Abstract
Purpose: Hyperthermia-induced activation of stress response proteins allows cells to withstand metabolic insults. In this study we set out to determine whether insulin secretion by pancreatic beta cells was affected by the acute inflammatory response, systemic inflammation-induced hyperglycaemia, and whole-body hyperthermia. Given that systemic-inflammation induces ER stress, we further examined whether hyperthermia can attenuate the extent of LPS-induced ER stress.
Materials and methods: Rats were randomised and divided into three treatment groups. Control rats received a 0.9% NaCl solution. Rats in the lipopolysaccharide (LPS) group received 7.5 mg of LPS/kg. Rats in the whole-body hyperthermia (WBH) + LPS group were exposed to 42 °C for 15 min, followed by injection with 7.5 mg of LPS/kg after 48 h. Glucose-potentiated insulin release and extent of ER stress were measured in beta cells.
Results: LPS inhibited glucose-induced insulin release from islet cells and induced the expression of Bip/GRP78, XBP-1, and CHOP transcripts. The inhibition of glucose-induced insulin release and induction of ER stress proteins by LPS was attenuated by WBH.
Conclusions: Our findings suggest that LPS-induced systemic inflammation decreased insulin release due to the effects of ER stress proteins on insulin secretion. Furthermore, the induction of ER stress proteins was prevented by pretreating rats with WBH. This may suggest that inhibiting the induction of ER stress proteins through WBH can restore insulin release in various disease states.
Introduction
Stress-induced hyperglycaemia is common in septic shock and trauma patients who are frequently treated in intensive care units of hospitals Citation[1]. As a result of hyperglycaemia, patients may experience a protective stress response to systemic inflammation Citation[2]. However, hyperglycaemia can be associated with worse outcomes and increased mortality in critically ill patients Citation[3], Citation[4]. Van den Berghe et al. demonstrated that tight glucose control offers significant benefits and reduces mortality in critically ill patients Citation[5].
The acute phase of systemic inflammation is characterised by the induction of a stress response that results in high circulating levels of glucagon, growth hormone, catecholamines, and glucocorticoids. These hormonal changes and increases in pro-inflammatory cytokines are important factors leading to hyperglycaemia Citation[6]. In addition, peripheral insulin resistance, increased hepatic glycogenolysis, and increased gluconeogenesis may redirect the energy supply to vital organs by increasing blood glucose levels. However, very little is known about the regulation of insulin secretion during septic shock.
The high rate of protein synthesis in pancreatic beta cells renders them susceptible to endoplasmic reticulum (ER) stress, a condition elicited by an imbalance in ER homeostasis that potentially influences insulin secretion by pancreatic islet cells Citation[7]. Furthermore, ER stress plays an important role in the pathogenesis of septic shock Citation[8] and might negatively regulate insulin secretion during septic shock. The induction of ER stress is accompanied by an increase in levels of BiP/GRP78, XBP-1, and C/EBP homologous protein (CHOP). Importantly, recent studies have demonstrated that these proteins are also implicated in insulin secretion Citation[9], Citation[10], Citation[11].
A fever is generally thought to be a beneficial response to infection, trauma, and other injuries Citation[12]. A moderate fever enhances the patient's defence system, thereby shortening disease duration and increasing the chance of survival Citation[13]. Somewhat analogous to a fever, the heat shock response is a highly conserved cellular defence mechanism that involves an up-regulation of molecular chaperones. In response to mild hyperthermia, activation of the heat shock response allows cells to withstand subsequent metabolic insults that would otherwise be lethal Citation[14]. Moreover, mild systemic hyperthermia can protect beta cells and their ability to regulate blood glucose concentrations in non-obese diabetic mice Citation[15]. Interestingly, heat shock protein 70 (Hsp70) has been shown to protect cells against ER stress Citation[16]. This finding underscores the multiple contexts in which heat shock proteins are cell protective. In the present study, we examined the effects of lipopolysaccharide (LPS)-induced systemic inflammation on insulin secretion and the ER stress response in pancreatic islets of rats. Furthermore, we explored the protective effects of whole body hyperthermia (WBH) in this context and found that the hyperthermia-mediated induction of stress response proteins improved insulin secretion and attenuated ER stress.
Methods
Treatment protocol
This study was approved by the Ethics Committee on Animal Research at the College of Medicine, Oita University, Oita, Japan. Male Wistar rats weighing 250–300 g (Kyudou, Saga, Japan) were housed with access to food and water ad libitum.
Animals were randomly assigned to one of three groups: (1) negative control group: rats were anaesthetised for 15 min, followed 48 h later by an intravenous (i.v.) bolus of 1 mL of 0.9% NaCl/kg; (2) untreated LPS group: rats were anaesthetised for 15 min (without WBH), followed 48 h later by an i.v. bolus of 7.5 mg of LPS/kg (Sigma, St Louis, MO); and (3) WBH-treated, LPS group: rats were exposed to 42 °C for 15 min, followed 48 h later by an i.v. bolus of 7.5 mg of LPS/kg.
WBH-treated rats were anaesthetised under 3% sevoflurane during the hyperthermia treatment, and then were subjected to body warming with a warm water blanket. Rectal temperatures were monitored continuously and body temperature was maintained at 42 °C. Following WBH treatment, rat body temperatures were maintained at 42 °C for 15 min.
LPS was administered through tail vein injection 48 h after heat exposure or sham anaesthesia. Rats under sevoflurane anaesthesia were sacrificed 12 h after LPS administration. Pancreatic tissue was quickly removed and processed as indicated below.
Transmission electron microscopy
Tissue from the tail region of each pancreas was immersed in 4% paraformaldehyde (1% CaCl2 and 7% sucrose) for 30 min at 4 °C. A razor blade was used to cut the fixed specimens into small pieces. Specimens were post-fixed in 2% osmium for 2 h at 4 °C, dehydrated through a graded ethanol series, and embedded in Epok 812 resin. Ultrathin sections (90–95 nm) were cut with a diamond knife on an ultramicrotome (Reichert-Nissei Ultracut S; Lecia, Vienna, Austria) and examined with a JEM-1200 EX II electron microscope (JEOL, Tokyo).
Isolation of rat pancreatic islets
Pancreatic islets were prepared by collagenase digestion. Following digestion, islets were incubated in Krebs-Ringer bicarbonate HEPES buffer (KRBH; 119 mM NaCl, 4.75 mM KCl, 5 mM NaHCO3, 2.54 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 20 mM HEPES, 3 mM glucose, pH 7.4) for isolation under a microscope. Islets were then incubated at 37 °C for 30 min and then immersed in KRBH supplemented with bovine serum albumin and 3 mM glucose. Islets were further incubated at 37 °C for 60 min. Insulin released into the medium was determined by enzyme-linked immunosorbent assay (ELISA).
Measurement of insulin secretion
Insulin concentration was determined using commercial kits (Shibayagi, Gunma, Japan) employing a sandwich ELISA method according to the manufacturer's protocols. Samples, negative controls, and diluted insulin standard markers were added to respective wells of 96-well plates precoated with monoclonal antibody specific to rat insulin. Absorbance values at 450 nm were determined using an ELISA reader.
Western blot analysis
Pancreatic specimens of the same treatment group were pooled and blood was cleared with saline. Pooled pancreas samples were homogenised, centrifuged at 10,000 rpm for 5 min, and the resultant supernatant was collected. Protein concentrations were measured using the BCA Protein Assay Reagent (Pierce, Rockford, IL).
Proteins were resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on a 10% gel. Immediately following SDS-PAGE, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) at 30 mA for 2 h. The membrane was blocked with 5% non-fat dry milk in TBS/Tween (25 mM Tris-HCl, 0.14 M NaCl, 2% Tween 20) at 4 °C overnight. Membranes were incubated with a 1:1000 dilution of monoclonal anti-Hsp70 (anti-Hsp72) antibody (StressGen Biotechnology, Temecula, CA) for 1 h. After three 10-minute washes, blots were incubated with secondary anti-rabbit IgG antibody (1:1000) for 1 h. Following three washes, proteins were revealed with enhanced chemiluminescence (ECL) (Amersham, Bucks) and blots were exposed to film (Hyperfilm ECL, Amersham).
Protein levels were measured by densitometry using the National Institutes of Health (NIH) Image software program (Research Services Branch, Bethesda, MD).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RNA was extracted from beta islet cells after rats were treated with LPS or saline using the acid guanidinium thiocyanate-phenol-chloroform method Citation[17]. RNA concentration was determined at an absorbance of 260 nm using a DU640 spectrophotometer (Beckman, Fullerton, CA).
RT-PCR was used to quantitatively assess changes in BiP/GRP78, CHOP, and XBP-1 transcript levels. First strand cDNA synthesis from 1 µg total RNA and subsequent PCR were conducted using a commercial RT-PCR kit (Toyobo, Tokyo) and the GeneAmp PCR System 2400 (Perkin-Elmer, Wellesley, MA). Primers were designed and PCR conditions optimised for each target. Specific primers used were 5′-CTGATCCGCAGCAG and 5′-GGATCTCTAAAACTAGAGGCT for XBP-1; 5′-GTTCTGCTTGATGTGTGTCC and 5′-TTTGGTCATTGGTGATGGTG for BiP/GRP78; 5′-GGAGAAGGAGCAGGAGAATGA and 5′-AGACAGACAGGAGGTGATGC for CHOP; and 5′-GTTCCGATGCCCCGAGGATCT and 5′-GCATTTGCGGTGCACGATGGA for beta-actin (control transcript). PCR products were confirmed by electrophoresis on 1.5% agarose gel stained with ethidium bromide.
Images of PCR products were scanned and analysed with the NIH Image 1.63 software program (Research Services Branch, Bethesda, MD). Intensities of bands were normalised to beta-actin.
Statistical analysis
Results are expressed as mean ± standard deviation (SD). Statistical analysis was performed using the Kruskal–Wallis and Mann-Whitney U tests. P-values less than 0.05 were considered significant.
Results
Effect of hyperthermia on Hsp72 levels in the pancreas
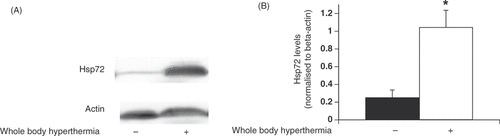
One primary aim was to examine the effects of WBH on the pancreas. We therefore harvested pancreatic tissue and determined Hsp72 protein levels in pancreatic tissue by Western blot analysis. We detected slight expression of Hsp72 in the pancreatic tissue from the negative control group. However, Hsp72 levels increased 48 h after WBH treatment (). These levels were quantified by densitometry. Relative intensity of Hsp72 levels in the negative control group was 0.255 ± 0.083. The WBH treatment group had significantly higher Hsp72 protein levels (1.046 ± 0.19) compared to that of the negative control (p < 0.05). We therefore confirmed that WBH increased Hsp72 protein levels in pancreatic tissue.
Figure 1. Changes in Hsp72 levels in pancreatic tissue after pretreatment with whole-body hyperthermia in rats. (A) Hsp72 expression in rat pancreatic tissue 48 h after WBH treatment at 42 °C or ambient temperature was detected by Western blot. (B) Signal intensities were based on densitometry using an image analyser. All data are expressed as the mean ± standard deviation (SD); * denotes a significant difference compared to the negative control group (p < 0.05).
Effect of WBH pretreatment on pancreatic tissue of rats treated with LPS
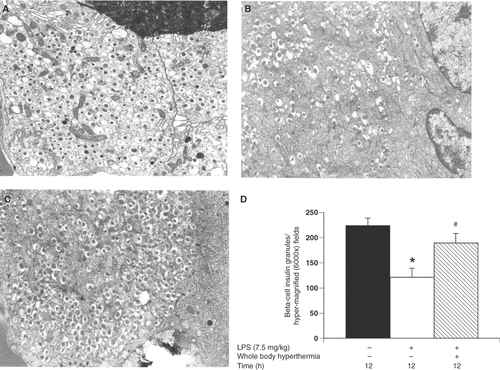
Pancreatic tissue specimens were obtained for electron microscopy 12 h after LPS administration with or without WBH pretreatment. Magnification was 6000× for all electron microscopy figures. In the negative control, we observed 224.7 ± 14.2 beta-cell insulin granules per hyper-magnified field. A significant decrease in the number of secretory granules was noted in the untreated LPS group (121.8 ± 17.8 beta-cell insulin granules per hyper-magnified field) compared to the negative control group ( and ; p < 0.05). In contrast, we observed a significant increase in secretory granules in the WBH LPS group (190 ± 18.5 beta-cell insulin granules per hyper-magnified field) compared to the untreated LPS group (; p < 0.05). It was apparent that LPS-induced systemic inflammation decreased insulin secretory granules in beta cells. On the other side, WBH pretreatment prevented the inhibition of a decrease in insulin secretory granules in a significant manner. As such, WBH has protective effects towards islet beta cells against LPS-induced systemic inflammation.
Figure 2. Evaluation of the effects of whole-body hyperthermia on pancreatic tissue in LPS-treated rats by electron microscopy. (A–C) High magnification of beta cell insulin granules. Pancreatic tissue specimens were obtained from the negative control group (A); untreated, LPS group (B); and WBH-treated, LPS group (C). (D) Sections from rats who were administered LPS for 12 h with or without WBH pretreatment. Sections from the three groups were blindly chosen and the number of insulin granules was counted in hyper-magnified fields. All data are expressed as the mean ± SD. # denotes a significant difference compared to the LPS group (p < 0.05); * denotes a significant difference compared to the negative control group (p < 0.05).
Effects of WBH pretreatment on insulin secretion levels in pancreatic beta cells
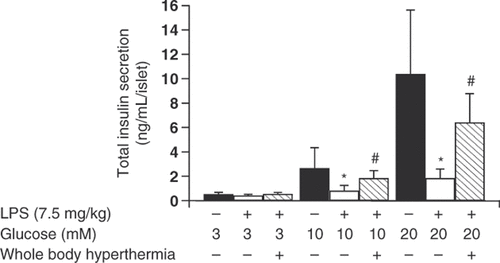
The untreated LPS group had significantly fewer insulin granules in the beta cells compared to the negative control group (). We examined whether insulin secretion was altered by pancreatic beta cells in preparation for a future study. Insulin secretion by pancreatic beta cells incubated in medium containing 3 mM glucose did not differ significantly across groups (insulin concentrations were 0.52 ± 0.17 ng/mL/islet for the negative control group, 0.43 ± 0.10 ng/mL/islet for the untreated LPS group, and 0.55 ± 0.22 ng/mL/islet for the WBH-treated LPS group) (). However, insulin secretion by pancreatic beta cells incubated in medium containing 10 mM glucose decreased significantly in the untreated LPS group compared to the negative control group (insulin concentrations were 2.66 ± 1.69 ng/mL/islet in the negative control group, and 0.82 ± 1.43 ng/mL/islet in the untreated LPS group; P < 0.05) (). WBH treatment prior to LPS administration diminished the inhibition of insulin secretion (insulin concentrations were 0.82 ± 1.43 ng/mL/islet in the untreated LPS group, and 1.84 ± 0.62 ng/mL/islet in the WBH-treated LPS group; P < 0.05) (). The same effect was observed in medium containing 20 mM glucose. Insulin secretion by pancreatic beta cells decreased significantly in the untreated LPS group compared to the negative control group (insulin concentrations were 10.37 ± 5.26 ng/mL/islet in the negative control group, and 1.85 ± 0.75 ng/mL/islet in the untreated LPS group; P < 0.05) (). WBH treatment prior to LPS administration diminished the inhibition of insulin secretion (insulin concentrations were 1.85 ± 0.75 ng/mL/islet in the untreated LPS group, and 6.42 ± 2.36 ng/mL/islet in the WBH-treated LPS group; P < 0.05). Pretreatment with WBH partially blocked insulin secretion suggesting that LPS-induced systemic inflammation inhibits insulin secretion from beta cells.
Figure 3. Insulin secretion from pancreatic beta cells of rats with or without whole-body hyperthermia pretreatment and LPS treatment. Insulin secretion from pancreatic beta cells of rats exposed to WBH or ambient temperatures prior to LPS administration. Insulin levels were determined using a rat insulin enzyme-linked immunosorbent assay (ELISA) kit. Harvested cells were incubated with KRB buffer containing the indicated concentrations of glucose. All data are expressed as the mean ± SD. # denotes a significant difference compared to the LPS group (p < 0.05); * denotes a significant difference compared to the negative control group (p < 0.05).
Effects of WBH pretreatment on BiP/GRP78, XBP-1, and CHOP transcript levels in pancreatic beta cells
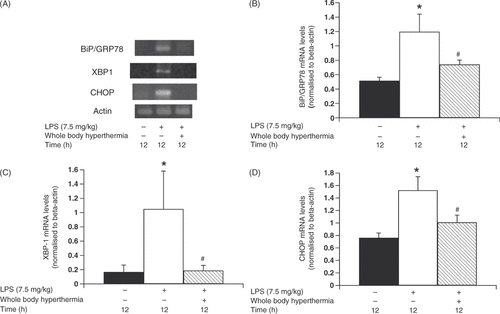
Insulin secretion has been found to be associated with ER stress proteins Citation[7]. We therefore examined the effects of LPS-induced systemic inflammation on ER stress protein levels. BiP/GRP78, XBP-1, and CHOP mRNA levels were measured using RT-PCR. Slight expression of BiP/GRP78 was revealed in the negative control group (relative intensity of BiP/GRP78 in the negative control group was 0.509 ± 0.16). We also observed a marked increase in mRNA levels in pancreatic beta cells 12 h after LPS administration. Relative intensity of BiP/GRP78 levels in the untreated LPS group was 1.056 ± 0.297; p < 0.05 compared to the negative control; ). This increase was more apparent in the group that was not pretreated with WBH prior to LPS administration (relative intensity of Hsp72 levels in the WBH-treated LPS group was 0.67 ± 0.191) (). Similarly, XBP-1 mRNA levels were significantly higher in the untreated LPS group compared to the negative control (p < 0.05 compared to the negative control and the untreated LPS group). Pretreatment with WBH also decreased XBP-1 expression (relative intensity of XBP-1 mRNA expression was 0.167 ± 0.098 in the negative control group, 1.049 ± 0.533 in the untreated LPS group, and 0.187 ± 0.073 in the WBH-treated LPS group) (). Moreover, CHOP mRNA levels were significantly higher in the untreated LPS group compared to the negative control group (p < 0.05 compared to the negative control group and the untreated LPS group). Pretreatment with WBH decreased CHOP expression (relative intensity of CHOP mRNA expression was 0.762 ± 0.077 in the negative control group, 1.524 ± 0.217 in the untreated LPS group, and 1.01 ± 0.116 in the WBH-treated LPS group) (). Pretreatment with WBH normalised ER stress proteins during systemic inflammation suggesting that LPS-induced systemic inflammation is up-regulated by ER stress-related proteins in islet beta cells.
Figure 4. Change in ER stress response gene levels in pancreatic beta cells due to whole-body hyperthermia treatment prior to LPS administration. Rats were treated with LPS following exposure to either 42 °C or ambient temperature. Pancreatic beta cells were harvested and transcripts were analysed by polymerase chain reaction (PCR) and quantified by densitometry. (A) Levels of BiP/GRP78, XBP-1, and CHOP transcripts in cells from LPS-treated rats were determined by RT-PCR. Signal intensities of BiP/GRP78 (B), XBP-1 (C), and CHOP (D) transcripts were based on densitometry using an image analyser. All data are expressed as the mean ± SD. # denotes a significant difference compared to the LPS group (p < 0.05); * denotes a significant difference compared to the negative control group (p < 0.05).
Discussion
Stress-induced hyperglycaemia is a common finding in acute illness and is frequently seen in non-diabetic patients in intensive care units. Patients with acute inflammation, systemic inflammation, and septic shock often experience hyperglycaemia and impaired glucose tolerance Citation[18]. Historically, patients with hyperglycaemia were treated using a strategy of benign neglect, with the belief that hyperglycaemia appeared due to an adaptive response to stress Citation[19]. Given this historical background, little is known about the mechanisms involved in the development of hyperglycaemia in critically ill patients, including those with septic shock. A potential mechanism may involve the actions of various hormones and cytokines which have been shown to influence insulin resistance Citation[20]. For instance, excess dextrose infusion is an often-overlooked contributor to hyperglycaemia.
To our knowledge, the present study is the first to demonstrate that LPS-induced systemic inflammation affects insulin secretion in pancreatic beta cells. Using a rat model, we demonstrated that LPS-induced systemic inflammation decreased the amount of secretory granules and inhibited insulin secretion by pancreatic beta cells. Importantly, WBH pretreatment blunted this effect. Furthermore, we found that insulin secretion was reduced in response to LPS-induced systemic inflammation when pancreatic beta cells were stimulated with glucose. These results suggest that systemic inflammation decreases not only glucose tolerance, but also insulin secretion.
The initial response to ER stress is the induction of proteins encoding ER chaperones, including BiP/GRP78, which function to increase the protein-folding capacity of the ER and prevent protein aggregation Citation[9]. ER stress is also relevant to pancreatic beta cell function and is implicated in the development of diabetes Citation[9], Citation[21]. A characteristic feature of pancreatic beta cells is a highly developed ER. For a cell specialised in insulin secretion, proper functioning of the ER is essential for cell survival.
In the current study, LPS administration in a rat systemic inflammation model led to the pronounced induction of BiP/GRP78. ER dysfunction would be expected to result in the accumulation of misfolded or unfolded proteins and elicit ER stress Citation[9]. Thus, our findings bridge ER stress and LPS-induced systemic inflammation.
XBP-1 is a transcriptional regulator of the unfolded protein response (UPR) Citation[22] that translocates into the nucleus upon ER stress and binds to promoters of chaperone genes. High expression of XBP-1 is indicative of UPR activation Citation[10], Citation[22]. In the present study, treatment with LPS resulted in a marked increase in XBP-1 transcript levels. Interestingly, WBH pretreatment prevented the LPS-mediated induction of XBP-1. These results suggest that unfolded proteins accumulate in the ER during LPS-induced systemic inflammation and that this accumulation can be prevented by WBH pretreatment.
In this study we demonstrated that expression of C/EBP homologous protein increased in response to LPS treatment in rat islets cells. A previous study demonstrated that the ER stress pathway mediated by CHOP is activated by LPS and is crucial for LPS-induced pathogenesis/inflammation Citation[23]. Furthermore, induction of CHOP had a negative impact on insulin secretion and CHOP deletion resulted in improved insulin secretion from islet cells Citation[11]. Indeed, we demonstrated that WBH pretreatment prevented the induction of CHOP in response to LPS and restored insulin secretion. These findings suggest that controlling the ER stress response can restore insulin secretion and might be beneficial in the context of LPS-induced systemic inflammation.
WBH pretreatment induced Hsp72 and attenuated the toxic effects of LPS despite the 48-h interval between WBH exposure and LPS administration. Main actions of HSPs and other anti-inflammatory molecules include hyperthermia-induced interactions with the host inflammatory response in processes such as glucocorticoid release or metabolism Citation[24] and gut permeability in response to endotoxin Citation[25]. In particular, Hsp72 protects cells against many acutely stressful conditions such as inflammation. Induction of Hsp72 results in stress tolerance and cytoprotection against an otherwise lethal exposure to inflammation-mediated cellular damage Citation[26]. Based on our results, we surmise that protection conferred by hyperthermia might be associated with the induction of Hsp72 rather than a direct thermal effect.
The current study establishes a relationship between hyperthermia-induced HSP synthesis and protection from LPS-induced systemic inflammation in pancreatic beta cells. In support of this we found that WBH was associated with an induction of Hsp72 levels, WBH treatment attenuated the LPS-induced ER stress response, and WBH restored insulin secretion after LPS administration. Previous studies have shown that insulin secretion and beta cell dysfunction are affected by ER stress Citation[27], the induction of HSPs both in vitro and in vivo is associated with cytoprotection Citation[28], Citation[29], and that induction of Hsp72 attenuates ER stress Citation[16]. These findings collectively suggest that the protective effect against ER stress that WBH confers may be related to HSP induction. We note, however, that we did not experimentally deplete Hsp72 in this study. Future studies will be needed to further clarify the relationship between ER stress and HSPs.
Conclusion
In conclusion we demonstrated that LPS decreased the number of secretory granules and inhibited insulin secretion by pancreatic beta cells. Furthermore, we found that WBH induced Hsp72 levels, thereby protecting pancreatic beta cells from LPS-mediated toxicity. WBH may lead to systemic endotoxaemia and precondition the host's response to subsequent challenges. These potential beneficial effects of hyperthermia require further investigation.
Acknowledgements
The authors wish to thank Hiroaki Kawasato and Aiko Yasuda for helpful advice on transmission electron microscopy methods.
Declaration of interest: The authors have no conflicts of interest to report.
References
- Frankenfield DC, Omert LA, Badellino MM, Wiles CE, 3rd, Bagley SM, Goodarzi S, Siegel JH. Correlation between measured energy expenditure and clinically obtained variables in trauma and sepsis patients. JPEN J Parenter Enteral Nutr 1994; 18: 398–403
- Mizock BA. Alterations in carbohydrate metabolism during stress: A review of the literature. Am J Med 1995; 98: 75–84
- Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med 2004; 30: 748–756
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: An independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87: 978–982
- van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patient. N Engl J Med 2001; 345: 1359–1367
- Mizock BA. Alterations in fuel metabolism in critical illness: Hyperglycaemia. Best Pract Res Clin Endocrinol Metab 2001; 15: 533–551
- Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance. Ann N Y Acad Sci 2006; 1083: 63–76
- Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem 2005; 138: 501–507
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002; 7: 335–345
- Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev 2003; 194: 29–38
- Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008; 118: 3378–3389
- Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. Role of fever in disease. Ann N Y Acad Sci 1998; 856: 224–233
- Jiang Q, Cross AS, Singh IS, Chen TT, Viscardi RM, Hasday JD. Febrile core temperature is essential for optimal host defense in bacterial peritonitis. Infect Immun 2000; 68: 1265–1270
- Dong HP, Chen HW, Hsu C, Chiu HY, Lin LC, Yang RC. Previous heat shock treatment attenuates lipopolysaccharide-induced hyporesponsiveness of platelets in rats. Shock 2005; 24: 239–244
- Capitano ML, Ertel BR, Repasky EA, Ostberg JR. Winner of the 2007 Society for Thermal Medicine Young Investigator Award. Fever-range whole body hyperthermia prevents the onset of type 1 diabetes in non-obese diabetic mice. Int J Hyperthermia 2008; 24: 141–149
- Gotoh T, Terada K, Oyadomari S, Mori M. Hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ 2004; 11: 390–402
- Ferreira DM, Zângaro RA, Villaverde AB, Cury Y, Frigo L, Picolo G, Longo I, Barbosa DG. Analgesic effect of He-Ne (632.8 nm) low-level laser therapy on acute inflammatory pain. Photomed Laser Surg 2005; 23: 177–181
- Gearhart MM, Parbhoo SK. Hyperglycemia in the critically ill patient. AACN Clin Issues 2006; 17: 50–55
- Mizock BA. Alterations in carbohydrate metabolism during stress: A review of the literature. Am J Med 1995; 98: 75–84
- Bessey PQ, Brooks DC, Black PR, Aoki TT, Wilmore DW. Epinephrine acutely mediates skeletal muscle insulin resistance. Surgery 1983; 94: 172–179
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the CHOP gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Investig 2002; 109: 525–532
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–891
- Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol 2006; 176: 6245–6253
- Alexandrova M, Farkas P. Stress-induced changes of glucocorticoid receptor in rat liver. J Steroid Biochem 1992; 42: 493–498
- Carter EA, Gonnella A, Tompkins RG. Increased transcellular permeability of rat small intestine after thermal injury. Burns 1992; 18: 117–120
- Subjeck JR, Shyy TT. Stress protein systems of mammalian cells. Am J Physol 1986; 250: 1–17
- Hettiarachchi KD, Zimmet PZ, Myers MA. Dietary toxins, endoplasmic reticulum (ER) stress and diabetes. Curr Diabetes Rev 2008; 4: 146–156
- Jacquier-Sarlin MR, Fuller K, Dinh-Xuan AT, Richard MJ, Polla BS. Protective effects of hsp70 in inflammation. Experientia 1994; 50: 1031–1038
- Kiang JG, Tsokos GC. Heat shock protein 70 kDa: Molecular biology, biochemistry, and physiology. Pharmacol Ther 1998; 80: 183–201