the fluxes of respiration, phosphorylation, and proton leak, respectively; Δψ, membrane potential. Averages from n = 3 independent experiments ± SEM. *Statistically significant difference, p < 0.05.
When analogous analysis was performed to compare the kinetics of oxidative phosphorylation modules at 37° and 42°C, different pattern of changes was obtained (, lower panels and ). The largest difference was strong activation of flux through the proton leak module at 42°C (). The flux through the proton leak module in state 3 (JL) increased more than 3-fold. The mean values in on the curve presenting kinetics at 42°C in the whole range of Δψ are lower than the points on the control curve. That implies that the activity of the respiratory module was slightly suppressed (). As a result of increased membrane leak and inhibition of the respiratory module, the value of Δψ in state 3 decreased by 10 mV (). It is important to note that although the kinetics of the phosphorylation module was not significantly affected (), the phosphorylation flux at Δψ value corresponding to state 3 was reduced by 28%.
Table II. Effect of increasing the temperature from 37°C to 42°C on the fluxes through the modules of oxidative phosphorylation and the membrane potential. Jo, Jp, JL, the fluxes of respiration, phosphorylation, and proton leak, respectively; Δψ, membrane potential. Averages from n = 3 independent experiments ± SEM. *Statistically significant difference, p < 0.05.
Temperature effects on ROS production and lipid peroxidation
The observed sudden changes in the membrane permeability at 42°C may be caused by several reasons–temperature-induced phase transition of membrane lipids, increase in the membrane fluidity, or damage to membrane structures due to lipid peroxidation induced by reactive oxygen species (ROS). Therefore we evaluated H2O2 generation and lipid peroxidation in rat-heart mitochondria incubated at different temperatures.
Data presented in indicate that hyperthermia indeed stimulated ROS production in isolated rat-heart mitochondria. However, the temperature dependence of H2O2 formation had a rather unexpected character with a clear maximum in the fever temperature range (40°C). The increase in temperature from 37° to 40°C was followed by a drastic increase (3.6-fold) in H2O2 production (). Further increase in the temperature caused a decrease in the rate of H2O2 generation. Nevertheless, it remained significantly higher than that at 37°C (2.6- and 2.1-fold at 42° and 45°C in comparison to that at 37°C, respectively). However, it is not possible to relate changes in membrane permeability to ROS production since at 42° and 45°C the rate of H2O2 production is lower than at 40°C, but the membrane permeability was progressively increasing at higher temperatures ().
Figure 4. Temperature dependence of ROS production (A) and lipid peroxidation (B) in isolated rat heart mitochondria. Averages from three independent experiments ± SEM; *Statistically significant difference, p < 0.05.
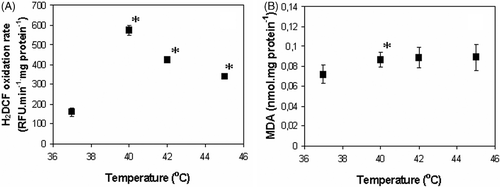
The changes in MDA amount indicated oxidative damage of mitochondrial lipids after only 3 min incubation at 40°C (). Significantly higher MDA amount (by 21%) was detected after incubation of mitochondria at 40°C as compared to 37°C. Increase in MDA amount at higher temperatures (by 22% and 23% at 42° and 45°C, respectively) was not statistically significant. That possibly is explainable by lower H2O2 production rate in comparison to 40°C (). These results indicate that the sudden increase in the inner membrane permeability to ions at 42°C () cannot be explained by a progressive oxidative damage of the membrane lipids.
We have performed the set of experiments for testing the possibility that under hyperthermic conditions rapid activation of the degradative enzymes such as phospholipase A2 or proteases could be responsible for accumulation of the products (such as free fatty acids) that could enhance uncoupling. The obtained results indicate that addition of fatty acid free BSA (several concentrations tested) slightly decreased state 2 respiration, however, did not reduce the uncoupling effect induced by hyperthermia (data not shown).
Reversibility of temperature effects on the mitochondrial function
To obtain more information about hyperthermia-induced changes in the membrane properties we evaluated the short-term recovery of mitochondrial functional parameters V2, V3 and Δψ. In this set of experiments, prior to the respiration measurements at 37°C, mitochondrial suspension was incubated in open vials for 0, 5, 10, 15 or 20 min at 37°, 42°, or 45°C (control–directly from ice). shows that V2 in control and after 5 min incubation at 37° and 42°C was similar–55 ± 6 and 60 ± 15 nmol · O min−1 · mg protein−1. Comparable values of V2 remained also after 10 and 15 min of incubation at these temperatures. At 37°C V3 was 355 ± 33 nmol · O min−1 · mg protein−1, and did not change after 5 min and longer incubation. At 42°C V3 was not significantly different from V3 at 37°C (350 ± 75 nmol · O min−1 · mg protein−1). At 37°C, Δψ was 161 ± 6 mV (state 2) and 143 ± 11 mV (state 3) (). After 5 min incubation at 37° and 42°C Δψ was similar and stable up to 15 min of incubation: in state 2–160 ± 5 and 163 ± 2 mV, in state 3–144 ± 6 and 149 ± 9 mV, after incubation at 37° and 42°C, respectively. Only after 20 min incubation at 42°C Δψ was slightly lower in both metabolic states.
Figure 5. Reversibility of temperature effects on the respiration rate (A) and Δψ (B). Measurements were performed in state 2 and 3 at 37°C after pre-incubation (0–20 min) at 37°, 42° and 45°C. Averages from n = 3 independent experiments ± SEM.
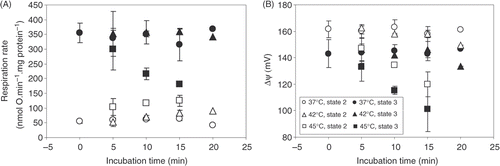
Our data indicate that changes in the mitochondrial functions observed at 42°C (, ) are completely reversible. In contrast, changes induced by hyperthermic incubation at 45°C are only partially reversible. Respiration rate in state 2 after 5 min incubation at 45°C was almost 2-fold higher in comparison to that in control conditions and after incubation at 37° and 42°C (104 ± 28 nmol · O min−1 · mg protein−1). The difference became statistically significant after incubation for 10 and 15 min in comparison to the same duration of incubation at 37° and 42°C (118 ± 4 and 125 ± 18 nmol · O min−1 · mg protein−1, respectively). Incubation at 45°C reduced mitochondrial ability to maintain Δψ in state 2 and 3 − Δψ value gradually decreased after 10 and 15 min incubations. From this we conclude that the changes in the inner mitochondria membrane properties occurring during the incubation at 45°C are only partially reversible and the permeability to ions remains increased after decreasing the temperature to 37°C. The irreversible changes determine a progressive inhibition of state 3 respiration–V3 decreased from 301 ± 70 nmol · O min−1 · mg protein−1 after 5 min incubation to 216 ± 20 nmol · O min−1 · mg protein−1 after 10 min incubation, and 180 ± 7 nmol · O min−1 · mg protein−1 after 15 min incubation at 45°C.
Taken together, our findings demonstrate that after pre-incubation at 45°C mitochondrial functions are only partly recovered in contrast to complete recovery after incubation at 42°C. This may be explained by faster progressive complex changes in the membrane fluidity, membrane lipid order or unknown interactions of the essential membrane components at 45°C. It is clear that these changes are not simply reversible and longer times are needed for the re-establishment of the initial membrane properties upon reversal to 37°C.
Discussion
The assessment of the response of heart mitochondria to hyperthermic condition may provide important information about molecular events that occur in cardiac cells under fever condition as well as upon hyperthermic treatment. Cardiac dysfunction and failure is the main cause of heat-related death Citation27. It is accompanied by a substantial injury of cardiomyocytes that is associated with changes in mitochondrial morphology and function Citation9, that might have crucial effects for cellular death or survival. In this study we have applied modular kinetic analysis to evaluate in more detail the hyperthermia-induced changes in the kinetics of the individual functional blocks (modules) of mitochondrial oxidative phosphorylation system.
Our study demonstrated for the first time that elevation of temperature in the febrile range (to 40°C) has a positive effect on the functions of heart mitochondria. However, exceeding the upper limit of that range (at 42°C) mitochondrial activity is severely challenged mostly because of the progressive loss of the inner membrane barrier function. The increase in the inner membrane permeability, uncoupling and decrease in the ADP/O ratio was observed by all authors Citation9, Citation13–15 in the supra-physiological range of hyperthermia. However, some discrepancies in the observed effects in the febrile range exist since some authors observe significant uncoupling Citation9, whereas others do not Citation13–15. Our results showed that the progressive increase in the membrane permeability to ions is not abolished by an inhibitor of the mitochondrial permeability transition pore cyclosporin A (data not shown). This indicates that other reasons than opening of the permeability transition pore are responsible for the increase in the membrane leak and uncoupling of oxidative phosphorylation in the supra-physiological range of temperatures.
There are numerous indications that cellular production of ROS is activated under hyperthermic conditions Citation29–31, leading to substantial changes in the redox status, depletion of reduced glutathione and accumulation of oxidative stress markers. Mitochondria are capable of increasing ROS generation upon stress-induced perturbations of electron transfer chain activity Citation32. However, until now more detailed information on the temperature dependence of mitochondrial ROS production was lacking. Here we showed that hyperthermia indeed significantly stimulates oxidation of H2DCF indicating increased formation of H2O2 in the mitochondrial matrix. However, ROS production and membrane peroxidation in isolated mitochondria was maximal in the febrile temperature range and decreased at higher temperatures. This observation indicates that a substantial damage of membrane barrier properties induced by supraphysiological hyperthermia was not induced by ROS. The question about the possible physiological relevance of the observed temperature dependence of mitochondrial ROS generation remains obscure, although an antiseptic defence function cannot be excluded.
The molecular mechanisms of cellular response to hyperthermia are very complex Citation33, Citation34. It has been demonstrated that temperature-induced change in physical membrane properties is an early and critical event in the chain of events of the cellular response Citation34, Citation35. We hypothesise that the sudden change in membrane permeability observed in our study at 42°C may be explained by altered membrane fluidity. Willis et al. Citation15 reported that hyperthermia-induced increase in membrane conductance in isolated liver mitochondria is related to an abrupt change in the order of the inner membrane that occurs when temperature rises above 42°C, as evaluated by the break point of Arrhenius plot of temperature dependence of 1,6-diphenyl-1,3,5-hexatriene polarisation. This could possibly explain the differences in the effects of febrile and supraphysiological temperatures on the membrane permeability observed in our experiments.
Benefits of fever or elevated body temperature between 37° and 41°C for healing different disease states, including infection by human immunodeficiency virus and cancer, are well documented Citation2. It has been shown that evolutionary conserved response to infections improves survival following infections because it enhances immune response Citation1 and decreases viability of infectious agents Citation2. Our results indicate that mitochondrial function is slightly improved in the febrile range of temperatures. Improvement in cellular ATP supply and increase in ROS production to some extent may be beneficial under conditions of fever. Nevertheless, analogous studies might provide basis for a rational selection of regimens for hyperthermic treatment in different tissues. Temperatures exceeding febrile temperatures are commonly used for hyperthermic treatment of tumours. However, the mechanisms of temperature cytotoxicity and reasons for the differences in the response of normal and cancerous tissue are poorly understood. Our data indicate that at a temperature that is only a few degrees above febrile temperature mitochondrial inner membrane becomes permeable to ions, ΔΨ and the efficiency of oxidative phosphorylation decreases. Therefore we conclude that at supra-physiological temperature mitochondrial functions are impaired and cellular energy supply severely declines. Although the rate of state 3 respiration seemingly remained the same (JO in ) at 37° and 42°C, mitochondrial ability to phosphorylate ADP was diminished due to uncoupling. Increase in the temperature from 42° to 45°C resulted in complete abolishment of ATP synthesis. Thus, at higher temperatures energy metabolism of cardiac cells may be rapidly and severely challenged, which may threaten the survival of the cells.
It remains to be established to what extent mitochondrial response in other normal and tumour cells is different and to what extent this difference may modify the response of the tissues to heating. In addition, our experiments are performed in vitro on isolated heart mitochondria, therefore it is very important to estimate whether mitochondrial response to heating in vivo is the same. A crowded cellular environment may increase heat capacity, and the presence of the molecular chaperones and other protective molecules may also substantially affect hyperthermia-induced changes in the membrane properties as well as in activity of enzymes and carriers located in the inner mitochondrial membrane.
Nevertheless, analogous studies as presented in this paper might provide the basis for a rational selection of regimens for thermal therapy in different tissues. It is known that the beneficial apoptotic pathway of cell death is energy dependent Citation36. One possible suggestion that might be raised from our data is that milder treatment conditions (i.e. mild hyperthermia) may be more beneficial, since it leads to less severe energy crisis and thus may be more preferable for the induction of apoptotic elimination of the pathways of tumour cells.
Acknowledgement
This work was supported by Lithuanian State Science and Studies Foundation (project no. T-34/06).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ostberg JR, Gellin C, Patel R, Repasky EA. Regulatory potential of fever-range whole body hyperthermia on Langerhans cells and lymphocytes in an antigen-dependent cellular immune response. J Immunol 2001; 167: 2666–2670
- Dickson JA, Calderwood SK. Thermosensitivity of neoplastic tissues in vivo. Hyperthermia in cancer therapy, FK Storm. GK Hall Medical Publishers, Boston 1983; 63–140
- Vertrees RA, Leeth A, Girouard M, Roach JD, Zwischenberger JB. Whole-body hyperthermia: A review of theory, design and application. Perfusion 2002; 17: 279–290
- Wierenga PK, Stege GJJ, Kampinga HH, Konings AWT. Intracellular free calcium concentrations in cell suspensions during hyperthermia. Eur J Cell Biol 1994; 63: 68–76
- Wieder ED, Fox MH. The role of intracellular free calcium in the cellular response to hyperthermia. Int J Hyperthermia 1995; 11: 733–742
- Matsumi N, Matsumoto K, Mishima N, et al. Thermal damage threshold of brain tissue: Histological study of heated normal monkey brains. Neurol Med Chir (Tokyo) 1994; 34: 209–215
- Haveman J, Van der Zee J, Wondergem J, Hoogeveen JF, Hulshof MC. Effects of hyperthermia on the peripheral nervous system: A review. Int J Hyperthermia 2004; 20: 371–391
- Fajardo LF. Pathological effects of hyperthermia in normal tissues. Cancer Res 1984; 44: 4826S–4835S
- Qian L, Song X, Ren H, Gong J, Cheng S. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones 2004; 9: 281–293
- Yuen WF, Fung KP, Lee CY, Choy YM, Kong SK, Ko S, Kwok TT. Hyperthermia and tumour necrosis factor-α induced apoptosis via mitochondrial damage. Life Sci 2000; 67: 725–732
- Ko S, Yuen WF, Fung KP, Lee CY, Choy YM, Cheng HK, Kwok TT, Kong SK. Reversal of TNF-α resistance by hyperthermia: Role of mitochondria. Life Sci 2000; 67: 3113–3121
- Nijhuis EHA, Le Gac S, Poot AA, Feiijnen J, Vermes I. Bax mediated mitochondrial membrane permeabilisation after heat treatment is caspase 2 dependent. Int J Hyperthermia 2008; 24: 357–365
- Lepock JR, Cheng KH, Al-Qysi H, Sim I, Koch CJ, Kruuv J. Hyperthermia-induced inhibition of respiration and mitochondrial protein denaturation in CHL cells. Int J Hyperthermia 1987; 3: 123–132
- Brooks GA, Hittelman KJ, Faulkner JA, Beyer RE. Temperature, skeletal muscle mitochondrial functions, and oxygen debt. Am J Physiol 1971; 220: 1053–1059
- Willis WT, Jackman MR, Bizeau ME, Pagliassotti MJ, Hazel JR. Hyperthermia impairs liver mitochondrial function in vitro. Am J Physiol Regul Integr Comp Physiol 2000; 278: 1240–1246
- Fell DA. Understanding the control of metabolism, K Snell. Portland Press, London, Miami 1997
- Dufour S, Rousse N, Canioni P, Diolez P. Top-down control analysis of temperature effect on oxidative phosphorylation. Biochem J 1996; 314: 743–751
- Hafner RP, Brown GC, Brand MD. Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the ‘top-down’ approach of metabolic control theory. Eur J Biochem 1990; 188: 313–319
- Mildaziene V, Baniene R, Nauciene Z, Marcinkeviciute A, Morkuniene R, Borutaite V, Kholodenko B, Brown GC. Ca2+ stimulates both the respiratory and phosphorylation subsystems in rat heart mitochondria. Biochem J 1996; 320: 329–334
- Gornal AG, Bardawill CJ, David MM. Determination of serum protein by means of the burette reaction. J Biol Chem 1949; 177: 751–766
- Kholodenko B, Zilinskiene V, Borutaite V, Ivanoviene L, Toleikis A, Praskevicius A. The role of adenine nucleotide translocator in regulation of oxidative phosphorylation in heart mitochondria. FEBS Lett 1987; 233: 247–250
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 1955; 217: 383–393
- Hawkins P, Parker D. The design, construction and evaluation of an inexpensive analyser for measuring glucose concentrations in serum, plasma and whole blood. Phys Med Biol 1973; 18: 570–576
- Available at http://www.colby.edu/cpse/equipment2/simple/algo.html
- Marcinkeviciute A, Mildaziene V, Crumm S, Demin O, Hoek JB, Kholodenko B. Kinetics and control of oxidative phosphorylation in rat liver mitochondria after chronic ethanol feeding. Biochem J 2000; 349: 519–526
- Zukiene R, Dapkunas A, Cizas P, Buzaite O, Nauciene Z, Baniene R, Zabarylo U, Minet O, Mildaziene V. Hyperthermia modulates the effect of Ca2+ overload on respiration and NAD(P)H fluorescence in rat heart mitochondria. Biologija (Vilnius) 2006; 3: 47–52
- Steenland K. Epidemiology of occupation and coronary heart disease: Research agenda. Am J Ind Med 1996; 30: 495–499
- Flanagan SW, Moseley PL, Buettner GR. Increased flux of free radicals in cells subjected to hyperthermia: Detection by electron paramagnetic resonance spin trapping. FEBS Letters 1998; 431: 285–286
- Zuo L, Christofi FL, Wright VP, Liu CY, Merola AJ, Berliner LJ, Clanton TL. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am J Physiol Cell Physiol 2000; 279: C1058–1066
- Arnaud C, Joyeux M, Garrel C, Godin-Ribuot D, Demenge P, Ribuot C. Free-radical production triggered by hyperthermia contributes to heat stress-induced cardioprotection in isolated rat hearts. Br J Pharmacol 2002; 135: 1776–1782
- Grasso S, Scifo C, Cardile V, Gulino R, Renis M. Adaptive responses to the stress induced by hyperthermia or hydrogen peroxide in human fibroblasts. Exp Biol Med (Maywood) 2003; 228: 491–498
- Barja G. Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: Implications for aging studies. Rejuvenation Res 2007; 10: 215–224
- Park HG, Han SI, Oh SY, Kang HS. Cellular responses to mild heat stress. Cell Mol Life Sci 2005; 62: 10–23
- Roti JLR. Cellular responses to hyperthermia (40–46°C): Cell killing and molecular events. Int J Hyperthermia 2008; 24: 3–15
- Despa F, Orgill DP, Neuwalder J, Lee RC. The relative thermal stability of tissue macromolecules and cellular structure in burn injury. Burns 2005; 31: 568–577
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell free extracts: Requirement for dATP and cytochrome C. Cell 1996; 86: 147–157