Abstract
Purpose: We aim to explore the role of macroautophagy in cellular responses to hyperthermia. Protein damage incurred during hyperthermia can either lead to cell death or may be repaired by polypeptide quality control pathways including: (1) the deterrence of protein unfolding by molecular chaperones and (2) proteolysis of the denatured proteins within the proteasome. A third pathway of protein quality control is triggered by formation of protein aggregates in the heat shocked cell. This is the macroautophagy pathway in which protein aggregates are transported to specialised organelles called autolysosomes capable of degrading the aggregates. The consequences for cell viability of triggering this pathway are complex and may involve cell death, although under many circumstances, including exposure of cells to hyperthermia, autophagy leads to enhanced cell survival. We have discussed mechanisms by which cells detect protein aggregates and recruit them into the macroautophagy pathway as well as the potential role of inhibiting this process in hyperthermia.
Conclusions: Directed macroautophagy, with its key role in protein quality control, seems an attractive target for a therapy such as hyperthermia that functions principally through denaturing the proteome. However, much work is needed to decode the mechanisms of thermal stress-mediated macroautophagy and their role in survival/death of cancer cells before recommendations can be made on targeting this pathway in combination with hyperthermia.
Introduction: Hyperthermia, protein quality and cell death
When cells are exposed to elevated temperatures, they are killed in a time- and temperature-dependent manner once a threshold thermal dose is achieved. This has led to the use of elevated temperatures in hyperthermia treatment of a range of cancers Citation[1], Citation[2]. Early studies established cell protein denaturation as the dominant target in thermal cell killing Citation[3]. However, the manner of tumour cell death after hyperthermia appears to be complex. Hyperthermia can lead to the induction of intrinsic programmed cell death pathways such as apoptosis, cause death by mitotic catastrophe and may block cell proliferation by stimulating replicative senescence Citation[4–6]. Initiating triggers for death in heat shocked cells include both (1) induction of physiological cascades and (2) thermal protein unfolding and aggregation Citation[7]. Necrosis can also be observed at extremely elevated temperatures Citation[8]. However, cells also possess homeostatic responses to decrease killing that involve cell cycle arrest and most notably the induction of a transcriptional programme leading to the production of molecular chaperones/heat shock proteins (HSP) Citation[1], Citation[9]. Such molecular chaperones are highly effective at preventing toxic damage to proteins that lead to denaturation and aggregation and can lead to protein refolding Citation[9]. Indeed it is well known that preconditioning cells with a priming heat shock leads to a heat-resistant, thermotolerant state in which cells express extremely elevated levels of HSP Citation[10–12].
The responses of cells to protein damage and unfolding involve multiple metabolic pathways that lead to either: (1) salvation of cells through repair/dismantling of damaged proteins or (2) sacrifice of the cells through the triggering of programmed cell death Citation[13]. Thermally unfolded proteins may be refolded in the cytoplasm by molecular chaperones. However, a second process may also be invoked in which the damaged proteins are broken down by the proteases embedded in the proteasome Citation[13]. Protein aggregates also accumulate in cells during stress as well as in the etiology of protein aggregation disorders such Parkinson's and Alzheimer's disease. Such aggregates compromise cell survival and can lead to apoptosis and loss of cognitive function in neurodegenerative diseases Citation[13]. As protein aggregates are too insoluble and excessively bulky for removal by either chaperone refolding or proteasomal degradation they are instead removed by a process known as autophagy. Autophagy was originally discovered as a component of the nutrient stress response and is described in more detail below (). A number of contrasting autophagy mechanisms have been described, including macroautophagy, microautophagy and chaperone-mediated autophagy Citation[14–17]. We will concentrate here on macroautophagy and its potential role in hyperthermia treatment of cancer. Macroautophagy was originally thought to be a pathway of programmed cell death in complex organisms, and molecular determinants of this process could be observed in dying cells Citation[18]. However, recent studies suggest that under many conditions this process may be beneficial to stressed cells and promote survival Citation[19]. Hyperthermia has been shown to induce macroautophagy under a range of conditions, and in most cases triggering of this pathway was associated with increased cell survival and decreased programmed cell death Citation[15], Citation[20–25]. Indeed, inhibition of macroautophagy with the specific inhibitor bafilomycin may increase the anti-tumour effects of hyperthermia Citation[21]. Clearly then, macroautophagy may play a significant role in tumour cell responses to hyperthermia, and much may be learned by a deeper appreciation of the mechanisms by which hyperthermia triggers this response.
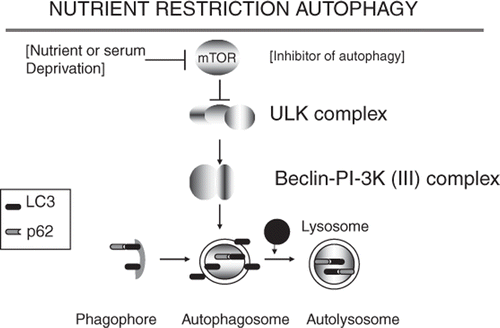
Figure 1. Nutrient restriction autophagy is triggered when the kinase mTOR is inhibited. mTOR inhibition leads to relief of repression of the ULK kinase cascade and formation of a Beclin1/PI-3 K (III) complex that triggers assembly of the phagophore and autophagosome. LC3 is induced and activated to the LC3-II form by conjugation to phosphatidylethanolamine during autophagy. LC3-II stimulates the formation of autophagosome and can bind to the protein aggregate binding protein p62/SQSTM1. Protein aggregates conjugated to p62 and LC3-II are then degraded when the autophagosome becomes associated with lysosomes to form the autolysosome.
Molecular determinants of macroautophagy in stress
Macroautophagy is classically triggered by nutrient stress and is induced when the major repressor of autophagy, the nutrient-sensing protein kinase mTOR becomes inhibited and mRNA translation is stopped Citation[14], Citation[15] (). This sets in motion the macroautophagy cascade and permits the cell to digest the proteins, lipids and DNA trapped in the autophagosome permitting liberation of nutrients for short-term survival of the cells. The pathway of macroautophagy, deduced originally in studies on yeast and Caenorhabditis elegans, is shown in . Inhibition of mTOR leads to dephosphorylation of the multiprotein ULK1 kinase complex, the gateway step in starvation-mediated autophagy Citation[26], Citation[27]. Although the exact targets of the ULK1 kinase complex are not clear, its dephosphorylation leads to the activation of the Beclin1/PI-3 kinase (III) complex, a step that initiates macroautophagy by generating highly phosphorylated phosphoinositide domains in membranes, thus permitting assembly of autophagy proteins into vesicles built from lipids derived from the Golgi Citation[28]. Open autophagophore structures are thus generated which can then be further processed into the double-membrane enclosed autophagosome in which protein aggregates can be packaged. Autophagosomes then fuse with intracellular lysosomes and the protease repertoire of the lysosome can be employed to digest the protein aggregates Citation[14]. One important factor in regulated macroautophagy is the ubiquitin-like protein atg-8 (also known as LC-3/GABARAP) Citation[15], Citation[29]. LC-3 is found attached to the internal and external membranes of autophagosomes and the internal membranes of autolysosomes Citation[29]. Such LC3, when processed to its active form LC3-II can be detected and bound by the proteins p62/SQSTM1 and NBR1. These proteins can also recognise ubiquitinated proteins and aggregates as well as LC-3 in the autophagosome Citation[30], Citation[31].
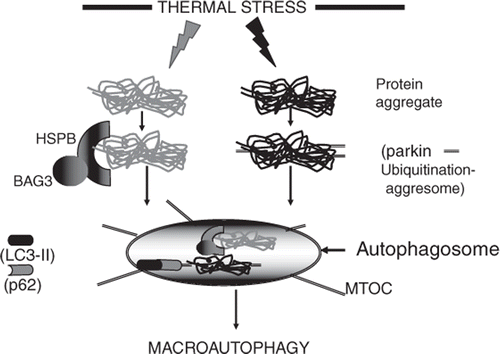
In addition to nutrient deprivation, cell stresses can also trigger macroautophagy. Such stress-induced macroautophagy was originally thought to be a pathway of programmed cell death after stress and indeed can be triggered simultaneously with apoptosis through common intermediates such as Bcl2 family proteins and p53 Citation[32–34]. These stress-mediated pathways generally intercede at the Beclin1 step defined in nutrient stress macroautophagy (). Much recent work indicates that stress-induced macroautophagy can, however, be cytoprotective and may moderate the effects of pro-apoptotic signalling Citation[18], Citation[35]. One way in which macroautophagy can increase cell survival is through the resolution of intracellular protein aggregates within the autolysosome. As mentioned above, cells can resolve denatured/damaged proteins by refolding them with their batteries of molecular chaperones or by degrading such polypeptides through the proteasome Citation[13]. However, these two processes are inefficient in dealing with protein aggregates as the pore of the proteasome is occluded by bulky protein aggregates Citation[13]. Hyperthermia leads to the accumulation of a large fraction of polyubiquitinated, aggregated proteins in cells Citation[36], Citation[37]. Protein aggregates can, however, be resolved after becoming ubiquitinated on multiple sites after interaction with the ubiquitin ligase parkin Citation[30], Citation[38] (). Aggregates marked by polyubiquitination can then interact with the motor protein dynein and histone deacetylase 6 to form aggresomes that are retrotransported within the cell to the microtubule organising centre (MTOC) adjacent to the nucleus Citation[30], Citation[39]. The aggresomes can then become enclosed in autophagosomes at the MTOC. It is thought that the protein p62/SQSTM1 may play a key role in this process, based on its ability to bind both UBL domain protein LC-3 and the polyubiquitinated proteins in the aggresome through its UBA domain Citation[30]. It can thus function as the ‘missing link’ between protein aggregation and macroautophagy. Interestingly, p62/SQSTM1 becomes consumed along with its polyubiquitinated client proteins within the autolysosome Citation[14], Citation[30]. Sorting of proteins targeted for degradation to the proteasome or autophagosome involves the nature of the binding site on ubiquitin; K-48 linked polyUb chains are in general recognised by proteasome receptors while K-63 linked chains are targeted to autophagy. The complexities of this process are described in a recent mini-review Citation[40].
Figure 2. Triggering of the macroautophagy response by stress induced protein aggregates. At least two pathways may be involved in sensing the presence of aggregates and directing them to the autophagosome. In the grey pathway, aggregates are sensed/bound by HSPB/BAG3 complexes and directed to the autophagosome. In the black pathway, aggregates are recognised by the ubiquitin E3 ligase parkin that sees aggregated proteins, polyubiquitinates them and leads to the formation of the aggresome. Aggresomes are translocated to the autophagosome located at the microtubule organising centre (MTOC). The polyubiquitin chains on the aggresome are then recognised by p62 which interacts with LC3 and triggers autophagy. Responses to stresses such as hyperthermia may involve a combination of the two pathways.
Heat shock triggering of macroautophagy
The next question is: how are the protein aggregates that are generated during hyperthermia sensed by the cell and targeted to the macroautophagy pathway? It has been shown that formation of protein aggregates can be induced by physiological stimuli. The stress-induced kinase MEKK1 leads to the nucleation of protein aggregates and can itself initiate the aggregation cascade Citation[41]. MEKK1 was found in the same insoluble fractions as aggregated proteins suggesting formation of complexes. MEKK1 may thus form part of the ‘aggregation sensor’.
It has also been shown that molecular chaperones can mediate the formation of the autophagosome. Heat shock protein 70 (Hsp70) and Hsp90, although contributing to a process known as chaperone-mediated autophagy, do not appear to play key roles in stress-induced macroautophagy Citation[13], Citation[17]. By contrast the small HSP (HSPB) family appear to play key roles in macroautophagy (). HspB8 forms a complex with the Hsp70 co-chaperone BAG3 that can prevent protein aggregate formation, increase the levels of LC3-II and stimulate macroautophagy Citation[42], Citation[43]. This complex is distinct from Hsp70-BAG3 complexes and binding to HspB8 involves dedicated domains within the BAG3 protein distinct from the Hsp70 binding site Citation[43]. It seems likely that HspB8-BAG3 complexes could thus target aggregated proteins to the autophagosome and stimulate macroautophagy. Interestingly, BAG3 inhibits Hsp70-mediated proteasomal degradation and may thus bias traffic of aggregates away from the proteasome and towards the autophagosome Citation[44], Citation[45]. More recently, a role for other members of the HSPB family have been discerned and HspB7 has been shown to be highly effective in preventing the toxicity of protein aggregates through the macroautophagy pathways Citation[46], Citation[47]. In contrast to HspB8, HspB7 appears to accomplish this without increasing the rate of macroautophagy and may utilise basal autophagy processes in resolution of aggregates. It should be noted that many of the experiments quoted here involve forced expression of aggregation-prone mutant proteins in cells rather than heat shock itself and it remains to be fully discerned how similar these processes are.
It is currently unclear how stress-induced aggregation is coupled to macroautophagy. It is possible that the presence of aggregated proteins might be detected by HSPB family molecules through their molecular chaperone properties, bound and delivered to the autophagosome. Such a mechanism would be homologous to the role of Hsp70 in sensing denatured proteins, coupling to the ubiquitin-E3 ligase CHIP and delivery of polyubiquitinated, denatured proteins to the proteasome for digestion Citation[48]. In each case a molecular chaperone is used to bind disordered proteins and deliver them to a pathway of destruction. This illustrates the versatility of the HSP in detecting protein unfolding and coupling denaturation to other cellular processes such as proteasome function and macroautophagic degradation generally through association with co-chaperones. It will be interesting to determine whether other enzymatic activities can be thus coupled to protein unfolding through molecular chaperones. One interesting possibility is the scaffold protein TTC5/STRAP that can bind Hsp90 as well as histone acetylases and other molecules active in transcriptional regulation Citation[49], Citation[50].
In addition to being detected by HSP, stress-induced aggregates may be sensed and polyubiquitinated by ligases such as parkin, packaged into aggresomes and delivered to autophagosomes by p62, or a combination of mechanisms may be involved Citation[39] (). Much remains to be learned regarding the interplay between the multiple arms of the stress response, the relative role of molecular chaperones or aggresomes. As mentioned above, members of the BAG family of co-chaperones may be involved in interplay between the various pathways as they can influence transcription of the HSP cohort, target Hsp70-CHIP complexes to the proteasome and promote interaction of HspB8 with the macroautophagy pathway Citation[45], Citation[51]. It is also unclear how the stress kinase MEKK1 is coupled to the response to protein aggregation Citation[41]. MEKK1 is a high molecular weight protein kinase that possesses ubiquitin ligase activity as well as kinase function and these properties could be involved in interaction with protein aggregates Citation[52].
Downstream of stress signalling after hyperthermia – Apoptosis or macroautophagy, life or death?
It is apparent that stress produces a number of effects on protein homeostasis that may compete in order to repair stress induced protein damage or lead to apoptosis. Pro-death pathways include stress-kinase mediated apoptosis as well as apoptosis due to accumulation of unrepaired protein aggregates Citation[7]. In addition, heat stress triggers the HSF1-mediated stress response as well as the activation of small heat shock protein HspB1 through the p38MAPK-signaling pathway each of which process leads to inhibition of the death pathways and enhanced folding of the proteome Citation[53], Citation[54]. Macroautophagy may be another component in this stress response network with its unique ability to target protein aggregates for degradation by lysosomal proteases. Indeed, it will be a challenge in the coming years to understand how the various arms of the stress response are coordinated in order to promote cell survival.
Macroautophagy and cancer treatment
Macroautophagy plays a somewhat ambiguous role in responses of tumour cells to some cancer therapies. This pathway may be triggered by some therapeutics in tumour cells harbouring mutations that inhibit the caspase-dependent apoptosis pathway Citation[55]. Indeed in such cells, macroautophagy has been described as the programmed death type II pathway, a default mechanism that permits cell death in the absence of the classical apoptosis cascade Citation[19]. However, in many contexts macroautophagy constitutes a pro-survival pathway, particularly in the acidic and hypoxic tumour microenvironment Citation[56]. Most of the available studies on hyperthermia suggest that autophagy reduces the cytotoxic effects of the therapy and may be a potential target in combination therapies Citation[21–24]. Autophagy can be inhibited by chemicals such as 3-methyladenine, hydroxychloroquine or bafilomycin A1 or reduced by targeting members of the autophagy-related gene family (ATG5, ATG6 (Beclin1), ATG10 and ATG12), by siRNA approaches Citation[19]. Combining inhibition of macroautophagy with hyperthermia by one of these approaches may thus be a promising approach to explore.
In conclusion therefore, directed macroautophagy, with its key role in protein quality control, seems a particularly attractive target in terms of a therapy such as hyperthermia that functions principally through denaturing the proteome Citation[57]. However, much work is needed on the mechanisms of thermal stress-mediated macroautophagy and its role in survival/death of cancer cells before recommendations can be made on targeting this pathway in combination with hyperthermia.
Acknowledgements
We thank the Department of Radiation Oncology, BIDMC, Harvard Medical School for continuous support and encouragement.
Declaration of interest: This work was supported by NIH research grants R01CA119045, R01CA047407, R01CA094397. The authors alone are responsible for the content and writing of the paper.
References
- Hahn GM. Hyperthermia and Cancer. Plenum Press, New York 1982
- van der Zee J, Gonzalez Gonzalez D, van Rhoon GC, van Dijk JD, van Putten WL, Hart AA. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: A prospective, randomised, multicentre trial. Dutch Deep Hyperthermia Group. Lancet 2000; 355: 1119–1125
- Westra A, Dewey WC. Heat shock during the cell cycle of Chinese hamster ovary cells in vitro. Int J Radiat Biol 1971; 19: 467–477
- Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY. Role of Hsp70 in regulation of stress-kinase JNK: Implications in apoptosis and aging. FEBS Lett 1998; 438: 1–4
- Gabai VL, Zamulaeva IV, Mosin AF, Makarova YM, Mosina VA, Budagova KR, Malutina YV, Kabakov AE. Resistance of Ehrlich tumor cells to apoptosis can be due to accumulation of heat shock proteins. FEBS Lett 1995; 375: 21–26
- Vidair CA, Doxsey SJ, Dewey WC. Thermotolerant cells possess an enhanced capacity to repair heat-induced alterations to centrosome structure and function. J Cell Physiol 1995; 163: 194–203
- Bellmann K, Charette SJ, Nadeau PJ, Poirier DJ, Loranger A, Landry J. The mechanism whereby heat shock induces apoptosis depends on the innate sensitivity of cells to stress. Cell Stress Chaperones 2010; 15: 101–113
- Mambula SS, Calderwood SK. Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int J Hyperthermia 2006; 22: 575–585
- Lindquist S, Craig EA. The heat shock proteins. Ann Rev Genet 1988; 22: 631–637
- Landry J, Bernier D, Chretien P, Nicole LM, Tanguay RM, Marceau N. Synthesis and degradation of heat shock proteins during the development and decay of thermotolerance. Cancer Res 1982; 42: 2457–2461
- Li GC, Werb Z. Correlation between the synthesis of heat shock proteins and the development of thermotolerance in CHO fibroblasts. Proc Natl Acad Sci USA 1982; 79: 3218–3222
- Subjeck JR, Sciandra JJ, Johnson RJ. Heat shock proteins and thermotolerance: A comparison of induction kinetics. Br J Radiol 1982; 55: 579–584
- Calderwood SK, Murshid A, Prince T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging – A mini-review. Gerontology 2009; 55: 550–558
- Klionsky DJ. The autophagy connection. Dev Cell 2010; 19: 11–12
- Yang Y, Xing D, Zhou F, Chen Q. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem Biophys Res Commun 2010; 395: 190–195
- Cuervo AM. Autophagy and aging: Keeping that old broom working. Trends Genet 2008; 24: 604–612
- Dice JF. Chaperone-mediated autophagy. Autophagy 2007; 3: 295–299
- Levine B, Yuan J. Autophagy in cell death: An innocent convict?. J Clin Invest 2005; 115: 2679–2688
- Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010; 6: 322–329
- Chen F, Wang CC, Kim E, Harrison LE. Hyperthermia in combination with oxidative stress induces autophagic cell death in HT-29 colon cancer cells. Cell Biol Int 2008; 32: 715–723
- Komata T, Kanzawa T, Nashimoto T, Aoki H, Endo S, Nameta M, Takahashi H, Yamamoto T, Kondo S, Tanaka R. Mild heat shock induces autophagic growth arrest, but not apoptosis in U251-MG and U87-MG human malignant glioma cells. J Neurooncol 2004; 68: 101–111
- Krmpot AJ, Janjetovic KD, Misirkic MS, Vucicevic LM, Pantelic DV, Vasiljevic DM, Popadic DM, Jelenkovic BM, Trajkovic VS. Protective effect of autophagy in laser-induced glioma cell death in vitro. Lasers Surg Med 2010; 42: 338–347
- Liu TT, Hu CH, Tsai CD, Li CW, Lin YF, Wang JY. Heat stroke induces autophagy as a protection mechanism against neurodegeneration in the brain. Shock 2008; 34: 643–648
- Swanlund JM, Kregel KC, Oberley TD. Autophagy following heat stress: The role of aging and protein nitration. Autophagy 2008; 4: 936–939
- Zhao Y, Gong S, Shunmei E, Zou J. Induction of macroautophagy by heat. Mol Biol Rep 2009; 36: 2323–2327
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–1991
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19: 5360–5372
- Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008; 19: 4762–4775
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34: 259–269
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12: 213–223
- Boya P, Kroemer G. Beclin 1: A BH3-only protein that fails to induce apoptosis. Oncogene 2009; 28: 2125–2127
- Zhang XD, Qin ZH, Wang J. The role of p53 in cell metabolism. Acta PharmacolSin 2010; 9: 1208–1212
- Zhang XD, Wang Y, Wu JC, Lin F, Han R, Han F, Fukunaga K, Qin ZH. Down-regulation of Bcl-2 enhances autophagy activation and cell death induced by mitochondrial dysfunction in rat striatum. J Neurosci Res 2009; 87: 3600–3610
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008; 4: 600–606
- Friant S, Meier KD, Riezman H. Increased ubiquitin-dependent degradation can replace the essential requirement for heat shock protein induction. EMBO J 2003; 22: 3783–3791
- Salomons FA, Menendez-Benito V, Bottcher C, McCray BA, Taylor JP, Dantuma NP. Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol Cell Biol 2009; 29: 1774–1785
- Chin LS, Olzmann JA, Li L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem Soc Trans 2010; 38: 144–149
- Simms-Waldrip T, Rodriguez-Gonzalez A, Lin T, Ikeda AK, Fu C, Sakamoto KM. The aggresome pathway as a target for therapy in hematologic malignancies. Mol Genet Metab 2008; 94: 283–286
- Clague MJ, Urbe S. Ubiquitin: Same molecule, different degradation pathways. Cell, 143: 682–685
- Meriin AB, Mabuchi K, Gabai VL, Yaglom JA, Kazantsev A, Sherman MY. Intracellular aggregation of polypeptides with expanded polyglutamine domain is stimulated by stress-activated kinase MEKK1. J Cell Biol 2001; 153: 851–864
- Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet 2005; 14: 1659–1669
- Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem 2009; 284: 5523–5532
- Doong H, Rizzo K, Fang S, Kulpa V, Weissman AM, Kohn EC. CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: Accumulation of poly-ubiquitinated Hsp90 client proteins. J Biol Chem 2003; 278: 28490–28500
- McCollum AK, Casagrande G, Kohn EC. Caught in the middle: The role of Bag3 in disease. Biochem J 2009; 425: e1–3
- Vos MJ, Zijlstra MP, Carra S, Sibon OC, Kampinga HH. Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy 2011; 7: 101–103
- Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM, Carra S, Sibon OC, Kampinga HH. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum Mol Genet 2010; 19: 4677–4693
- Calderwood SK. Molecular chaperones and the ubiquitin proteasome system in aging. The Ubiquitin Proteasome System in the Central Nervous System, M Di Napoli, C Wojcik. Nova, New York 2007; 537–552
- Xu D, La Thangue NB. Strap: A versatile transcription co-factor. Cell Cycle 2008; 7: 2456–2457
- Xu D, Zalmas LP, La Thangue NB. A transcription cofactor required for the heat-shock response. EMBO Rep 2008; 9: 662–669
- Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006; 440: 551–555
- Enzler T, Chang X, Facchinetti V, Melino G, Karin M, Su B, Gallagher E. MEKK1 binds HECT E3 ligase Itch by its amino-terminal RING motif to regulate Th2 cytokine gene expression. J Immunol 2009; 183: 3831–3838
- Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 1996; 18: 567–577
- Wu C. Heat shock transcription factors: Structure and regulation. Ann Rev Cell Dev Biol 1995; 11: 441–469
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2: 897–909
- Rouschop KM, Wouters BG. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med 2009; 9: 417–424
- Ritchie KP, Keller BM, Syed KM, Lepock JR. Hyperthermia (heat shock)-induced protein denaturation in liver, muscle and lens tissue as determined by differential scanning calorimetry. Int J Hyperthermia 1994; 10: 605–618