Abstract
Neurodegenerative diseases including amyotrophic lateral sclerosis, Parkinson's disease, Alzheimer's disease, and polyglutamine (polyQ) diseases are thought to be caused by protein misfolding. Heat shock proteins (HSPs), which function mainly as molecular chaperones, play an important role in the folding and quality control of proteins. The histopathological hallmark of neurodegenerative diseases is accumulation and/or inclusions of the disease-causing proteins in residual neurons in targeted regions of the nervous system. The inclusions combine with many components of molecular chaperone pathways and ubiquitin-proteasome, raising the possibility that misfolding and altered degradation of mutant proteins may be involved in the pathogenesis of neurodegenerative diseases. Overexpression of HSPs has been reported to reduce the number and size of inclusions and accumulation of disease-causing proteins, and ameliorate the phenotypes in neuronal cell and mouse models. Hsp90 inhibitors also exert therapeutic effects through selective proteasome degradation of its client proteins. Elucidation of its pathophysiology using animal models has led to the development of disease-modifying drugs, i.e., Hsp90 inhibitor and HSP inducer, which inhibit the pathogenic process of neuronal degeneration. These findings may provide the basis for development of an HSP-related therapy for neurodegenerative diseases.
Introduction
In chronic neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), Alzheimer's disease (AD), and polyglutamine (polyQ) diseases, abnormal accumulation and inclusions of disease-causing protein are a commonly observed characteristic. Under pathological conditions, the level of such misfolded proteins may exceed the protective machinery of the cell to either maintain them in a soluble form or degrade them, resulting in their accumulation and inclusion. ALS is an adult-onset motor neuron disease and most forms of ALS are sporadic, but ∼10% of patients have an inherited familial form of the disease of dominant causative mutations in the gene encoding copper/zinc superoxide dismutase 1 (SOD1) or 43 kDa TAR DNA-binding protein (TDP-43). Ubiquitinated protein aggregates found in many patients with sporadic or familial ALS are mainly composed of SOD1 or TDP-43 proteins Citation[1]. The α-synuclein gene is mutated in rare familial forms of PD, and α-synuclein is a major component of the pathological protein aggregates characteristic of the disease, known as Lewy bodies and Lewy neuritis Citation[2]. PolyQ diseases are caused by the expansion of a trinucleotide CAG repeat encoding glutamine in the causative genes, and to date, nine disorders have been identified as polyQ diseases Citation[3]. Many molecular biological studies have been undertaken to elucidate the pathogenesis of these diseases and to develop treatment methods. Among these treatments, pharmacological induction of molecular chaperones may ameliorate these disorders. Here we highlight the research findings from which the concept of the chaperone-dependent pathophysiology of neurodegenerative diseases has emerged, and discuss disease-modifying therapeutic approaches targeting molecular chaperones.
Involvement of the molecular chaperone pathway and the ubiquitin-proteasome system in the histopathology of neurodegenerative disorders
In patients with neurodegenerative diseases, neurons are selectively depleted in the nervous system. A pathological hallmark of most neurodegenerative diseases is the presence of nuclear and cytoplasmic inclusions. For example, nuclear inclusions (NIs) containing the mutant androgen receptor (AR) are detected in residual motor neurons in the brainstem and spinal cord Citation[4–6] as well as in the skin, testis and some other visceral organs in patients with spinal and bulbar muscular atrophy (SBMA), one of the polyQ diseases Citation[7], Citation[8]. These NIs are detectable by antibodies that recognise many components of the molecular chaperone pathway and the ubiquitin-proteasome system (UPS), implying that these proteins are involved in neurodegeneration in these diseases Citation[9–11]. Heat shock proteins (HSPs) are classified into the following families according to molecular size: Hsp100, Hsp90, Hsp70, Hsp60, Hsp40, and small HSPs Citation[12]. These molecular chaperones are either constitutively expressed or inducibly synthesised after cellular stress. HSPs play important roles in suppressing protein misfolding by synergistically assisting misfolded proteins in the refolding process, as well as maintaining correct folding, assembly, and intracellular transport of newly synthesised proteins (). However, several studies suggest that misfolded proteins interfere with the protective cellular responses against cytotoxic stress Citation[13]. Transfected cells expressing truncated polyQ-expanded AR showed delayed induction of Hsp70 after heat shock compared with that in transfected cells expressing the full-length AR Citation[14]. Progressive reductions in the expression of Hsp70, Hsp40 and α B-crystallin in the brain lesion of a mouse Huntington's Disease (HD) model were reported Citation[15–17], and such reductions were also observed in SBMA model mice Citation[18]. Reduced expression of heat shock cognate protein (Hsc)-70 was found in PD patients Citation[19], reduced expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7 (SCA7) Citation[20], and reduced expression of Hsp27 in ALS-SOD1 mice Citation[21]. In keeping with the importance of Hsp70 in defining neuronal susceptibility to degeneration, an inverse correlation between the relative amount of Hsp70 in neuronal subtypes and susceptibility to mutant Huntington-induced cell death was reported in a mouse model of HD Citation[22]. Taken together, impairment of the capability of HSP induction is implicated in the pathogenesis of neuron degeneration in neurodegenerative diseases ().
Figure 1. Diagram showing the roles of chaperones in folding of proteins and degradation of misfolded proteins. It has been suggested that a pathogenic process leads to formation of misfolded conformations of the target protein in neurodegenerative diseases. Correct protein folding is an essential biological process, and to achieve this, many proteins interact with molecular chaperones in the cellular environment. The Hsp70 class of molecular chaperones recognises the unfolded substrate early in the folding process, and maintains the polypeptide in the soluble conformation. To facilitate protein folding, Hsp70 interacts with its co-chaperones that regulate its chaperone activity. A major class of Hsp70 co-chaperone proteins is the Hsp40 family. Both Hsp70 and the Hsp40 family of chaperones have been shown to interact with the E3 ubiquitin ligases, particularly the quality-control E3 ligases, and are implicated in the biology of neurodegenerative disorders involving protein misfolding. Quality-control E3 ligases like CHIP could specifically target misfolded mutant disease proteins for proteasomal degradation and thereby could potentially slow disease progression.
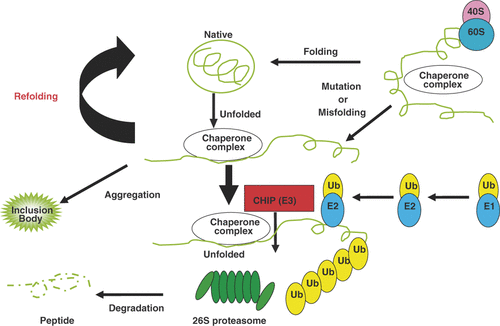
Table I. Changes in chaperone expression in neurodegenerative diseases.
Induction of heat shock proteins
Accumulation of mutant proteins has been considered to be central to the pathogenesis of neurodegenerative diseases including polyQ diseases. It has been postulated that expanded polyQ confers monomeric protein conformational change from random coil to β-sheet, leading to formation of polyQ oligomer Citation[23]. The misfolded monomer and oligomer that are resistant to denaturing detergents possess toxic property by means of interacting with normal cellular proteins Citation[24]. Direct inhibition of polyQ oligomerisation by Congo red has been demonstrated to exert therapeutic effects in a mouse model of HD Citation[25]. Oligomerisation of causative protein has thus been implicated in the pathogenic process of neurodegeneration, whereas formation of inclusion body, or matured amyloid fibrils, appears to possess cytoprotective property Citation[26], Citation[27]. Based on this hypothesis, HSPs have been drawing a great deal of attention, because they inhibit oligomer assembly and thereby mitigate its toxicity Citation[28]. These HSPs have been proposed to prevent the initial conformation conversion of mutant polyQ-containing protein from a random coil to a ß-sheet, leading to attenuation of toxic oligomer formation Citation[13]. For example, HSPs, particularly Hsp70, were shown to suppress aggregate formation and cellular toxicity in polyQ disease Citation[18], Citation[45–47], ALS Citation[34] and PD models Citation[35], Citation[36]. Hsp104 also reduced α-synuclein aggregation and dopaminergic degeneration in a rat model of PD Citation[37]. Moreover, Hsp70 overexpression enhanced degradation of polyQ-expanded proteins via its interaction with the UPS Citation[32], Citation[38]. CHIP (carboxyl terminus of Hsc70-interacting protein) might be one such coupling factor between the Hsp70 chaperone system and the machinery responsible for degrading mutant proteins Citation[39], Citation[40]. Thus, enhancement of chaperone expression under preserved UPS function is a reasonable clinical approach for the treatment of pathogenic mutant or modified protein-mediated neurodegenerative diseases ().
Favourable effects obtained by genetic modulation of HSPs suggest that pharmacological induction of molecular chaperones might be a promising approach for the treatment of neurodegenerative diseases. Hsp90 inhibitors and celastrol function as HSP inducers, resulting in the dissociation of HSF-1 from the Hsp90 complex and subsequent HSF-1 trimerisation, thereby leading to HSP activation Citation[41], and exerting potentially neuroprotective effects in models of polyQ diseases Citation[15], Citation[33], Citation[45–47], tauopathies Citation[45–47], ALS Citation[48], Citation[49] and PD Citation[50–52]. Geranylgeranylacetone (GGA), an acyclic isoprenoid compound with a retinoid skeleton, has been shown to strongly induce HSP expression in various tissues Citation[53]. This compound has been used as an oral anti-ulcer drug. Oral administration of GGA up-regulated the levels of Hsp70, Hsp90, and Hsp105 via activation of heat shock factor-1 in the central nervous system and inhibited nuclear accumulation of the pathogenic AR protein, resulting in amelioration of polyQ-dependent neuromuscular phenotypes of the SBMA mouse model Citation[18]. Thus, enhancement of cellular defences using Hsp90 inhibitors and GGA is a reasonable clinical approach for the treatment of neurodegenerative diseases ().
Table II. Compounds that induce chaperone expression.
Hsp90-dependent pathogenesis
Heat shock protein 90, a cytosolic molecular chaperone, is involved in the maturation and activation of a number of proteins, known as client proteins. Hsp90 functions in multi-component complexes of chaperone proteins including Hsp70, Hop, Cdc37, and p23 Citation[54]. Two main Hsp90 complexes are thought to exist: one complex is a stabilising form with Cdc37 and p23 and this complex stabilises Hsp90 client proteins, while the other complex is a proteasome-targeting form with Hsp70 and Hop and it directs Hsp90 client proteins to proteasome degradation Citation[55–58]. Hsp90 inhibitors inhibit the ATP-dependent progression of the Hsp90 complex toward the stabilising form and shift it to the proteasome-targeting form, resulting in proteasomal degradation of the Hsp90 client proteins, particularly their mutant versions Citation[56]. Another possibility is that Hsp90 client proteins may also be recognised initially by the Hsp40/Hsp70 complex with CHIP, and transferred to the Hsp90 complex by Hop Citation[59]. There remain two fates for the client proteins: refolding or ubiquitin-dependent proteasomal degradation. For example, dephosphorylation and refolding of p-tau is initially facilitated by an Hsp90/P23/Pin1 complex, however, when refolding is not accomplished, p-tau is transferred to the Hsp70/CHIP complex, and polyubiquitination mediates their degradation Citation[59]. Steroid receptors and numerous oncoproteins belonging to the Hsp90 client protein family are selectively degraded in the UPS by Hsp90 inhibitors, and 17-allylamino-17-demethoxygeldanamycin (17-AAG) and 17-dimethylamino-17-demethoxy-geldanamycin (17-DMAG) are now under clinical trials as novel molecular-targeted agents for a wide range of malignancies Citation[60]. We previously examined the effectiveness of 17-AAG and 17-DMAG in a mouse model of SBMA Citation[41], Citation[61]. These compounds significantly reduced the amount of the insoluble high-molecular-weight complex of mutant AR as well as the amount of soluble monomer of mutant AR in the spinal cord and skeletal muscle, and significantly reduced the amount of diffuse nuclear-accumulated AR. Alternatively, Hsp90 inhibitors may inhibit aggregation of mutant AR by inducing Hsp70 and Hsp40 expression, as described above. Of interest is the finding that Hsp90 inhibitors preferentially targeted mutant AR rather than wild-type AR to proteasomal degradation. A high association between p23 and AR containing expanded-polyQ tract renders the mutant AR more sensitive to Hsp90 inhibitors than wild-type AR Citation[41].
On the other hand, hsp90 inhibitors were reported to degrade the Hsp90 client proteins via macroautophagy (referred to here as autophagy) or chaperone-mediated autophagy (CMA). Macroautophagy is a process in which cells form double-membrane vesicles, called autophagosomes, around a portion of cytoplasm. These autophagosomes ultimately fuse with lysosomes, resulting in degradation of their substrates. CMA is selective for specific cytosolic proteins that contain a KFERQ-like motif Citation[62]. This motif is recognised by Hsc70, which transfers their substrates to the lysosomal membrane where they are translocated into the lysosomal lumen and degraded via binding to the receptor lysosome-associated membrane protein 2A (LAMP2A) Citation[63]. The IkB kinase (IKK), an essential activator of NF-κB, was selectively degraded by autophagy following geldanamycin (GA) treatment Citation[64]. Inactivation of ubiquitination or proteasome failed to block IKK degradation, however, inhibition of autophagy by an autophagy inhibitor or knockout of Atg5, a key component of the autophagy pathway, significantly rescued IKK from GA-induced degradation Citation[64]. GA increased total cellular proteolysis through an induction of CMA, which was not 3-MA sensitive Citation[65]. Another Hsp90 inhibitor, apratoxin A (oz-apraA), a cyclodepsipeptide isolated from a marine cyanobacterium, also promoted the degradation of epidermal growth factor receptor (EGFR), which has a KFERQ-like motif in the kinase domain, through CMA Citation[66]. Mutation of this motif prevented EGFR degradation by CMA and promoted its proteasomal degradation in oz-apraA–treated cells. Oz-apraA modulates Hsp90 chaperone complexes and enhances the interaction of Hsp90 client proteins and Hsc70, which can then be recognised by LAMP-2A for degradation via CMA Citation[66]. On the contrary, not an autophagic inhibitor but a proteasome inhibitor was able to prevent the degradation of p35 and mutant tau over a treatment with the Hsp90 inhibitors Citation[67]. These observations suggest that a subset of Hsp90 client proteins may be degraded via autophagy following Hsp90 inhibition.
In the case of neurodegenerative diseases, phosphorylated tau would be one of the target proteins of Hsp90 inhibitors, since Hsp90 inhibitors significantly reduce the total amount of phosphorylated tau Citation[45], Citation[46], Citation[59] due to a dose- and time-dependent degradation of p35 by Hsp90 inhibitors Citation[67], and also reduce aggregated form of tau Citation[45], Citation[67]. In AD, tau hyperphosphorylation is suggested to be a pathogenic process caused by aberrant activation of several kinases, in particular cyclin-dependent protein kinase 5 (Cdk5), leading to phosphorylation of tau on pathogenic sites. Reduction of p35 levels by Hsp90 inhibition affected the activity of the Cdk5/p35 complex Citation[67]. Leucine-rich repeat kinase 2 (LRRK2) would be another target protein of Hsp90 inhibitors Citation[68]. Although the majority of PD cases are sporadic, some are inherited, including those causing LRRK2 mutations. LRRK2 forms a complex with Hsp90 and its co-chaperone Cdc37, and inhibition of Hsp90 disrupts the association of Hsp90 with LRRK2 and leads to proteasomal degradation of LRRK2. Hsp90 inhibitors preferentially reduced the amount of the mutant LRRK2 by integrating into the Hsp90 chaperone complex compared with the wild-type LRRK2, and rescued the axon growth retardation caused by overexpression of the LRRK2 G2019S mutation in neurons Citation[68]. On the other hand, the steady expression level of α-synuclein, a common constituent of Lewy bodies (LBs) and Lewy neurites (LNs) in PD, was not affected by Hsp90 inhibition Citation[50]. We regard this general versatility as very important for the development of Hsp90 inhibitors as a treatment for neurological disorders. The strategy behind Hsp90 inhibitors differs from previous strategies employed against neurodegenerative diseases, which unavoidably allowed abnormal protein to remain and placed much value mainly on inhibition of protein aggregation. We consider that the ability to facilitate degradation of disease-causing or disease-related proteins by modulation of Hsp90 function would be of value when applied to neurodegenerative diseases. There is no doubt that reduction of the amount of the main culprit protein would have a curative effect against various neurodegenerative diseases. In fact, one therapeutic approach that directly reduces the level of abnormal protein by RNA interference, has already proven beneficial in various mouse models of polyQ diseases and amyotrophic lateral sclerosis Citation[69–71].
Hsp90 inhibitor-induced degradation requires well-preserved proteasome function; however, the question of whether the UPS is impaired in patients with neurodegenerative diseases has been raised with respect to this UPS-dependent therapy Citation[72]. The UPS is responsible for the turnover of most soluble proteins and plays an essential role in degrading short-lived regulatory proteins and damaged or misfolded proteins Citation[73]. It is generally accepted that the UPS is involved in the pathology of neurodegenerative diseases, since many components of the UPS and molecular chaperones are known to co-localise with neuronal inclusions Citation[29], Citation[74]. The function of the UPS has been evaluated in various assays that provided divergent results, ranging from impaired Citation[75–78] to preserved Citation[79], Citation[80] UPS function. If the function of the UPS was impaired, Hsp90 inhibitors would not be able to exert their pharmacological effect on neurodegenerative diseases. It is possible that mutant polyQ-expanded proteins may directly affect proteasome function within the specific subcellular compartment of synaptic UPS activities Citation[81], although it has been demonstrated that neuronal dysfunction can develop without significant impairment of the UPS in a mouse model of SCA7 Citation[82]. Consistent with this are data showing that proteasome impairment did not contribute to the pathogenesis of HD in a mouse model Citation[83]. Taking these data together, we consider that treatment with Hsp90 inhibitors, which would enhance a self-clearing system of target disease-causing proteins via the UPS, is a reasonable therapeutic strategy against neurodegenerative diseases.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Lagier-Tourenne C, Cleveland DW. Rethinking als: The fus about tdp-43. Cell 2009; 136: 1001–1004
- Lesage S, Brice A. Parkinson's disease: From monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009; 18: R48–59
- Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet 2005; 6: 756–765
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med 2003; 9: 768–773
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 1998; 44: 249–254
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002; 35: 843–854
- Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Hashizume Y, Fischbeck KH, Sobue G. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol 1998; 153: 695–701
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in sbma patients. Brain 2005; 128: 659–670
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha–synuclein toxicity in a drosophila model for parkinson's disease. Science 2002; 295: 865–868
- Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, Rechsteiner M, Riess O, Landwehrmeyer GB. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: Redistribution and differential recruitment of 26s proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol 2002; 51: 302–310
- Wang J, Farr GW, Zeiss CJ, Rodriguez-Gil DJ, Wilson JH, Furtak K, Rutkowski DT, Kaufman RJ, Ruse CI, Yates JR, et al. Progressive aggregation despite chaperone associations of a mutant sod1-yfp in transgenic mice that develop als. Proc Natl Acad Sci U S A 2009; 106: 1392–1397
- Macario AJ, Conway de Macario E. Sick chaperones, cellular stress, and disease. N Engl J Med 2005; 353: 1489–1501
- Wyttenbach A. Role of heat shock proteins during polyglutamine neurodegeneration: Mechanisms and hypothesis. J Mol Neurosci 2004; 23: 69–96
- Cowan KJ, Diamond MI, Welch WJ. Polyglutamine protein aggregation and toxicity are linked to the cellular stress response. Hum Mol Genet 2003; 12: 1377–1391
- Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet 2004; 13: 1389–1405
- Zabel C, Chamrad DC, Priller J, Woodman B, Meyer HE, Bates GP, Klose J. Alterations in the mouse and human proteome caused by huntington's disease. Mol Cell Proteomics 2002; 1: 366–375
- Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, Nukina N. Mutant huntingtin reduces hsp70 expression through the sequestration of nf-y transcription factor. EMBO J 2008; 27: 827–839
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 2005; 102: 16801–16806
- Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm 2004; 111: 1543–1573
- Tsai HF, Lin SJ, Li C, Hsieh M. Decreased expression of hsp27 and hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7. Biochem Biophys Res Commun 2005; 334: 1279–1286
- Maatkamp A, Vlug A, Haasdijk E, Troost D, French PJ, Jaarsma D. Decrease of hsp25 protein expression precedes degeneration of motoneurons in als-sod1 mice. Eur J Neurosci 2004; 20: 14–28
- Tagawa K, Marubuchi S, Qi ML, Enokido Y, Tamura T, Inagaki R, Murata M, Kanazawa I, Wanker EE, Okazawa H. The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci 2007; 27: 868–880
- Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci U S A 2002; 99: 5596–5600
- Li M, Chevalier-Larsen ES, Merry DE, Diamond MI. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J Biol Chem 2007; 282: 3157–3164
- Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003; 421: 373–379
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431: 805–810
- Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol 2004; 11: 1215–1222
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005; 6: 11–22
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in sca1. Nat Genet 1998; 19: 148–154
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones hsp70 and hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 2000; 275: 8772–8778
- Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible hsp70 chaperone suppresses neuropathology and improves motor function in sca1 mice. Hum Mol Genet 2001; 10: 1511–1518
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, et al. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci 2003; 23: 2203–2211
- Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med 2007; 85: 1421–1428
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in als mice. Nat Med 2004; 10: 402–405
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem 2004; 279: 25497–25502
- Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against mptp- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem 2005; 94: 995–1004
- Lo Bianco C, Shorter J, Regulier E, Lashuel H, Iwatsubo T, Lindquist S, Aebischer P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of parkinson disease. J Clin Invest 2008; 118: 3087–3097
- Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet 2002; 11: 515–523
- Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Perez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, et al. Chip protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 2006; 281: 26714–26724
- Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. Chip overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci 2007; 27: 5115–5126
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-aag, an hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med 2005; 11: 1088–1095
- Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of huntington's disease. Hum Mol Genet 2001; 10: 1307–1315
- Agrawal N, Pallos J, Slepko N, Apostol BL, Bodai L, Chang LW, Chiang AS, Thompson LM, Marsh JL. Identification of combinatorial drug regimens for treatment of huntington's disease using drosophila. Proc Natl Acad Sci U S A 2005; 102: 3777–3781
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 2008; 283: 26188–26197
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A 2003; 100: 721–726
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, et al. Chip and hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 2004; 13: 703–714
- Benussi L, Ghidoni R, Paterlini A, Nicosia F, Alberici AC, Signorini S, Barbiero L, Binetti G. Interaction between tau and alpha-synuclein proteins is impaired in the presence of p301l tau mutation. Exp Cell Res 2005; 308: 78–84
- Batulan Z, Taylor DM, Aarons RJ, Minotti S, Doroudchi MM, Nalbantoglu J, Durham HD. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis 2006; 24: 213–225
- Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis 2005; 2: 246–254
- Auluck PK, Bonini NM. Pharmacological prevention of parkinson disease in drosophila. Nat Med 2002; 8: 1185–1186
- Auluck PK, Meulener MC, Bonini NM. Mechanisms of suppression of {alpha}-synuclein neurotoxicity by geldanamycin in drosophila. J Biol Chem 2005; 280: 2873–2878
- Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of parkinson's disease. J Mol Biol 2005; 351: 1081–1100
- Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 1996; 111: 345–357
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003; 228: 111–133
- Sullivan W, Stensgard B, Caucutt G, Bartha B, McMahon N, Alnemri ES, Litwack G, Toft D. Nucleotides and two functional states of hsp90. J Biol Chem 1997; 272: 8007–8012
- Neckers L. Heat shock protein 90 inhibition by 17-allylamino-17- demethoxygeldanamycin: A novel therapeutic approach for treating hormone-refractory prostate cancer. Clin Cancer Res 2002; 8: 962–966
- Egorin MJ, Zuhowski EG, Rosen DM, Sentz DL, Covey JM, Eiseman JL. Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (nsc 330507) in cd2f1 mice1. Cancer Chemother Pharmacol 2001; 47: 291–302
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the vhl tumor suppressor proceed through distinct chaperone pathways. Cell 2005; 121: 739–748
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, et al. The high-affinity hsp90-chip complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 2007; 117: 648–658
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med 2002; 8: S55–61
- Tokui K, Adachi H, Waza M, Katsuno M, Minamiyama M, Doi H, Tanaka K, Hamazaki J, Murata S, Tanaka F, et al. 17-dmag ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an sbma model mouse. Hum Mol Genet 2009; 18: 898–910
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273: 501–503
- Cuervo AM, Dice JF. Lysosomes, a meeting point of proteins, chaperones, and proteases. J Mol Med 1998; 76: 6–12
- Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of ikappab kinase (ikk). Cell Res 2006; 16: 895–901
- Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005; 1: 141–145
- Shen S, Zhang P, Lovchik MA, Li Y, Tang L, Chen Z, Zeng R, Ma D, Yuan J, Yu Q. Cyclodepsipeptide toxin promotes the degradation of hsp90 client proteins through chaperone-mediated autophagy. J Cell Biol 2009; 185: 629–639
- Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc Natl Acad Sci U S A 2007; 104: 9511–9516
- Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci 2008; 28: 3384–3391
- Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, Paulson HL, Yang L, Kotin RM, Davidson BL. Rnai suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 2004; 10: 816–820
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL. Rna interference improves motor and neuropathological abnormalities in a huntington's disease mouse model. Proc Natl Acad Sci U S A 2005; 102: 5820–5825
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of sod1 through rna interference retards disease onset and progression in a mouse model of als. Nat Med 2005; 11: 423–428
- La Spada AR, Weydt P. Targeting toxic proteins for turnover. Nat Med 2005; 11: 1052–1053
- Hegde AN, Upadhya SC. The ubiquitin-proteasome pathway in health and disease of the nervous system. Trends Neurosci 2007; 30: 587–595
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003; 40: 427–446
- Jana NR, Zemskov EA, Wang G, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated n-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 2001; 10: 1049–1059
- Seo H, Sonntag KC, Isacson O. Generalized brain and skin proteasome inhibition in huntington's disease. Ann Neurol 2004; 56: 319–328
- Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell 2005; 17: 351–365
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in huntington's disease. Nature 2007; 448: 704–708
- Ding Q, Lewis JJ, Strum KM, Dimayuga E, Bruce-Keller AJ, Dunn JC, Keller JN. Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. J Biol Chem 2002; 277: 13935–13942
- Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J, Lucas JJ. Neuronal induction of the immunoproteasome in huntington's disease. J Neurosci 2003; 23: 11653–11661
- Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ. Impaired ubiquitin-proteasome system activity in the synapses of huntington's disease mice. J Cell Biol 2008; 180: 1177–1189
- Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 2005; 14: 679–691
- Bett JS, Goellner GM, Woodman B, Pratt G, Rechsteiner M, Bates, GP. Proteasome impairment does not contribute to pathogenesis in r6/2 huntington's disease mice: Exclusion of proteasome activator reg{gamma} as a therapeutic target. Hum Mol Genet 2006; 15: 33–44